
Abstract
AMG X, a human neutralizing monoclonal antibody (mAb) against a soluble human protein, caused thrombocytopenia, platelet activation, reduced mean arterial pressure, and transient loss of consciousness in cynomolgus monkeys after first intravenous administration. In vitro, AMG X induced activation in platelets from macaque species but not from humans or baboons. Other similar mAbs against the same pharmacological target failed to induce these in vivo and in vitro effects. In addition, the target protein was known to not be expressed on platelets, suggesting that platelet activation occurred through an off-target mechanism. AMG X bound directly to cynomolgus platelets and required both the Fab and Fc portion of the mAb for platelet activation. Binding to platelets was inhibited by preincubation of AMG X with its pharmacological target or with anti-human Fc antibodies or by preincubation of platelets with AMG X F(ab′)2 or human immunoglobulin (IVIG). AMG X F(ab′)2 did not activate platelets. Thus, platelet activation required both recognition/binding of a platelet ligand with the Fab domain and interaction of platelet Fc receptors (i.e., FcγRIIa) with the Fc domain. These findings reflect the complexity of the mechanism of action of mAbs and the increasing awareness of potential for unintended effects in preclinical species.
Introduction
Acute thrombocytopenia mediated by antibodies is a common adverse effect of medications. Antibody-mediated platelet destruction due to drug administration has been definitively demonstrated for approximately 50 marketed compounds (both small and large molecules) and is likely for another 15 molecules (George et al. 1998). Both preexisting and newly formed endogenous antibodies are implicated in immune destruction of platelets.
There are six major mechanisms by which drug-related antibodies mediate platelet destruction, as described by Aster (2007, used by permission), including (1) drug binds covalently to platelet membrane glycoprotein and acts as hapten (e.g., penicillin); (2) drug binds noncovalently to platelet membrane glycoprotein and produces either a compound epitope or induces a conformational change that is antigenic (e.g., quinidine); (3) drug induces autoantibodies that do not require the presence of a drug to bind to platelets (e.g., gold salts); (4) drug binds to a normal protein associated with platelets to form immunogenic complexes, and antibodies against these protein-platelet complexes form immune complexes that activate platelet (e.g., heparin); (5) drug binds to the platelet fibrinogen receptor, which induces conformational changes that are recognized by the antibody (e.g., eptifibatide); and (6) drug binds to its intended target on platelets and is recognized by antibodies that bind to drug-coated platelets (e.g., abciximab). Although drug-induced immune thrombocytopenia usually occurs after multiple drug exposures, it also can occur after a first dose due to naturally occurring antibodies. In addition, use of targeted antiplatelet agents, such as ticlopidine hydrochloride (Ticlid) and clopidogrel bisulphate (Plavix), are associated with thrombotic thrombocytopenia purpura, characterized by platelet aggregation/activation and thrombocytopenia (Zakarija et al. 2009; Mangalpally and Kleiman 2011).
Large molecules most frequently associated with thrombocytopenia include heparin and abciximab. Heparin-induced thrombocytopenia occurs when antibodies bind to noncovalent complexes of platelet factor 4 and heparin on the platelet surface, creating immune complexes that activate platelet via their FcγRIIa receptors (Warkentin 2004). Abciximab, which binds the platelet glycoprotein IIIa to prevent thrombosis, results in thrombocytopenia when antibodies bind a specific murine sequence of abciximab after the drug has bound to platelets (Aster 2007).
Monoclonal antibodies (mAbs) are a major class of successful biologic therapeutics and are generally well tolerated in preclinical species and humans. Rare adverse events are usually specifically related to the targeted molecule or pathway (Kamba and McDonald 2007). The administration of mAbs has also been associated with infusion reactions, which can occur immediately following administration and include symptoms such as fever, fainting, flushing, and itching (Lenz 2007). The underlying causes postulated for infusion reactions include syndromes such as cytokine release (Suntharalingam et al. 2006), anaphylactic (Type IV sensitivity) or anaphylactoid reactions, and antidrug antibody–mediated reactions (Type III hypersensitivity; Winkler et al. 1999; Baert et al. 2003; Descotes and Gouraud 2008; Peeters, Balfour, and Arnold 2008; Corren et al. 2009).
Effects of mAbs on platelets, described as potentially off-target, have been infrequently reported in the literature. Mild to severe decreases in platelets as off-target effects were observed during nonclinical in vivo testing of mAbs against IL-13, IgE, sclerostin, and an unspecified cell-based target (“Omalizumab” 2002; Martin et al. 2008; Everds et al. 2011; Rudmann et al. 2011). Some mAbs developed for platelet research directly bind and activate platelets in a manner that requires both the Fc and Fab portion of the mAb (Horsewood et al. 1991). The Fab domain binds to its cognate antigen, and the Fc domain can bind to the IgG receptor FcγRIIa (CD32) on the same or adjacent platelets (Slupsky et al. 1992; Perutelli and Mori 1993). These studies demonstrate that mAbs can act as platelet agonists, causing outside-in signaling and recruitment of additional proactivation pathways.
Platelets can be activated by agonists of a wide range of membrane proteins. Activation of platelets generally results in downstream synergistic and autocrine amplification of activation pathways. During activation, platelets secrete the contents of dense bodies and alpha granules. Among other functions, the contents of dense bodies (i.e., adenosine diphosphate [ADP] and serotonin) serve to amplify activation and constrict blood vessels, respectively. Contents of alpha granules serve to amplify interaction and adhesion among platelets, the vasculature, and leukocytes. P-selectin (CD62P) is translocated to the platelet membrane after activation and serves to tether platelets. Agonists such as ADP, thrombin, or phorbol 12-myristate 13-acetate (PMA) induce platelet activation and aggregation in vitro. Platelet activation causes a conformational change of αIIbβ3 (CD41/CD61; glycoprotein [GP] IIb/IIIa), the fibrinogen receptor, by inside-out signaling. The altered conformation changes the affinity of αIIbβ3 from low to high, allowing stabilization and growth of the platelet aggregate by binding of recognition domain–containing glycoproteins such as fibrinogen, von Willebrand factor, vitronectin, or fibronectin. Platelet activation is assessed by activation-specific conformational changes in αIIbβ3 (detected by the first procaspase activating compound [PAC-1] antibody; Shattil 1995) and surface CD62P (Schlossmann and Neupert 1995) using flow cytometry. Platelet aggregation is assessed in vitro by evaluating decreases in turbidity of a platelet suspension or by increases in electrical impedance caused by clot formation (Born 1962; Michelson et al. 2009).
Platelet activation in vivo can result in systemic cardiovascular events primarily because of release of serotonin from dense bodies and by activation of the complement and coagulation cascades. Activated platelets are rapidly removed from circulation, likely because of sequestration in vascular beds and phagocytosis by neutrophils. Expression of membrane CD62P and phosphatidyl serine are thought to be important signals in removal of platelets from circulation (Maugeri et al. 2009).
Platelets of humans and nonhuman primates have similar surface molecules. Although not many amino acid sequences have been reported for nonhuman primate platelet receptors, antiplatelet drugs developed for humans are generally pharmacologically active in nonhuman primates. In addition, numerous research reagents against surface molecules on human platelets cross-react with those of cynomolgus monkeys and other nonhuman primates.
In a nonclinical toxicity study, AMG X, a human neutralizing mAb directed against a soluble human plasma protein, not expressed on platelets, was administered once weekly for 5 wk to cynomolgus monkeys (N = 5/sex/group) by subcutaneous (SQ) injection at dose levels of 0 (vehicle control), 30, 100, or 300 mg/kg or by intravenous (IV) bolus injection at 300 mg/kg. AMG X is pharmacologically active against the target from both humans and cynomolgus monkeys with similar subnanomolar potency. On the first day of dosing, 5 of the 10 animals at 300 mg/kg IV fainted/lost consciousness during or soon after dosing. No other fainting episodes occurred on subsequent days of IV or on any day of SQ administration, including day 1. Clinical pathology at 7 days after the fourth dose and prior to the fifth dose indicated decreased platelet counts (means were ∼70–75% of concurrent vehicle control means), and the presence of large platelets was noted in all groups dosed with AMG X. No other AMG X–related changes were noted in any animal on study at day 30, and there were no histological changes in megakaryocytes after four weekly doses of AMG X. All animals tested negative for anti–AMG X antibodies prior to dosing and during the 5-wk treatment period. Because of high serum AMG X concentrations noted within the 5-wk treatment period, the immunoassay’s ability to detect anti–AMG X binding antibodies may have been impaired; however, there was no impact on the toxicokinetic profile in these animals. Based on these observations, additional in vivo and in vitro studies were conducted to understand the mechanism for AMG X–mediated platelet decreases and fainting.
Results from these studies demonstrated that AMG X activated macaque platelets by a mechanism unrelated to its pharmacological target. In addition, AMG X activation and aggregation were specific to macaque platelets and were not observed in baboon or human platelets. The studies also demonstrated that AMG X bound directly to cynomolgus platelets and that AMG X–induced activation of platelets required interactions through both the Fc and Fab domains.
Materials and Methods
Chemicals and Reagents
AMG X, AMG A, AMG B, and AMG C are xenomouse-derived human monoclonal antibodies (IgG2) against the same soluble human plasma protein (“target”). Compared with AMG X, these three mAbs varied in primary amino acid sequences in the variable regions but had comparable potency in cell-based assays and competed with AMG X (and with each other) for binding to the target (data not shown). Anti-target mAbs (70 mg/mL) and the isotype-matched control mAb, antistreptavidin (anti-SA; 40 mg/mL), were supplied in a frozen liquid formulation in the vehicle control material (9% [w/v] sucrose solution, buffered with 10 mM sodium acetate, adjusted to pH 5.2, and stabilized with 0.004% [w/v] polysorbate 20).
ADP (cat. No. 384; Chronolog, Havertown, PA), recombinant human thrombin (cat. No. 27-0846-01; Amersham Biosciences, Arlington Heights, IL), ristocetin (cat. No. 396; Chronolog, Havertown, PA), and PMA (cat. No. P1585; Sigma, St. Louis, MO) were purchased from commercial sources. AMG X-F(ab′)2 was generated by pepsin digestion using standard procedures (Andrew and Titus 2003).
Antibodies
CD41a-APC (cat. No. 559777; mouse anti-human GPIIb) and CD62P-PE (cat. No. 550561; mouse anti-human P-selectin) were purchased from BD Biosciences (San Jose, CA). According to the manufacturer, these antibodies cross-react with cynomolgus, rhesus, pigtail, and baboon GPIIb and P-selectin, respectively. Anti-human IgG-FITC (A80-319F) was purchased from Bethyl Laboratories (Montgomery, TX). Intravenous immunglobulin (IVIG) was purchased from Octapharma AG (cat. No. A6120278431, A622C8431, A625A8431, 4190358431, 4250498431, A624B8431, A625B8431, A626D8431, A626E8431; Lachen, Switzerland).
Animals and Husbandry
Chinese origin male cynomolgus monkeys (Macaca fascicularis) 2 to 6 years old (2.1 to 6.2 kg) were cared for in accordance with the Guide for the Care and Use of Laboratory Animals (Institute for Laboratory Animal Resources [ILAR] publication, 1996, NRC Press). Animals were housed at an indoor, Association for Assessment and Accreditation of Laboratory Animal Care International–accredited facility in species-specific housing. All study protocols and animal housing were approved by the Institutional Animal Care and Use Committee (IACUC).
Certified Primate Diet (PMI No. 5048; Richmond, IN) was provided daily in amounts appropriate for the age and size of the animals. Tap water was available ad libitum to each animal via an automatic watering device. The animals were given additional supplements as a form of environmental enrichment and were given various cage-enrichment devices. Animals were maintained on a 12:12 h light:dark cycle in rooms at 18°C to 29°C and relative humidity of 30% to 70%. In cardiovascular assessment telemetry studies, animals were naive to large molecules; general toxicity studies used naive animals. All animals were negative for simian retrovirus (SRV).
Investigative Study Design
Three single-dose investigative studies were conducted in male cynomolgus monkeys. Although no gender differences in systemic exposure to AMG X were noted in the 5-wk study, males were chosen for subsequent studies based on the higher incidence of transient loss of consciousness.
In the first study, male cynomolgus monkeys received a single dose of the vehicle control article (n = 2) or AMG X (n = 4-5) at 30, 100, or 300 mg/kg via SQ injection (interscapular region). Additional cynomolgus monkeys received a single IV bolus injection (saphenous vein) of the vehicle control (n = 2) or AMG X at 15 (n =1), 100 (n = 5), or 300 mg/kg (n = 9). The animals were evaluated for clinical signs, changes in body weight, clinical pathology parameters, serum drug concentrations, complement analysis, and platelet parameters as described below.
In a second study, male cynomolgus monkeys received the vehicle control or AMG A, AMG B, or AMG C (n = 2) at 100 mg/kg via IV bolus injection (saphenous vein). The animals were evaluated for clinical signs, changes in body weight, clinical pathology parameters, serum drug concentrations, and platelets.
In a cardiovascular study, telemeterized male cynomolgus monkeys received the vehicle control article on day 1 via SQ (n = 4) or IV (n = 6) bolus administration. On day 3 (IV) or day 8 (SQ), the same monkeys received a single-dose administration of AMG X at 300 mg/kg via SQ or IV bolus administration. The animals were evaluated for clinical signs and cardiovascular parameters for up to 24 h (IV) or 73 h (SQ) after dosing.
In all studies, the vehicle control and AMG X, AMG A, AMG B, or AMG C were administered at a dose volume of 4.3 mL/kg, and the duration of dosing was approximately 2 to 3 min.
Blood Sampling
In the first single-dose study with AMG X, blood for hematology and specialized testing (platelet aggregation and complement activation) was collected predose (week –1) and at 0.083 and 0.25 (5 and 15 min, respectively, IV route only), 8, 12 (SQ route only), 24, 36 (SQ route only), 72, and 168 h postdosing via venipuncture of a peripheral vein (femoral, saphenous, or cephalic). Complement (C3a and C5a) was evaluated predose and at 0.083, 0.25, 8, 36, and 168 h postdosing by the IV route.
In the second single-dose study, blood was collected for hematology at predose (week –1) and at 0.25, 8, 24, 72, and 168 h after the vehicle control, AMG A, AMG B, or AMG C dose.
Hematology
Blood was collected from vehicle control– and test article–treated monkeys into a tube containing EDTA. The following hematology parameters were measured using a Bayer Advia 120 Automated Hematology Analyzer (Siemens Healthcare, Deerfield, IL): red blood cell (RBC) count, hemoglobin concentration, hematocrit, mean corpuscular volume (MCV), mean corpuscular hemoglobin concentration, mean corpuscular hemoglobin, red cell distribution width, reticulocyte count, platelet count, mean platelet volume, platelet crit (PCT), mean platelet component, platelet component distribution width, mean platelet dry mass, and white blood cell counts, including automated differentials. Blood smears were evaluated microscopically for platelet morphology.
Bioanalytical, Toxicokinetic, and Dynamic Analyses
In the first single-dose study, blood (0.5-1 mL each) was collected from control- and test article–treated cynomolgus monkeys from all animals at predose and at approximately 1, 3, 8, 12, 24, 36, 72, and 168 h postdose for SQ groups and at predose and at approximately 0.25, 1, 3, 8, 24, 72, and 168 h postdose for IV groups. In the second single-dose study, blood was collected at predose and at 0.25, 8, 24, 36, 72, and 168 h postdose for animals dosed with AMG A, AMG B, and AMG C. Blood samples were processed to serum by centrifugation and stored frozen at –60°C or colder until analysis. Drug concentration was determined according to the validated analytical procedure by electrochemiluminescent immunoassay. The nominal assay range was 50.0 to 50,000 ng/mL, and the assay lower limit of quantification was 50.0 ng/mL.
Individual concentration-time data were analyzed with noncompartmental methods using WinNonlin (Enterprise Version 5.1.1, 2006, Pharsight Corp, Mountain View, CA) within the Pharsight Knowledgebase Server (PKS, v.3.1a, 2006). Individual animal platelet counts as a percentage of predose and time-matched postdose mean control group data were plotted against serum AMG X concentrations to explore the correlation between toxicokinetics and dynamics. A sigmoidal Emax pharmacodynamic model was used to quantify the relationship between individual platelet count change from baseline and serum AMG X concentration. E, E0, and Emax are the observed, baseline, and maximum pharmacodynamic effects (E), respectively; C is the corresponding measured serum AMG X concentration, and γ is an exponent describing the number of drug molecules that combine with each target receptor:
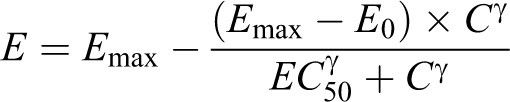
Complement Analysis
Approximately 0.7 mL of whole blood (WB) from cynomolgus monkeys was collected in a tube containing EDTA and processed to plasma. Plasma was shipped frozen to the Complement Laboratory of the National Jewish Center for Immunology (Denver, CO) and analyzed for C5a by a competitive radioimmunoassay and C3a by enzyme-linked immunosorbent assay as described in Henry et al. (1997).
Cardiovascular Assessment in Telemetered Conscious Cynomolgus Monkeys
Surgical procedures were performed using standard aseptic techniques and under general anesthesia, and animals received appropriate postoperative veterinary care, including appropriate analgesia. Male cynomolgus monkeys were implanted with an electrocardiogram (ECG) and pressure transmitter (model No. TL11 M2-D70-PCT; Data Sciences International, Minneapolis, MN) into the abdomen and sutured to the abdominal wall. The ECG leads of the transmitter were arranged in an approximate Lead II configuration. A pressure catheter was placed in an artery and advanced into the abdominal aorta. The animals were implanted (>2 wk) prior to study initiation and acclimated to the study room for at least 2 wk prior to dosing. A group of six cynomolgus monkeys received a single-dose of the vehicle control (day 1) and then AMG X (day 3 or 8) as described above. On each dosing day, arterial blood pressure, heart rate, ECG, and intra-abdominal body temperature were recorded for at least 90 min prior to dosing, during dose administration, and continuously for at least 7 h after dosing. After the end of the continuous data collection, telemetry data were recorded for at least the last 15 min of every hour through at least 24 h (IV) or 73 h (SQ) postdose.
Cardiovascular endpoints, including arterial blood pressures, heart rate, and ECG parameters (QT interval, corrected QT interval, PR interval, and QRS duration), were measured pretest (baseline) and following dosing; clinical signs were also monitored during these times. Data Sciences International (DSI, Minneapolis, MN) Dataquest OpenART telemetry equipment was used to generate and acquire the telemetry data input, and the data were analyzed with PONEMAH (P3P; Ponemah Physiology Platform-Plus, Cleveland, OH) software.
In Vitro Experimental Plan
Phlebotomy
Blood was collected from nonhuman primates and human donors who were not exposed to aspirin, ibuprofen, or other anti-inflammatory analgesics in the preceding 7 d. Phlebotomy was conducted using methods to reduce clotting (i.e., using a 19-gauge or 21-gauge needle, light tourniquet technique, and slow flow into Beckton-Dickinson BD Vacutainer [San Jose, CA] tubes containing anticoagulant and discarding the first 2–5 mL of blood collected). The anticoagulant was acid citrate dextrose for activation studies and 3.2% sodium citrate for aggregation studies. Blood samples were used within 2 to 4 h of collection.
Preparation of Platelet-Rich Plasma (PRP)
WB collected in 3.2% sodium citrate was centrifuged at 170 × g for 15 min at room temperature (RT); the rotor was allowed to stop without braking. PRP was gently transferred with a polypropylene pipette to a new polypropylene plastic tube. The remaining sample was centrifuged at 2,000 × g for 10 min at RT, and the supernatant, platelet-poor plasma (PPP), was transferred to a new tube. PPP was added to PRP to a final platelet concentration of 250,000 cells/µL as determined on an Advia 120 hematology analyzer.
Preparation of Washed Platelets (wPLT)
PRP was diluted in five volumes of citrate wash buffer (11 mM glucose, 128 mM NaCl, 4.3 mM NaH2PO4, 7.5 mM Na2HPO4, 4.8 mM sodium citrate, 2.4 mM citric acid, 0.35% [w/v] bovine serum albumin [BSA], pH 6.5) and pelleted (600 × g). Platelets were resuspended in modified HT (mHT) buffer (10 mM HEPES, 137 mM NaCl, 2.8 mM KCl, 1 mM MgCl2, 12 mM NaHCO3, 0.4 mM Na2HPO4, 0.35% [w/v] BSA, 5.5 mM glucose, pH 7.4) in a volume equal to the original PRP volume.
Measurement of Platelet Activation
Control or test articles were added to 20 µL WB, mHT diluted WB (20% WB), or wPLTs in a final volume of 25 µL and incubated for 20 min at RT. The control or test articles included various concentrations of AMG X (0.001–14 mg/mL), 8 mg/mL anti-streptavidin mAb (human IgG2 isotype control), one of three positive control agonists: recombinant human thrombin (1.0–2.0 U/mL [human blood] or 10–20 U/mL [nonhuman primate blood], 20 µM ADP, or 5 µM 4-PMA) or 5 µL of the vehicle control. After incubation, anti-CD41a-APC (for identification of platelets) and anti-CD62P-PE (for identification of activation), diluted in mHT, were added, each at 10 µg/mL final concentration, and incubated for 20 min at RT. Four hundred microliters of lyse/fixative solution (2% formalin, 10 mM HEPES, 0.15 mM NaCl, pH 7.4) was added to each sample, and samples were stored at 4°C until analyzed on a flow cytometer (FacsCalibur; Beckton-Dickinson). Platelets were identified by gating on CD41a+ and excluding events with low forward scatter to remove platelet-derived microparticles. Activated platelets were identified by an increase in mean fluorescence intensity of CD62P compared to resting platelets. Data were analyzed using FlowJo software (version 7.3; Treestar Inc., Ashland, OR).
Detection of Anti-target Antibody Binding to Platelets
Twenty microliters of mHT diluted WB (20% WB) or wPLTs was incubated with 5 µL anti-target mAb for 20 min at RT, washed with phosphate-buffered saline (PBS)/1% fetal bovine serum (FBS), and centrifuged for 6 min at 0.6 rcf. Platelets were resuspended in 25 µL mHT and stained with anti-CD41a-APC/CD62P-PE (10 µg/mL) and anti-human IgG-FITC (40 µg/mL). The reaction mixtures were incubated for 20 min at RT, washed with PBS/1% FBS, and resuspended in 200 µL lyse/fix (2% formaldehyde in 10 mM HEPES, 0.15 mM NaCl, pH to 7.4). Samples were analyzed on a flow cytometer as described above. AMG X and AMG A were tested for binding to platelets.
Inhibition of AMG X–Induced Platelet Activation
Fifteen microliters of wPLTs were incubated with 5 µL AMG X F(ab′)2, IVIG, or mHT buffer for 10 min at RT followed by incubation with 5 µL AMG X for 20 min at RT. In some experiments, AMG X or Isotype was preincubated with target protein or anti-huFc antibodies for 10 min at RT, and this mixture was added to 20 µL wPLTs for 20 min at RT. Platelets were stained with anti-CD41a-APC and CD62P-PE and analyzed by flow cytometry as described above. Bound AMG X was detected as described above in the “Detection of Anti-target Antibody Binding to Platelets” section.
Measurement of Platelet Aggregation
Aggregation was measured using an aggregometer (Chrono-log 560vs Platelet Optical Aggregometer; Chrono-log Corporation, Havertown, PA) according to the manufacturer’s instructions. Briefly, 250 µL PRP was carefully pipetted into a cuvette containing a stir bar and placed in the aggregometer for 5 min at 37°C with stirring at 1,200 rpm. A baseline measurement was made and set at 0% light transmission. Changes in turbidity were measured after adding test articles or controls in a volume not exceeding 30 µL. AMG X (30 µL; 7.5, 2.5, 0.8, or 0.25 mg/mL final concentration), anti-streptavidin isotype control (30 µL; 4.3 mg/mL final concentration), or a positive control agonist, ADP (30 µL; 20 µM final concentration) was added. Changes in light transmission were then recorded continuously for 8 to 40 min.
Serotonin Release Assay
WB or PRP from cynomolgus macaques was stimulated with AMG X or other agonists for 40 min at RT. Cells were then removed by spinning at 2,500 × g for 10 min. The resulting PPP samples were kept at –80°C until assayed for serotonin using an enzyme-linked immunoassay kit according to the manufacturer’s instructions (LND, Nordhorn, Germany).
Thromboxane B2 Release Assay
wPLTs in mHT buffer were incubated at RT with varying concentrations of vehicle control, AMG X, or human thrombin for 40 min. Platelets were removed by centrifugation at 2,500 × g for 10 min. The resulting supernatants were assayed for thromboxane B2 (a hydrogenated form of thromboxane A2) using an immunoassay kit according to the manufacturer’s instruction (Assay Designs, Ann Arbor, MI).
Results
IV Administration of AMG X Results in Transient Loss of Consciousness and Marked Thrombocytopenia
AMG X was administered once weekly for 5 wk to cynomolgus monkeys (n = 5/sex/group) by SQ injection at dose levels of 0 (vehicle control), 30, 100, or 300 mg/kg or by IV bolus injection at a dose level of 300 mg/kg. Transient loss of consciousness/fainting and flushing was noted only after the first dose in 5 of 10 monkeys either during injection or soon after dosing at 300 mg/kg IV. No clinical signs were noted in monkeys receiving AMG X via SQ injection at all dose levels up to and including 300 mg/kg. Decreased platelet counts and the presence of large platelets were observed in all dose groups via IV or SC injection, when measured 7 d after the fourth weekly dose (data not shown). Clinical pathology samples were not collected at intermittent time points postdosing. All animals were negative for anti–AMG X antibodies prior to dosing and during the 5-wk treatment period (data not shown). Based on these data, additional in vivo studies were conducted to determine the dose response for transient loss of consciousness as well as the platelet count nadir and potential mechanisms for platelet changes.
In single-dose studies, male cynomolgus monkeys received the vehicle control article or AMG X at 30, 100, or 300 mg/kg via SQ injection. Additional cynomolgus monkeys received a single IV bolus injection (saphenous vein) of the vehicle control article or AMG X at 15, 100, or 300 mg/kg. Consistent with the multidose study described above, a single IV bolus injection of AMG X at 300 mg/kg resulted in a transient loss of consciousness, unresponsiveness, hypoactivity, and reddened (flushing) skin during dose administration. Flushing at the time of IV dosing at ≥15 mg/kg and slight to severe ecchymosis at the blood collection sites at ≥100 mg/kg were also observed. Again, in contrast to IV dosing, no loss of consciousness or other clinical signs were observed after single SQ administration of doses up to 300 mg/kg (highest dose tested).
Moderately (at 15 mg/kg AMG X) to markedly (at 100 and 300 mg/kg AMG X) decreased platelet counts and platelet crit were observed at 15 min after IV dosing, followed by a gradual recovery to prestudy counts at 168 h postdose recovery (Figure 1A; Table 1). At doses of 100 and 300 mg/kg, degranulation of platelets was also noted at time points up to 24 h as measured by mean platelet component and microscopic evaluation of blood smears. Increased platelet production at later time points was evidenced by increased mean platelet volume and microscopically observed giant platelets (data not shown). The decreases in platelet counts likely contributed to ecchymosis at injection/phlebotomy sites. Complement fractions C3a and C5a, as determined by enzyme-linked immunosorbent assay and competitive radioimmunoassay, respectively, were not affected by IV dosing of AMG X (data not shown), suggesting that complement activation secondary to platelet activation did not contribute to the in vivo findings. After single SQ administration of AMG X at 30 to 300 mg/kg, minimal to moderate decreases in platelet counts (Figure 1B; Table 2) and alterations in other platelet parameters occurred with no clinical signs. Platelet counts generally reached a nadir at 36 h after subcutaneous dosing, were similarly reduced at 72 h after dosing, and had recovered by 168 h after dosing.
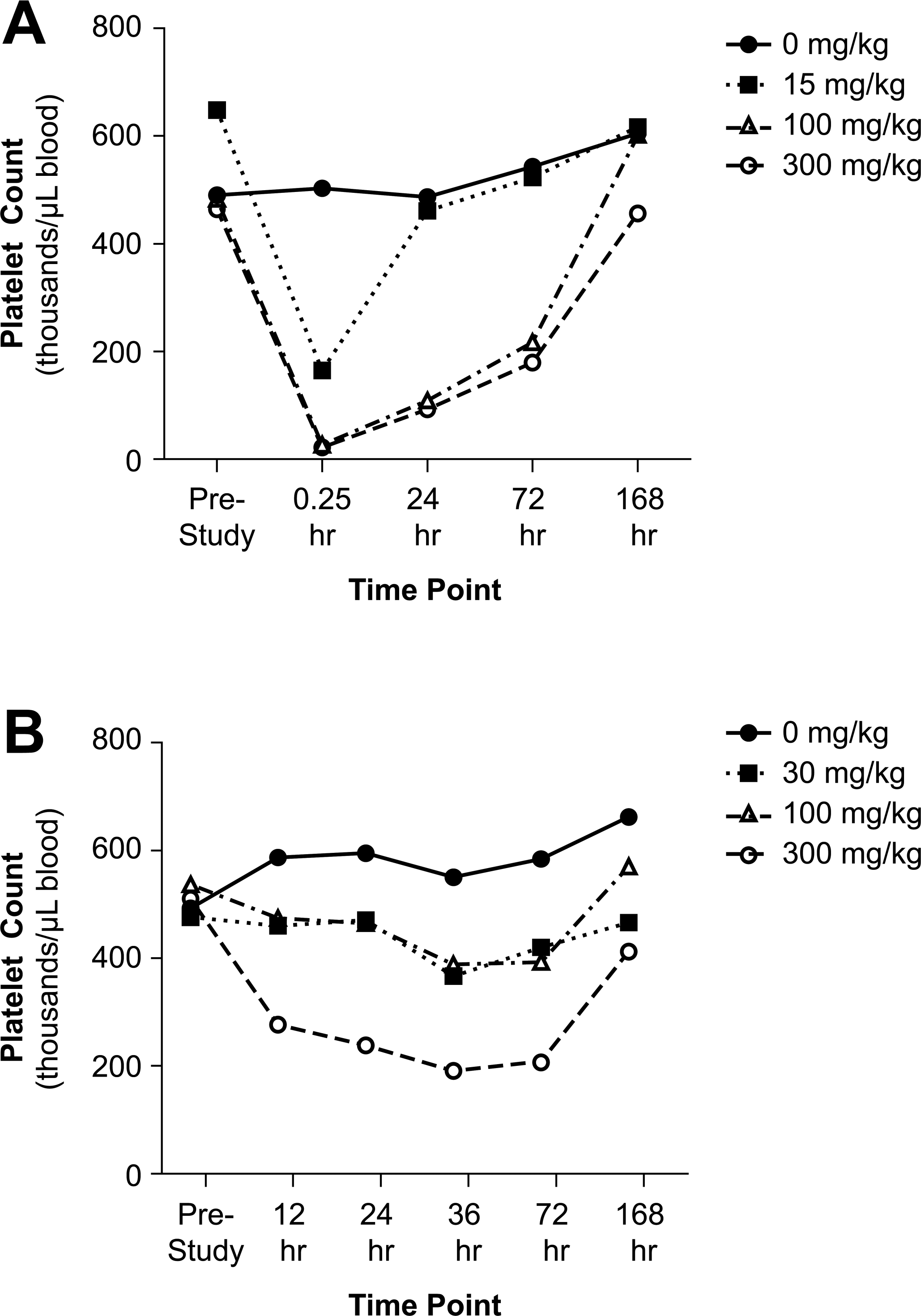
Intravenous (IV) administration of AMG X resulted in marked acute thrombocytopenia in cynomolgus monkeys, while subcutaneous (SQ) dosing resulted in minimal to moderate decreased platelet counts. For graphing purposes, the data from the first and second in vivo investigative studies are combined when the drug was administered at the same amount and by the same route. (A) Male monkeys received a single IV bolus injection (saphenous vein) of the vehicle control (0 mg/kg; closed circles) or AMG X at 15, 100, and 300 mg/kg (open square, open triangle, open circle symbols, respectively). (B) Male monkeys received a single-dose administration via SQ injection (interscapula region) of the vehicle control (0 mg/kg; closed circles) or AMG X at 30, 100, or 300 mg/kg (open square, open triangle, open circle symbols, respectively). Blood samples were collected prior to the initial dose (prestudy) and at the indicated time points postdosing. Mean data are shown with n = 2 to 4 for each group, except for the 15-mg/kg IV dose group (n = 1 at all time points) and the 300-mg/kg IV dose group (n = 1 at the 0.25- and 24-h time points due to excessive ecchymosis in that dose group).
AMG X results in marked acute thrombocytopenia via IV dosing in cynomolgus monkeys (first investigative study).
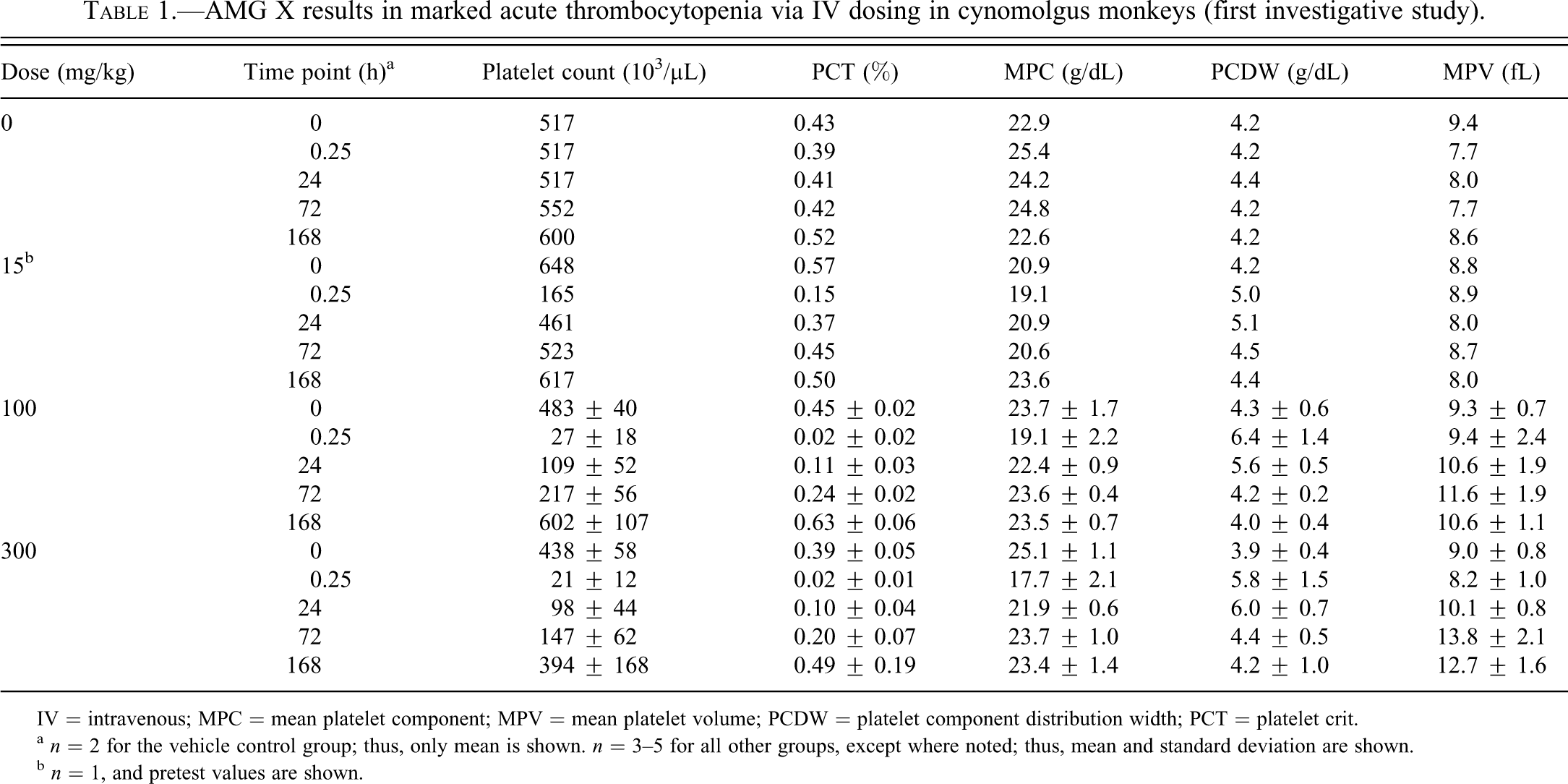
IV = intravenous; MPC = mean platelet component; MPV = mean platelet volume; PCDW = platelet component distribution width; PCT = platelet crit.
a n = 2 for the vehicle control group; thus, only mean is shown. n = 3–5 for all other groups, except where noted; thus, mean and standard deviation are shown.
b n = 1, and pretest values are shown.
AMG X results in marked acute thrombocytopenia via IV dosing in cynomolgus monkeys, with minimally to moderately reduced platelet counts after SQ dosing (second investigative study).
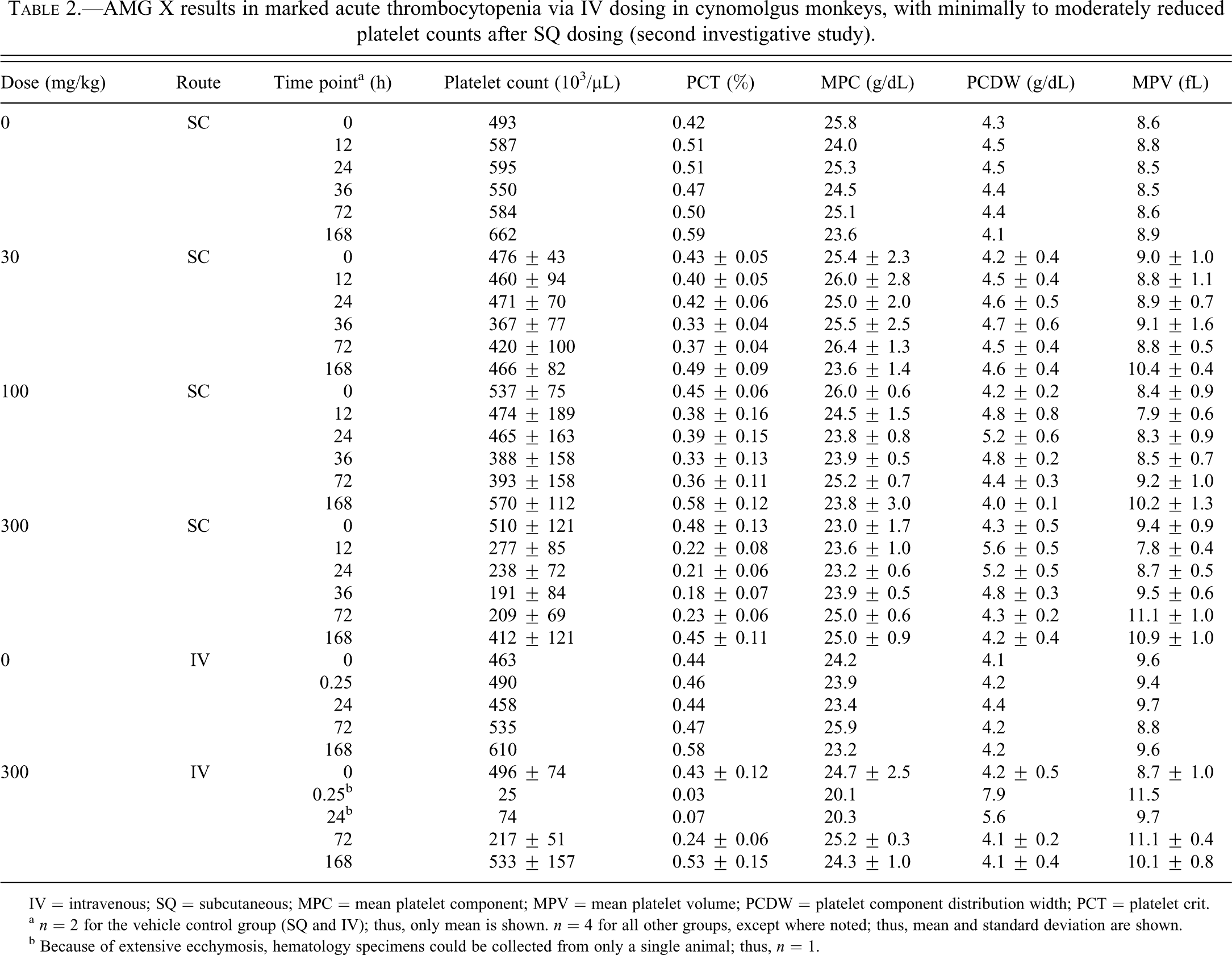
IV = intravenous; SQ = subcutaneous; MPC = mean platelet component; MPV = mean platelet volume; PCDW = platelet component distribution width; PCT = platelet crit.
a n = 2 for the vehicle control group (SQ and IV); thus, only mean is shown. n = 4 for all other groups, except where noted; thus, mean and standard deviation are shown.
b Because of extensive ecchymosis, hematology specimens could be collected from only a single animal; thus, n = 1.
AMG X-Mediated Transient Loss of Consciousness and Marked Thrombocytopenia Is Not Observed with Other mAb Antagonists against the Same Target
A single IV bolus dose study in male cynomolgus monkeys was conducted to examine whether other human mAb antagonists (AMG A, AMG B, and AMG C) against the same pharmacological target and with the same isotype (IgG2) would exhibit similar acute clinical signs and thrombocytopenia as had been observed with AMG X. In contrast to in vivo findings observed with AMG X after IV administration, no clinical signs or changes in platelet parameters were observed in cynomolgus monkeys treated with a single IV bolus dose of the vehicle control article or any of three other human mAbs at 100 mg/kg. Platelet counts were 94% to 104% of the pretest value for the vehicle control animals and 87% to 111% of respective pretest values for animals dosed with mAbs. These data demonstrated that the in vivo findings were specific to AMG X and therefore concluded to be unrelated to its pharmacological target.
Administration of 300 mg/kg AMG X IV Lowered Arterial Pressure in Cynomolgus Monkeys in a Time Frame That Coincided with Loss of Consciousness
To understand the possible etiology for the transient loss of consciousness observed in monkeys during IV administration of AMG X, a single-dose cardiovascular safety pharmacology study was conducted in conscious telemetry-instrumented male cynomolgus monkeys to assess acute cardiovascular changes associated with drug administration. An IV dose of 300 mg/kg of AMG X resulted in transient loss of consciousness during dosing that was associated with decreases in systolic blood pressure, diastolic blood pressure, mean arterial pressure, and heart rate (Figure 2, and data not shown). Compared with the predosing period (no restraint: –60 to –15 min in Figure 2), physical restraint for IV dosing of vehicle control elevated mean arterial pressure by approximately 50 to 60 mm Hg and elevated heart rate by 113 bpm (data not shown). Following vehicle control treatment, arterial pressure recovered to predosing levels after 30 min (Figure 2). After IV administration of AMG X, arterial pressure was rapidly lowered compared with vehicle control, demonstrating that AMG X elicited an acute hypotensive reaction. In addition, transient but marked reduction in heart rate (peak change: 77 bpm) was also observed. The effects on arterial pressure and heart rate occurred immediately after dosing and lasted only a few minutes. In addition, all animals administered 300 mg/kg AMG X had frequent premature ventricular complexes during the 5-min period immediately after start of dose administration (data not shown). In comparison, a single SQ dose of 300 mg/kg AMG X to conscious telemetry-instrumented cynomolgus monkeys had no treatment-related effects on clinical signs, arterial blood pressure, or heart rate during the 73-h observation period after dosing (data not shown).
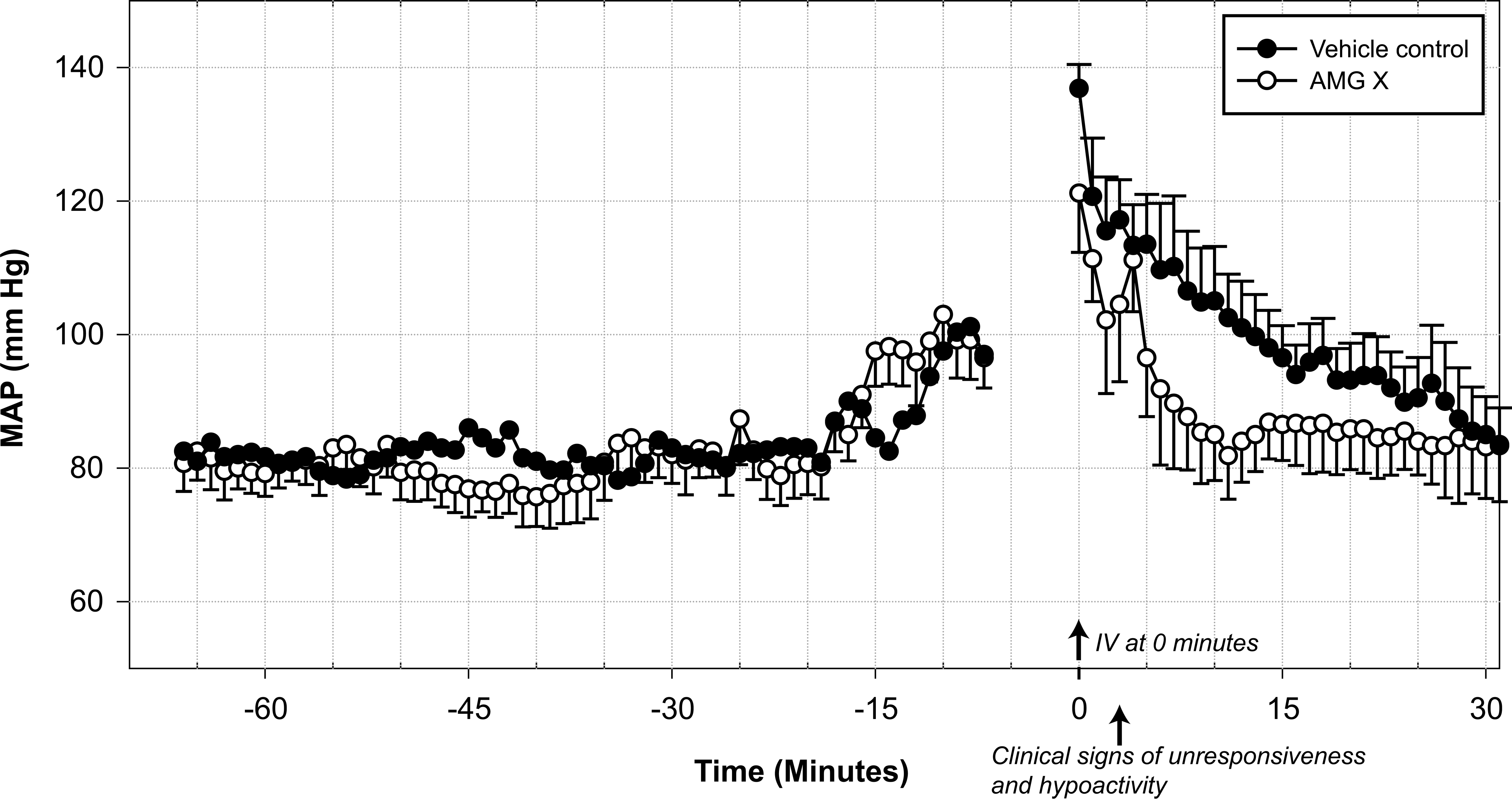
Intravenous (IV) administration of AMG X at 300 mg/kg lowered mean arterial pressure (MAP) in restrained cynomolgus monkeys. Six male telemetered monkeys received either a single IV bolus injection containing vehicle control or AMG X (300 mg/kg), and each animal was its own control. MAP values are mean ± SEM (1-min averages) taken during the predosing and dosing phase of the study. Infusion of vehicle control or AMG X occurred from 0 to 3 min; no data were collected during the animal restraint period (–7 to 0 min).
Dose-proportional peak (Cmax) and cumulative (AUC) exposures were observed after both IV (15 to 300 mg/kg) and SQ (30 to 300 mg/kg) administration of AMG X
Individual serum AMG X concentrations peaked between 0.25 to 3 h and 24 to 72 h postdosing after IV and SQ administration, respectively. AMG X concentrations then declined slowly in a biphasic pattern in both dosing routes. The mean serum AMG X concentration-time profiles are presented in Figure 3A. Dose-proportional peak (Cmax) and cumulative (AUC) exposures were observed after both IV (15 to 300 mg/kg) and SQ (30 to 300 mg/kg) administration (Table 3). The estimated time-zero (t0) serum AMG X concentration value (C0) indicated initial distribution of the drug was limited to the systemic circulation after IV administration. Both serum concentration-time profiles and the estimated toxicokinetic parameters of AMG X after either IV or SQ administration were similar to those previously observed in repeat-dose studies in male and female cynomolgus monkeys (data not shown).
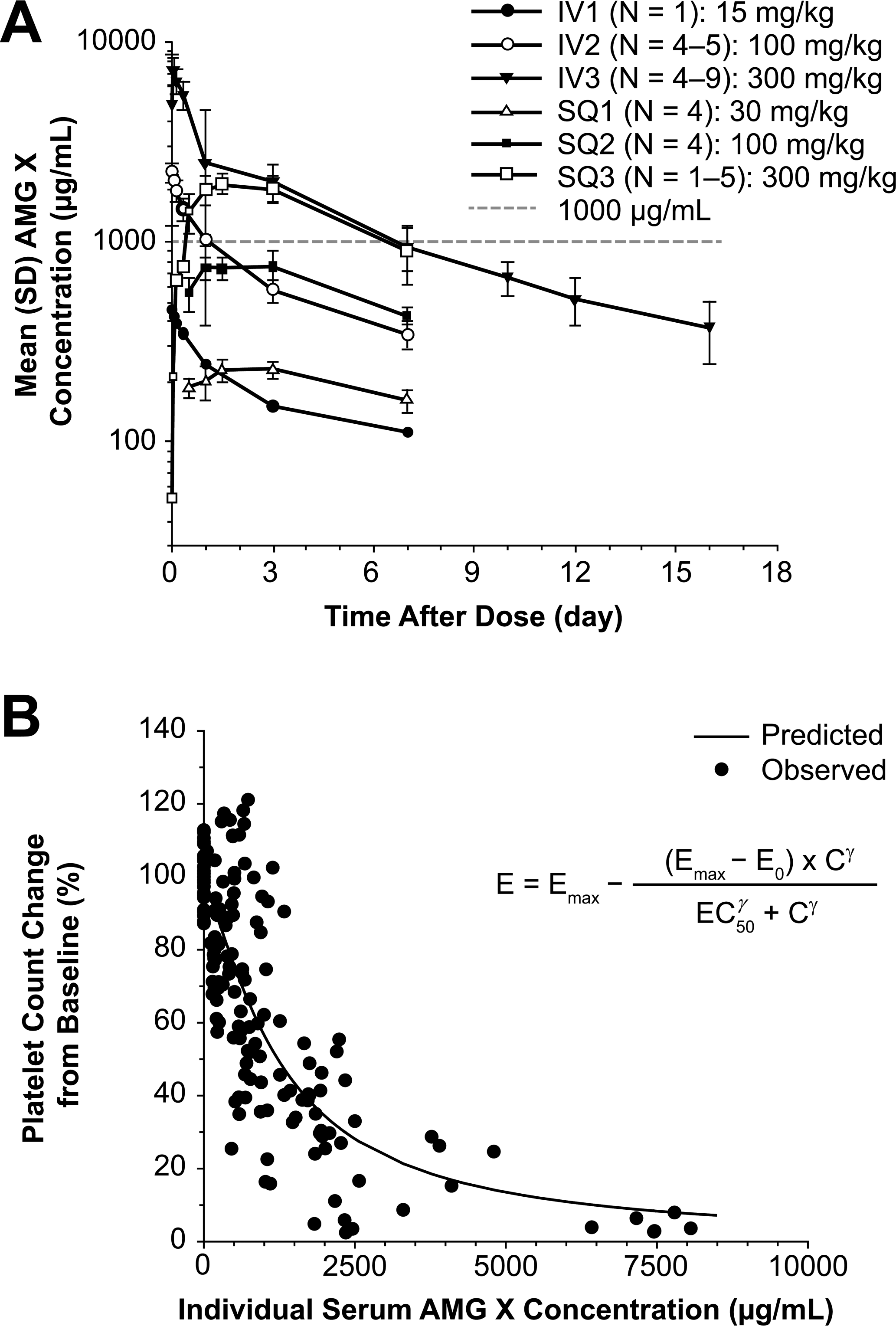
The sigmoidal Emax pharmacodynamic model adequately described the direct quantitative relationship between platelet count percentage change from baseline and the corresponding serum AMG X concentration. (A) Mean (SD) serum AMG X concentration-time plots. (B) The observed and sigmoidal Emax model predicted platelet count percentage change from predose baseline and time-matched postdose mean vehicle control baseline compared with serum AMG X concentration plots.
Mean toxicokinetic parameters of AMG X in male cynomolgus monkeys.
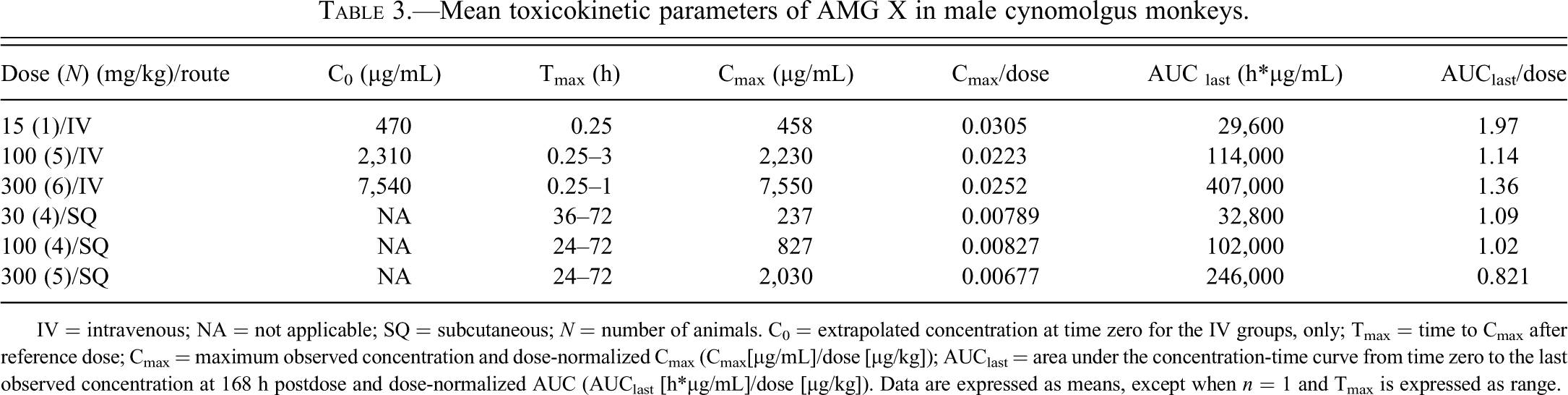
IV = intravenous; NA = not applicable; SQ = subcutaneous; N = number of animals. C0 = extrapolated concentration at time zero for the IV groups, only; Tmax = time to Cmax after reference dose; Cmax = maximum observed concentration and dose-normalized Cmax (Cmax[µg/mL]/dose [µg/kg]); AUClast = area under the concentration-time curve from time zero to the last observed concentration at 168 h postdose and dose-normalized AUC (AUClast [h*µg/mL]/dose [µg/kg]). Data are expressed as means, except when n = 1 and Tmax is expressed as range.
During 24 h after IV dosing, decreases in platelet counts were proportional to the dose of AMG X administered. At 100 mg/kg IV, platelets recovered to pretest values (483 × 103 platelets/µL) by 168 h postdosing (602 × 103 platelets/µL) at serum AMG X concentrations similar to those that caused moderately decreased platelets immediately after IV dosing (15 mg/kg). For example, the mean serum concentration in the 100-mg/kg IV-treated group at 168 h postdosing, when platelets had recovered, was 346 µg/mL. In contrast, the serum concentration in the 15-mg/kg IV-treated animal at 0.25 h postdosing was 458 µg/mL with moderately decreased platelets (165 × 103 platelets/µL). These data demonstrate that gradual recovery from thrombocytopenia occurred even in the presence of elevated drug concentrations, albeit at a later time (1 wk after dosing). Figure 3B shows that the sigmoidal Emax pharmacodynamic model adequately described the direct quantitative relationship between platelet count percentage change from baseline and the corresponding serum AMG X concentration. The model estimated EC50 value for percentage platelet count decrease compared with baseline was 1,270 µg/mL (Emax, E0, and γ estimated to be 97.7%, 0.00281%, and 1.33, respectively), indicating that profound thrombocytopenia may occur only at extremely elevated serum AMG X concentration levels.
AMG X Induced Activation of Macaque Platelets In Vitro but Not Human or Baboon Platelets
To test the hypothesis that platelet activation by AMG X was responsible for the observed hematology changes and to determine the possible relevance to humans, in vitro studies were conducted using WB from cynomolgus monkeys and two other macaque species (rhesus and pigtail), as well as baboon and human donors. Various concentrations of AMG X (0.3–14.0 mg/mL), isotype control (8 mg/mL), positive controls, or an equivalent volume of vehicle control were added to WB samples, and activation was measured by flow cytometry. Platelets were identified by constitutive expression of CD41a, and activation was measured as an increase in CD62P surface expression (i.e., upregulation). AMG X induced activation of platelets in cynomolgus WB in a concentration-dependent manner, causing CD62P upregulation at concentrations ≥0.9 mg/mL, while no such activation was observed with either the human IgG2 isotype or vehicle control–treated samples (Figure 4A, E). AMG X also activated platelets in a concentration-dependent manner in WB from rhesus or pigtail macaques at concentrations ≥2.7 mg/mL for rhesus and ≥0.3 mg/mL for pigtail (Figure 4B, C). In contrast, AMG X did not activate platelets in human or baboon WB at concentrations up to 14 mg/mL, the highest concentration tested (Figure 4D, E). Platelets in WB samples from all five species tested were capable of responding since positive control agonists such as thrombin, ADP, and PMA each elicited a canonical platelet activation response. Although responses to ADP and PMA were consistently strong, those noted with thrombin were more variable.
The ability of AMG X to cause cynomolgus platelet activation was further confirmed by the measurement of serotonin and thromboxane A2 release. The addition of AMG X to cynomolgus WB, but not human WB or PRP, resulted in a concentration-dependent release of serotonin (Table 4, and data not shown). Cynomolgus platelets incubated with AMG X also released thromboxane A2 (TXA2), which was measured indirectly by detection of its hydrogenated form, thromboxane B2 (TXB2). The incubation of AMG X with cynomolgus wPLTs, but not human wPLTs, resulted in an increased release of TXB2 (Table 4, and data not shown).
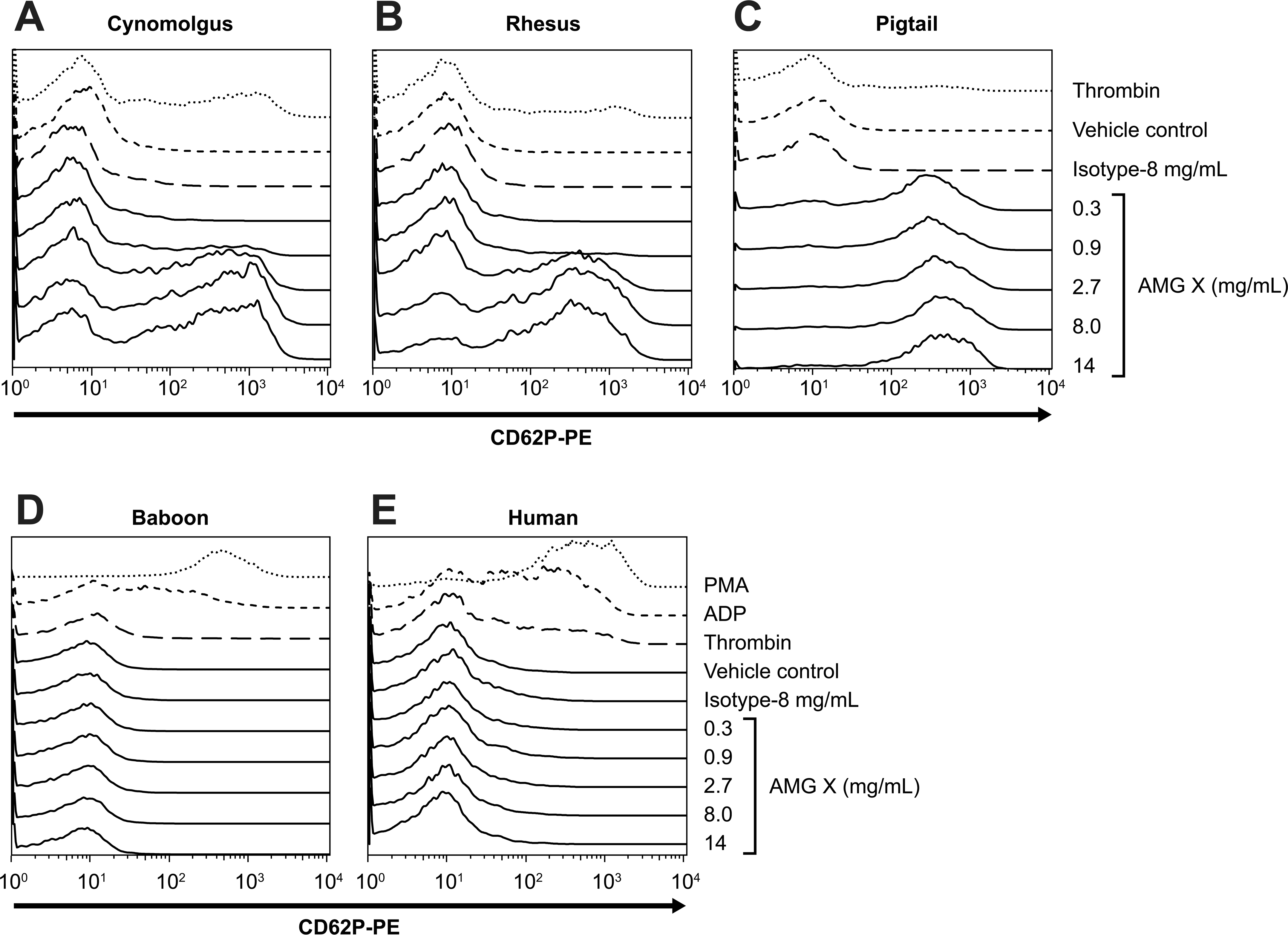
AMG X induced activation in vitro of macaque, but not human or baboon, platelets. AMG X (0.3–14.0 mg/mL), isotype control (8.0 mg/mL), vehicle control, thrombin (1 U/mL-human, 10 U/mL-nonhuman primate), PMA (1 uM), and ADP (10 uM) were added to whole blood from three macaque donors, one human and one baboon donor, and assayed for platelet activation. Platelets were identified using flow cytometry based on forward scatter and CD41a expression, and CD62P expression (platelet activation) was determined. The x- and y-axes indicate fluorescence intensity and percentage of maximum, respectively.
In vitro stimulation of cynomolgus platelets with AMG X results in the release of serotonin and TXB2.
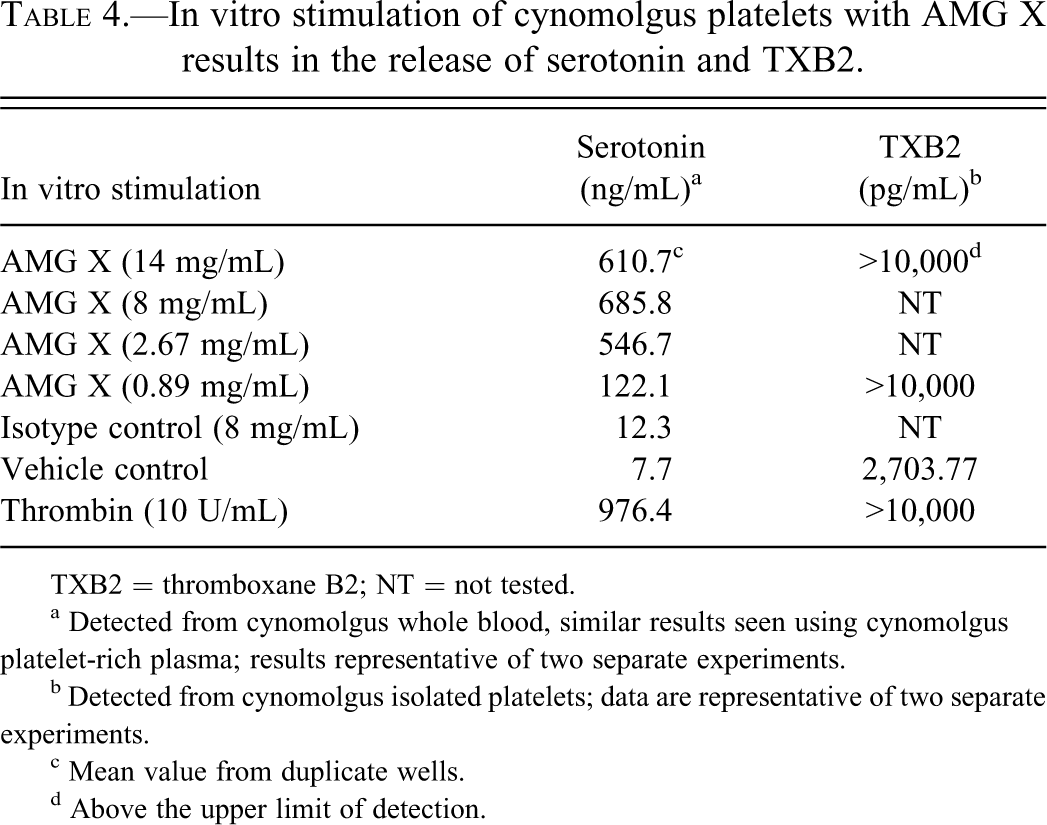
TXB2 = thromboxane B2; NT = not tested.
a Detected from cynomolgus whole blood, similar results seen using cynomolgus platelet-rich plasma; results representative of two separate experiments.
b Detected from cynomolgus isolated platelets; data are representative of two separate experiments.
c Mean value from duplicate wells.
d Above the upper limit of detection.
AMG X–Induced Aggregation In Vitro of Macaque but Not Human or Baboon Platelets
Upon activation, platelets undergo shape change and form aggregates, which are measured as a decrease in turbidity and an increase in light transmission (reviewed in Shah and Ma 2007). The ability of AMG X to induce platelet aggregation was investigated in PRP from cynomolgus, rhesus, and pigtail as well as baboon and human donors. Aggregation was detectable in cynomolgus, rhesus, and pigtail PRP at AMG X concentrations ≥2.8 mg/mL (cynomolgus and rhesus) or ≥0.8 mg/mL (pigtail; Figure 5A–C). The platelet agonist ADP induced aggregation in every sample from all five species, while the isotype control did not induce aggregation in any sample. AMG X–induced aggregation was concentration dependent in cynomolgus, rhesus, and pigtail macaque PRP but did not occur in baboon or human PRP (Figure 6A–E).
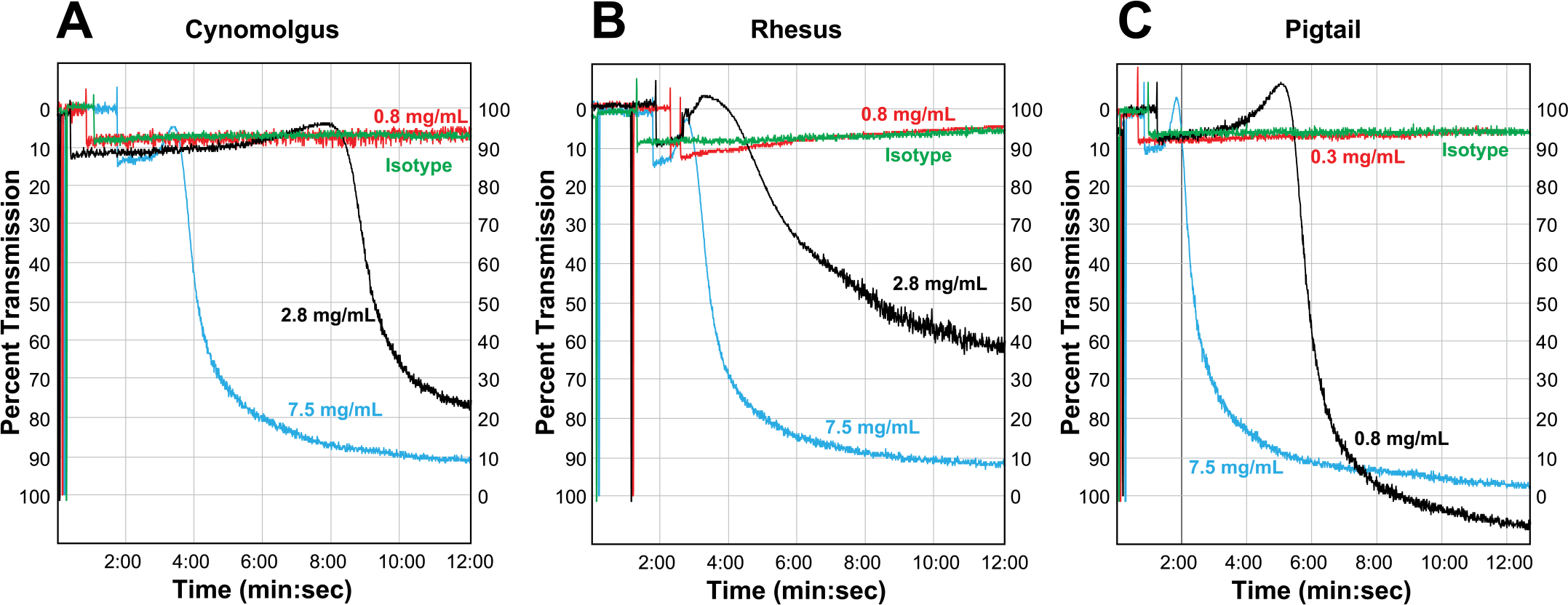
AMG X–induced aggregation in vitro of macaque platelets. AMG X (0.3–7.5 mg/mL) and isotype control (4.3 mg/mL) were added to platelet-rich plasma from three macaque donors (cynomolgus, rhesus, and pigtail) and assayed for platelet aggregation as indicated by increased light transmission.
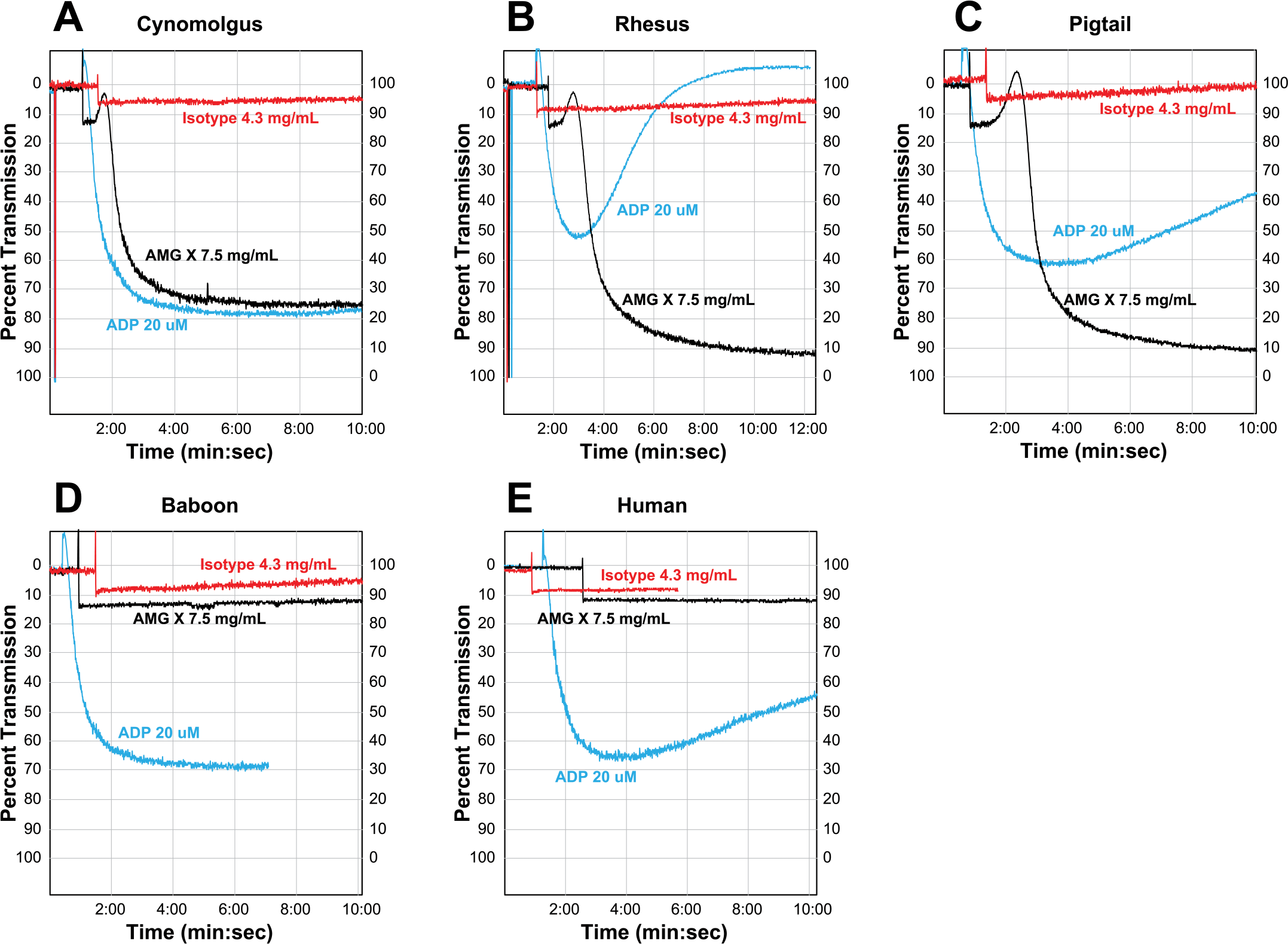
AMG X–induced aggregation in vitro of macaque but not human or baboon platelets. AMG X (7.5 mg/mL), isotype control (4.3 mg/mL), and adenosine diphosphate (20 uM) were added to macaque, human, and baboon platelet-rich plasma and assayed for platelet aggregation as indicated by increased light transmission.
AMG X activation of macaque platelets correlated with binding to platelets and was not observed with other human mAbs against the same pharmacological target
To determine whether platelet activation was specific to the molecular target, three other human mAbs (AMG A, AMG B, and AMG C) against the same pharmacological target were investigated. Compared with AMG X, these three mAbs varied in primary amino acid sequences in the variable regions but had comparable potency in cell-based assays and competed with AMG X (and with each other) for binding to the target (data not shown). All four mAbs (14.0 mg/mL), isotype control (8 mg/mL), or an equivalent volume of vehicle control were added to WB from macaque, baboon, or human donors, and CD62P expression on platelets was measured by flow cytometry. AMG A, AMG B, and AMG C each did not induce platelet activation in WB samples from any of the macaque species (Figure 7A, and data not shown). No activation of baboon or human platelets was observed by any of the four mAbs, including AMG X (Figure 7B, and data not shown). These results also suggest that platelet activation induced by AMG X in macaque samples was due to an off-target effect.
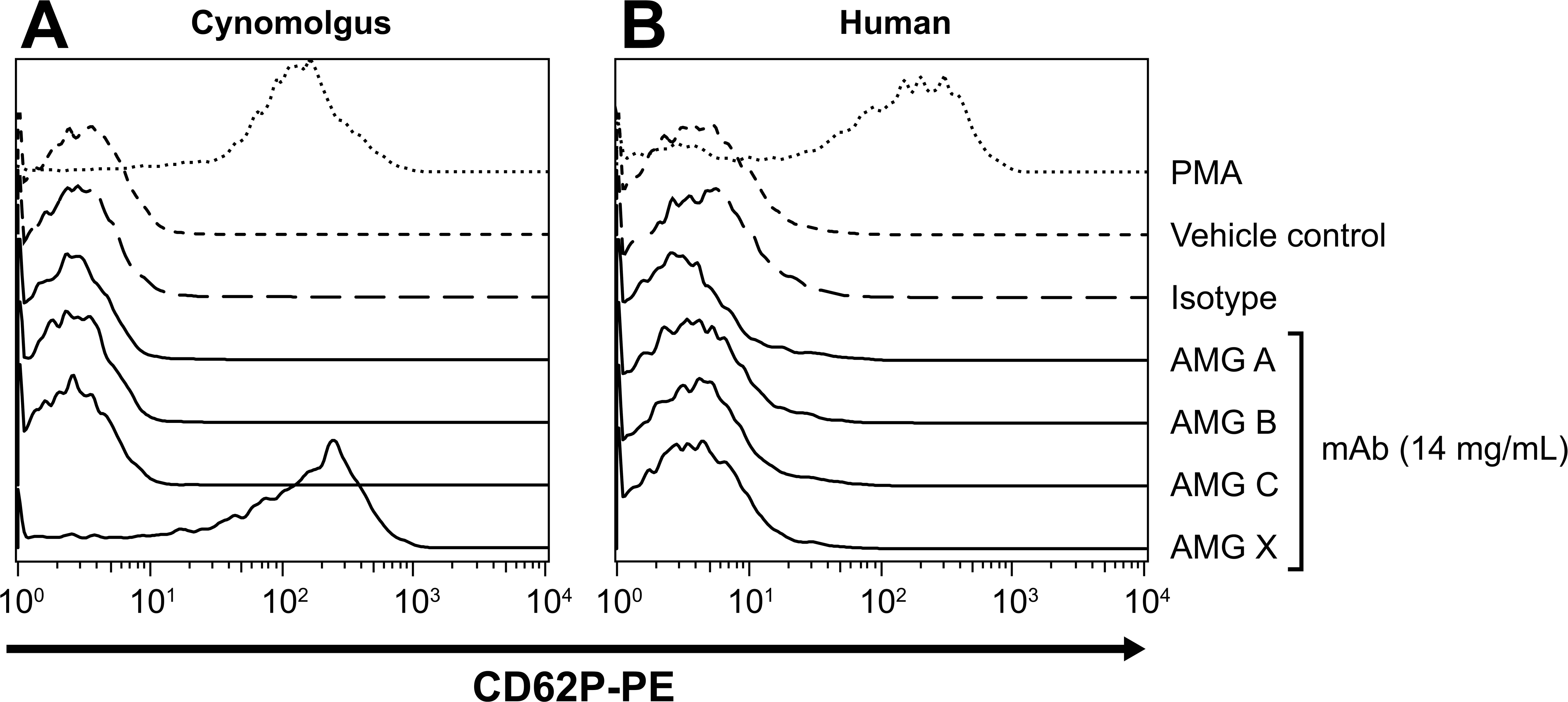
AMG X–induced macaque platelet activation via an off-target mechanism. AMG X and three other monoclonal antibodies against the same pharmacological target (14.0 mg/mL), isotype control (8.0 mg/mL), and phorbol 12-myristate 13-acetate (1 μM) were added to whole blood from a cynomolgus monkey and a human donor and assayed for platelet activation. Platelets were identified using flow cytometry based on forward scatter and CD41a expression, and platelet activation was measured by CD62P expression. The x- and y-axes indicate fluorescence intensity and percentage of maximum, respectively.
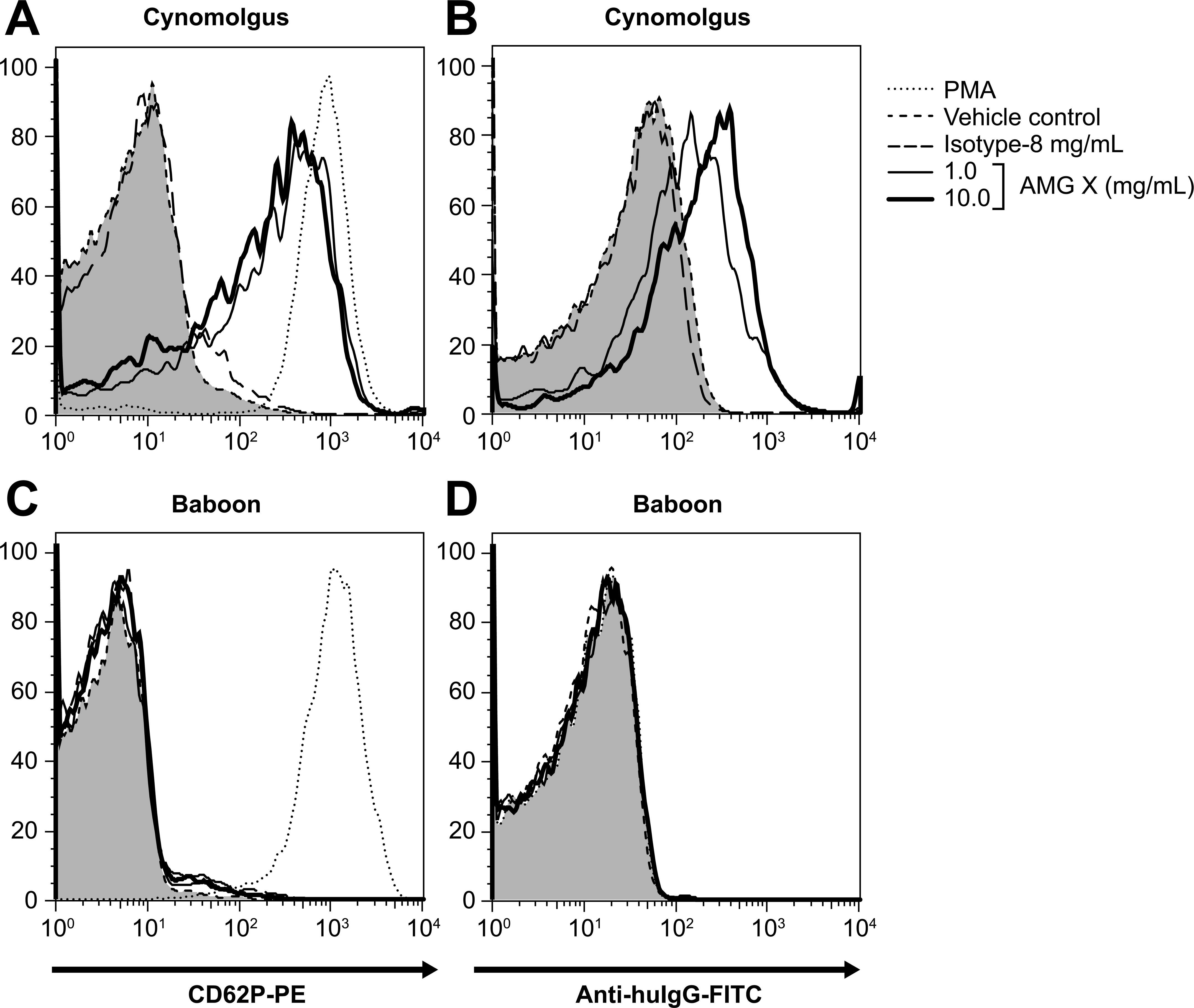
The correlation of platelet binding and activation was investigated with AMG X and AMG A. AMG X bound to and activated cynomolgus platelets (20% WB) at ≥0.9 mg/mL. In contrast, AMG A neither bound to nor activated cynomolgus platelets (20% WB) at any concentration tested (up to 14 mg/mL; Figure 8A, B). Furthermore, AMG X neither bound to nor activated baboon platelets in WB at concentrations up to 10 mg/mL, in contrast to the effects in cynomolgus platelets (Figure 9A–D). Binding of AMG X to human platelets could not be measured because of a high level of nonspecific staining with the secondary Ab (anti-human IgG). Together, these data indicate that AMG X activation of macaque platelets correlates with its binding to platelets.
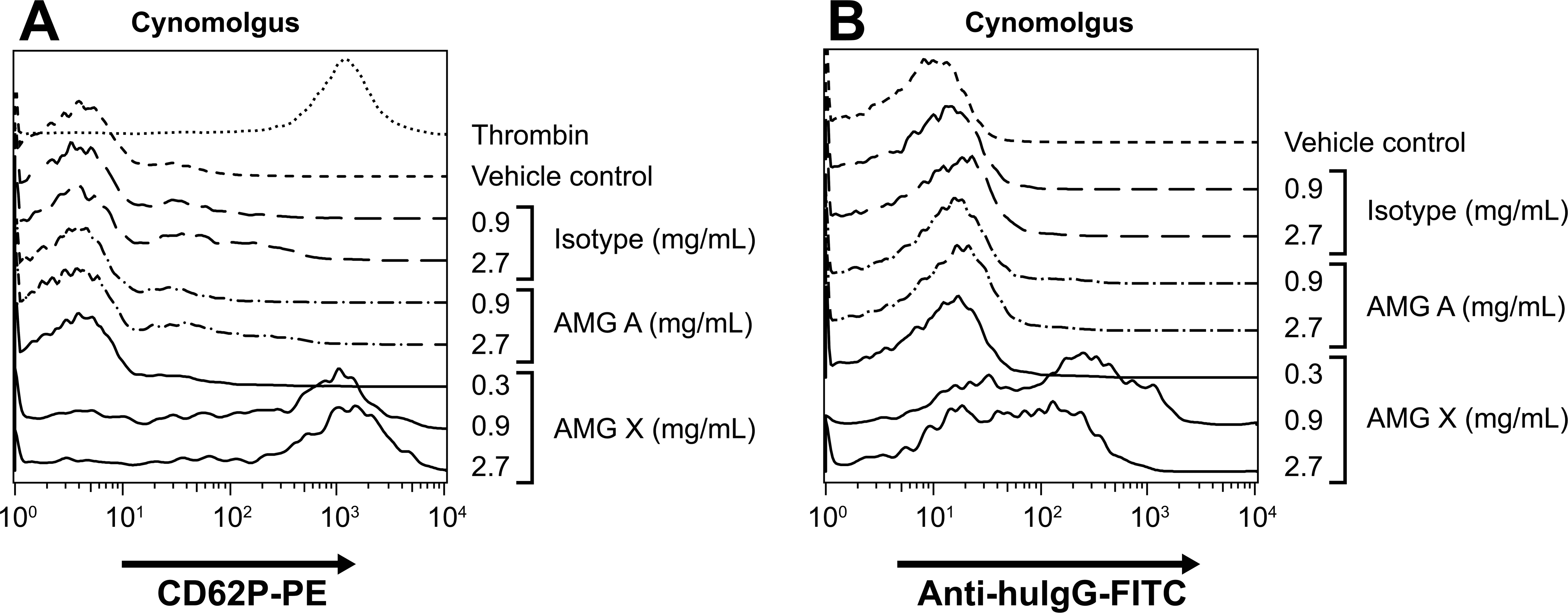
AMG X, but not AMG A, bound to macaque platelets. AMG X (0.3-2.7 mg/mL), AMG A (0.9–2.7 mg/mL), and isotype control (0.9–2.7 mg/mL) were added to 1/5 diluted cynomolgus monkey whole blood and assayed for platelet activation and binding of the anti–target molecule. Platelets were identified based on forward scatter and CD41a expression. Platelet activation (CD62P expression) and binding of AMG X (detected by anti hu-IgG) to platelets were determined. The x- and y-axes indicate fluorescence intensity and percentage of maximum, respectively.
AMG X binds to macaque platelets but does not activate or bind to baboon platelets. AMG X (1–10 mg/mL), isotype control (8.0 mg/mL), vehicle control, and phorbol 12-myristate 13-acetate (1 μM) were added to whole blood from a cynomolgus monkey and a baboon donor and assayed for platelet activation and binding of anti-target Ab. Platelets were identified based on forward scatter and CD41a expression. CD62P expression (platelet activation), and binding of AMG X (detected by anti hu-IgG) to platelets was determined. Tinted histograms correspond to vehicle control–treated platelets. The x- and y-axes indicate fluorescence intensity and percentage of maximum, respectively.
AMG X–Induced Platelet Activation Required Both F(ab′)2 and Fc Domains
Cynomolgus wPLTs were used in a series of experiments to determine what portion(s) of AMG X were required for binding to and activation of platelets. Various molar ratios of AMG X and recombinant target protein (human) were preincubated together and added to cynomolgus wPLTs. Preincubation of AMG X with equimolar amounts or fivefold molar excess amounts of the target protein inhibited both binding and activation (Figure 10A, B). Thus, binding through the complementarity determining region (CDR) is required for AMG X to activate cynomolgus platelets.
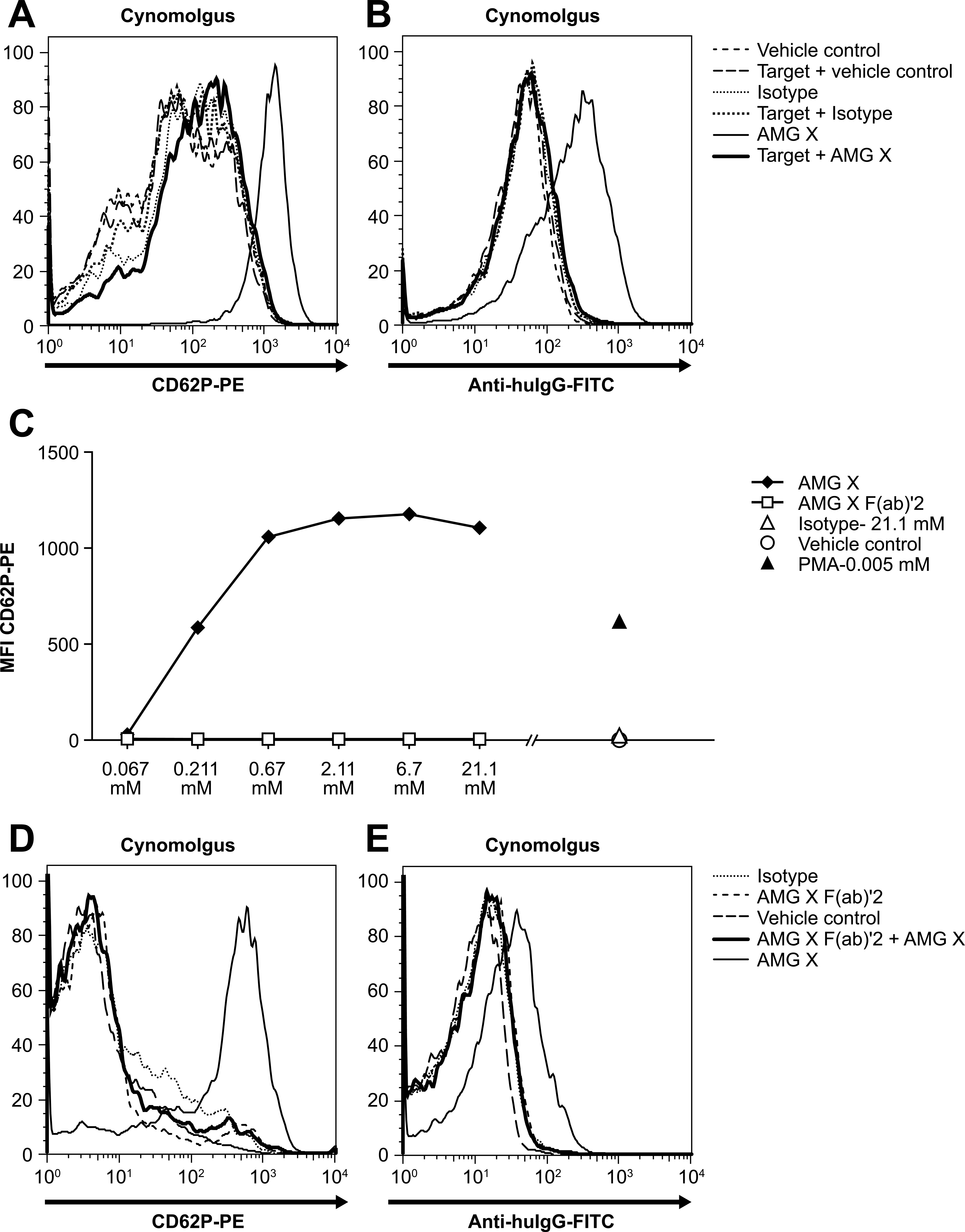
(A, B) AMG X–induced platelet activation required both Fab and Fc domains. AMG X (2.68 mM), isotype control (2.68 mM), or vehicle control was preincubated with the target protein (13.4 mM; 10 min room temperature [RT]), added to cynomolgus washed platelets (wPLTs), and assayed for binding to and activation of platelets. Platelets were identified via flow cytometry based on forward scatter (FSC) and CD41a expression. Platelet activation was measured by CD62P expression, and AMG X binding was detected with anti hu-IgG. Tinted histograms correspond to vehicle control–treated platelets. The x- and y-axes indicate fluorescence intensity and percentage of maximum, respectively. (C) AMG X F(ab′)2 does not activate cynomolgus platelets. AMG X (0.067–21.1 mM), AMG X Fab′2 (0.067–21.1 mM), isotype control (21.1 mM), and phorbol 12-myristate 13-acetate (5 μM) were added to cynomolgus wPLTs and assayed for platelet activation. Platelets were identified based on FSC and CD41a expression, and CD62P expression (platelet activation) was determined. (D, E) Preincubation of platelets with AMG X F(ab′)2 inhibits AMG X–induced platelet activation and binding of AMG X to cynomolgus platelets. Cynomolgus wPLTs were incubated with AMG X F(ab′)2 (8.43 mM; 10 min RT) followed by AMG X (2.11 mM), isotype control (0.67 mM), or vehicle control. Excess Ab was washed away, and platelets were assayed for platelet activation and AMG X binding. Platelets were identified based on FSC and CD41a expression, and CD62P expression (platelet activation) and AMG X binding, detected by anti hu-IgG, were determined. Tinted histograms correspond to vehicle control–treated platelets. The x- and y-axes indicate fluorescence intensity and percentage of maximum, respectively.
Platelets express a single Fc receptor, FcγRIIa (CD32), which can bind IgG2 with low affinity (Capel et al. 1994). Several studies have implicated CD32 as a major contributor to platelet activation either directly or indirectly through association with other platelet receptors and internal signaling (Anderson and Anderson 1990). To assess the contribution of the Fc portion of AMG X to platelet activation, an F(ab′)2 fragment of AMG X was generated. Various concentrations of AMG X F(ab′)2 and AMG X (67 µM to 21.1 mM) were tested for activation. Unlike intact AMG X, AMG X F(ab′)2 did not induce activation of cynomolgus wPLTs, even at concentrations as high as 21.1 mM (3 mg/mL), whereas AMG X caused maximal activation of cynomolgus wPLTs at concentrations of ≥0.67 µM (0.1 mg/mL; Figure 10C). In addition, preincubation of cynomolgus wPLTs with AMG X F(ab′)2 inhibited AMG X–induced platelet activation and binding of AMG X to cynomolgus platelets (Figure 10D, E). These data demonstrate the necessity of CDR-mediated binding for subsequent platelet activation. Blocking the Fc of AMG X by preincubation of AMG X with anti-human Fc Abs (polyclonal and monoclonal) or blocking CD32 on platelets by preincubation of cynomolgus wPLTs with IVIG also inhibited AMG X–induced platelet activation (data not shown). Together, these data indicate that interactions via both the CDR and Fc domains of AMG X are required for cynomolgus platelet activation.
Discussion
IV administration of ≥100 mg/kg of AMG X to cynomolgus monkeys resulted in rapid and profound thrombocytopenia, lowered arterial pressure, and transient loss of consciousness and flushing. Given the profound impact of drug-induced changes in platelets, investigative studies focused on the potential direct effects of AMG X on platelet function. The findings from these investigative studies conducted in vivo and in vitro demonstrated that the monoclonal antibody, AMG X, activated macaque platelets by a mechanism unrelated to its pharmacological target, which is not expressed on platelets. AMG X activation and aggregation were not observed in baboon or human platelets. The studies also demonstrated that AMG X bound directly to cynomolgus platelets and that both the Fc and Fab domains of AMG X were required for activation. The platelet molecule to which AMG X bound has not been identified.
Platelet activation in vivo can result in systemic cardiovascular events primarily because of the release of serotonin from dense bodies and by activation of the complement and coagulation cascades (Wiggins et al. 1985). Approximately 96% of serotonin in mammals is stored in dense bodies of platelets. In vivo, exposure to AMG X resulted in decreased platelet counts and granularity, suggesting release of alpha granules and dense bodies, but no activation of complement was detected. Serotonin release could not be measured in vivo because of technical complications with serum sample integrity; however, AMG X was able to induce serotonin release from platelets in vitro. Serotonin is a potent vasoactive amine that elicits a variety of cardiovascular effects, including modulation of arterial pressure and heart rate, through the activation of vascular serotonin receptors or cardiovascular and vagal reflex pathways (Zucker and Cornish 1980; Veelken et al. 1993; Ramage and Villalon 2008). Intravenous infusion of serotonin or serotonergic agonists in animals (e.g., rats, dogs, and primates) causes hypotension, bradycardia, and cardiac arrhythmia (Bachman and Katz 1977; Zucker and Cornish 1980; Veelken et al. 1993, 1998). Cardiovascular changes secondary to serotonin release likely underlie the flushing and loss of consciousness observed in cynomolgus monkeys given intravenous AMG X. Although platelet activation can also affect cardiovascular parameters secondary to complement activation (Henry 1997
Activated platelets are rapidly removed from circulation, likely due to sequestration in vascular beds and phagocytosis by neutrophils. Expression of membrane CD62P, shown to be induced by AMG X in vitro, and phosphatidyl serine are thought to be important signals in removal of platelets from circulation (Maugeri et al. 2009).
After IV administration of AMG X, platelet counts decreased rapidly and proportionally to dose. After SQ administration, decreases in platelet counts were more gradual and less pronounced than those after IV administration. Platelet counts were approximately 5% of predose at peak exposure to AMG X (15 min postdose; first time point evaluated) after 100 mg/kg IV (AMG X concentration of 2,230 µg/mL; platelet counts of 27 × 10e3/µL) but were much less affected (37–46% of predose) at peak exposure after 300 mg/kg SQ even though Cmax values were similar (AMG X concentration of 2030 µg/mL; platelet counts of 191–238 × 10e3/µL). This finding suggests that the platelet effects are more profound when platelets are exposure to an abrupt, high concentration of AMG X. Activation of virtually all platelets at a Cmax of 2,230 µg/mL after IV dosing is consistent with in vitro data showing that activation of platelets in WB, and aggregation of platelets in PRP, occurred at AMG X concentrations ≥0.9 and 2.8 mg/mL, respectively. These data were also consistent with the pharmacodynamic model–predicted EC50 value for percentage platelet count decrease over baseline (1,270 µg/mL). Immediate activation and release of granule contents from platelets, as occurred in vivo with IV dosing and in vitro, may result in autocrine and paracrine amplification of platelet activation cascades, causing many more platelets to be activated than would occur if platelets were being gradually exposed to AMG X and activated (e.g., with SQ dosing). This could explain why, despite similar Cmax values, clinical signs, including cardiovascular changes, and platelet effects were different after IV and SQ dosing. Alternatively, translocation of AMG X from subcutaneous tissues to the vasculature may alter a property of AMG X such that it is less able to activate platelets. These hypotheses are consistent with the observations that at 1 wk (168 h) after 300 mg/kg IV or SQ administration, exposures to AMG X were still at ∼1,000 µg/mL, but platelet counts had recovered to approximate pretest values. Alternative reasons for the recovery of platelets at 1 wk after SQ or IV dosing despite continuing high concentrations of drug include lower susceptibility of younger platelets to effects of AMG X, accelerated platelet production compensating for low platelet counts, or alterations in cell surface proteins of platelets produced by megakaryocytes exposed to AMG X. In addition, circulating AMG X might have been structurally altered after 1 wk in circulation so that it could no longer bind to platelets to cause activation.
Activation of platelets by AMG X was direct and likely occurred by an off-target mechanism. The pharmacological target of AMG X is not expressed on platelets, as demonstrated by the lack of binding of AMG A to platelets and analysis of existing platelet transcriptomes and proteomes (Gnatenko et al. 2003; Bugert et al. 2003; McRedmond et al. 2004; Rox et al. 2004; Macaulay et al. 2005; Dittrich et al. 2008; Jones et al. 2009). AMG X bound to cynomolgus platelets through its CDR, and binding correlated with direct activation. Other mAbs (AMG A, AMG B, and AMG C), each of which compete with AMG X for binding to the pharmacological target and exhibit comparable potency in cell-based assays, did not induce macaque platelet activation (M. cynomolgus, M. nemestria, and M. mulatta). All four mAbs share an identical Fc primary amino acid sequence, suggesting that the differences in binding/activation of macaque platelets by these mAbs are due to differences in the CDR sequences. Further work is required to determine which CDR residues may be mediating platelet activation.
The data presented here are consistent with the hypothesis that AMG X binds via its Fab domains to an unknown platelet molecule as well via its Fc to FcγRIIa on platelets, resulting in sufficient cross-linking to cause activation. Platelets across many and diverse species express highly conserved proteins and pathways. For example, agonists such as ADP, epinephrine, thrombin, and collagen activate and aggregate platelets from numerous species. In addition, a number of reagents, including mAbs, cross-react with both human and macaque platelets. Despite this, the off-target binding of AMG X appears to be specific for a platelet surface protein that is similar among closely related macaque species but sufficiently different from the human and baboon orthologs such that AMG X binds and activates macaque, but not human or baboon, platelets. Alternatively, AMG X may bind to a protein expressed on macaque platelets but not expressed on human or baboon platelets.
The requirement for binding of platelets via both Fab and Fc domains of AMG X for platelet activation was empirically demonstrated. Activation of platelets by AMG X was inhibited by the following preincubations: AMG X with its pharmacological target or anti-human Fc antibodies, and platelets with AMG X F(ab′)2 or human IVIG. In addition, AMG X F(ab′)2 alone was insufficient to activate platelets. Primate platelets express only the activating IgG receptor FcγRIIa (Gewirtz et al. 1992; Cassel et al. 1993; Mahan et al. 1993). Monomeric IgG has very low affinity for FcγRIIa, but dimeric and higher-order IgG complexes have much higher affinity (Luo et al. 2009). The dependence of platelet activation on an intact Fab domain is consistent with the inability of monomeric IgG to activate platelets by binding to FcγRIIa alone.
Activation of platelets by mAbs has most commonly been reported with nontherapeutic mAbs specifically directed against platelet membrane proteins. Most mAbs that activate platelets do so through interactions via both the Fab domain (to a surface protein) and the Fc domain (to FcγRIIa; Horsewood et al. 1991; Perutelli and Mori 1993). The interactions of the CDR portion of these mAbs with platelets are likely to be of high affinity. The high concentration of AMG X required for in vitro activation suggests a low-affinity interaction. However, despite the putative low-affinity interaction, markedly decreased platelets (∼25% of the predose value) occurred after a relatively low single IV dose of 15 mg/kg AMG X in cynomolgus monkeys, which approximated the anticipated human clinical dose (data not shown).
To date, the platelet molecular target of AMG X binding has not been identified. The fact that AMG X activated macaque but not human or baboon platelets provided an avenue to try to identify the platelet molecule or activation pathway involved in the off-target effect of AMG X. Studies using inhibitors to various platelet activation pathways (e.g., αIIbβ3, P2Y12, P2X1, GP1a, GP1b, GPVI, PGH2/TXA2R, PAFR, and PAR1) did not identify a specific pathway required for AMG X–induced activation. Immunoprecipitation of platelet extracts from cynomolgus and baboon platelets using AMG X identified candidate proteins, but attempts to definitively demonstrate AMG X binding to any individual protein were unsuccessful, possibly because of a low-affinity interaction. However, antibodies to either CD41 or CD9, cell surface proteins involved in platelet activation, each competed with AMG X for binding to and/or activation of platelets (data not shown). Further investigation is required to determine whether these data are relevant to the in vitro and in vivo activation of platelets by AMG X.
Possible off-target effects on cynomolgus platelets due to therapeutic mAbs have been rarely reported. In reported cases, effects occur acutely after dosing and range from mild to severe. Anti-human IL-13 mAb (IgG1) caused a slight decrease in platelet counts (mean was approximately 60% of control) after a single dose in both normal and allergic macaques (Martin et al. 2008). The effects on platelets were nonprogressive, reversible, and thought to be due to peripheral destruction of platelets. No human data with this mAb have been published. Omalizumab, a humanized antibody drug approved for patients with moderate-to-severe allergic asthma, caused dose-related reversible decreases in platelet counts in monkeys at high doses (“Omalizumab” 2002). In vitro studies failed to show direct binding to or activation of human or nonhuman platelets by omalizumab nor inhibition of usual platelet aggregation responses. Mild decreases in peripheral blood platelet counts have been observed in more omalizumab-exposed human subjects compared with control subjects (“Omalizumab” 2002). A mAb developed against sclerostin caused profound thrombocytopenia in rats but not in cynomolgus monkeys because of off-target binding to rat megakaryocytes and platelets (Rudmann et al. 2012). In addition, a mAb against an unnamed target caused rapid profound thrombocytopenia, sometimes with mild to marked decreases in red cell mass, after a single dose in cynomolgus monkeys at ≥50 mg/kg via the SC or IV route (Everds et al. 2011). The effect on platelets and red cells was considered to be secondary to activation of splenic macrophages. This mAb induced phagocytosis of platelets by cynomolgus, but not human, peripheral blood monocytes, and was not administered to humans. In each of the cases described above, presumed off-target effects of mAbs were first observed with in vivo studies.
In vitro platelet activation studies conclusively showed that AMG X activated macaque platelets and that this activation was not observed for baboons and humans. The sequelae of platelet activation in cynomolgus monkeys included toxicities that created challenges for development of the molecule, even though the effects were considered to be specific to macaques. The lack of platelet activation observed with other mAbs against the same pharmacological target provided opportunities to develop a mAb without potential platelet liabilities. Although platelet activation is thought to be a rare off-target effect of mAbs, simple prescreening of drug candidates for platelet activation can be useful in choosing among potential mAb candidates for development. During candidate selection, it is desirable to exclude from further development those mAbs in which a concern exists for significant off-target toxicity, even if considered irrelevant for humans.
Unexpected effects of monoclonal antibodies are rarely reported (“Omalizumab” 2002; Martin et al. 2008; Bumbaca et al. 2011; Everds et al. 2011). Establishing that unexpected effects are due to off-target mechanisms can be challenging and requires a weight-of-evidence approach. Lines of evidence can include (1) lack of similar effects with mAbs of similar amino acid sequence that compete for binding to the intended target; (2) lack of similar findings with small molecules against the same target; (3) lack of effects in relevant genetically modified animals; (4) lack of expression (confirmed by mRNA or protein) of the intended target in cell(s) exhibiting mAb binding and/or toxicity; (5) identification of the molecule that the mAb is binding to, resulting in an unexpected effect; (6) knowledge of the pathophysiology underlying the unexpected effects; and (7) similarity of the spectrum of unexpected effects with that associated with a known clinical condition. Identification of the mechanism underlying unexpected effects of mAbs can be difficult and may present regulatory challenges for nonclinical and clinical development.
In summary, AMG X caused off-target activation of macaque platelets in vivo and in vitro. In vivo, platelet activation after IV bolus dosing resuted in immediate profound thrombocytopenia and clinical signs consistent with release of serotonin. In vitro, platelet activation was shown to require binding of the Fab domain of AMG X to a macaque platelet protein and binding of the Fc domain to FcγRIIa. AMG X activation of platelets in vitro resulted in platelet aggregation, release of serotonin, and increased surface expression of CD62P, which occurred in a macaque-selective and mAb-specific manner. These findings, and the referenced case studies, reflect the complexity of the mechanism of action of mAbs and the increasing awareness of potential for unintended effects in preclinical species.
Footnotes
Acknowledgments
The authors would like to thank the following individuals for technical assistance in the preparation of this article and/or consultation: Richard H. Aster, Linda Carlock, Samuel Chuang, David R. Doherty, John Valliere-Douglass, Glenn Elliot, Patricia Giclas, Gayle Kwon, Alan LaRochelle, Nianyu Li, Andrea Mitchell, and Sonal K. Patel.
All authors are employed by Amgen, Inc.