
Abstract
ZAP-70 is a critical molecule in the transduction of T cell antigen receptor signaling and the activation of T cells. Upon activation of the T cell antigen receptor, ZAP-70 is recruited to the intracellular ζ-chains of the T cell receptor, where ZAP-70 is activated and colocalized with its substrates. Inhibitors of ZAP-70 could potentially function as treatments for autoimmune diseases or organ transplantation. In this work, we present the design, optimization, and implementation of a screen for inhibitors that would disrupt the interaction between ZAP-70 and the T cell antigen receptor. The screen is based on a fluorescence polarization assay for peptide binding to ZAP-70.
Keywords
Introduction
ZAP-70 (zeta-associated protein of 70 kDa) plays an essential role in T cell signaling. Its misregulation can lead to immune-related diseases. The overactivation of ZAP-70 can result in autoimmune diseases, while its absence can cause severe combined immune deficiency (SCID). Overexpression of ZAP-70 in chronic lymphocytic leukemia (CLL) is correlated with a poor prognosis. A small-molecule inhibitor of ZAP-70 could potentially be used as a treatment for autoimmune disease and organ transplant rejection. Traditional ATP-competitive kinase inhibitors that specifically target the active site of ZAP-70 have not yet been found. 1
The interaction of ZAP-70 with the CD3 ζ-chain of the T cell antigen receptor plays a critical role in T cell signal transduction by connecting extracellular ligand binding to intracellular signaling events. Recognition of immunoreceptor tyrosine-based activation motifs (ITAMs) on the T cell receptor by the tandem SH2 unit of ZAP-70 facilitates T cell activation in two ways. First, recruitment of ZAP-70 to the ζ-chain localizes ZAP-70 to the plasma membrane and in proximity to its substrates, the scaffolding proteins, LAT and SLP-76. Second, ITAM binding to ZAP-70 relieves autoinhibition of the kinase by effecting an intramolecular domain reorientation that is not compatible with formation of the autoinhibited conformation. Blocking ZAP-70 recruitment to phosphorylated ITAMs has been validated as a means of inhibiting T cell signaling, as overexpression of the isolated tandem SH2 domains of ZAP-70 has a dominant-negative effect that blocks downstream signaling.2–4
ZAP-70 assumes an inactive autoinhibited state in the absence of ITAM.5,6 In this conformation, the tandem SH2 domains of ZAP-70 pack against the distal side of the kinase domain, and the conformation of the tandem SH2 domains is nearly identical to that seen in the crystal structure of the isolated domains. 7 The inter-SH2 linker, also referred to as interdomain A, forms the bulk of the interaction between the tandem SH2 domains and the kinase domain. The SH2-kinase linker, also referred to as interdomain B, contributes a number of hydrophobic residues that interact with the kinase domain and the inter-SH2 linker to form a hydrophobic core.
We sought to exploit the insight gleaned from structural and functional studies of ZAP-70 to discover inhibitors of the interaction of ZAP-70 with the ζ-chain. Toward this end, we developed a fluorescence polarization (FP)–based assay that detects the binding of an ITAM-derived phosphopeptide to ZAP-70. The fluorescently labeled peptide consists of the ITAM sequence (CGNQLpYNELNLGRREEpYDVLD) in the ζ- chain and is henceforth denoted 2pY. In the absence of an inhibitor, the bound peptide would experience slow molecular tumbling, resulting in a high FP value. The presence of an inhibitory compound that disrupts peptide binding leads to a decreased FP value. This assay was applied to a high-throughput screen against a library of 132,842 compounds at the Small Molecule Discovery Center (SMDC) at the University of California, San Francisco (UCSF). Bona fide inhibitors of this interaction could potentially function by directly competing with 2pY binding or by stabilizing the autoinhibited conformation and allosterically preventing 2pY binding. We discovered several compounds that block the function of the ZAP-70 tandem SH2 domains by covalent modification, and these compounds have been described in a previous publication. 8
Materials and Methods
Molecular Cloning and Protein Purification
A ZAP-70 construct spanning residues 1–606 was prepared as described previously. 6 A biotin-tagged ZAP-70 construct was created by appending a biotinylation tag (sequence: GLNDIFEAQKIEWHE) to the C terminus of the ZAP-70 (1–606) protein sequence, and the ZAP-70 expression cassette was subcloned into pFastbackHTb. This construct was expressed and purified as described previously. 6 The purified protein was then subjected to in vitro biotinylation (Avidity, LLC, Aurora, CO) and repurified by size exclusion chromatography (Superdex 200 16/60, GE Lifesciences, Pittsburgh, PA). Cloning and purification of the tandem SH2 domains of ZAP-70 was performed as previously described. 9
Peptide Preparation and Labeling
ITAM-derived peptides used in the FP assay and time-resolved fluorescence resonance energy transfer (TR-FRET) assay were prepared by the Howard Hughes Medical Institute (HHMI) University of California (UC), Berkeley, Core Facility. Tetramethylrhodamine (TAMRA)-labeled ITAM 2pY peptide used in the FP assay consisted of the following sequence: CGNQLpYNELNLGRREEpYDVLD. 2pY peptide was incubated with twofold molar excess of TCEP to reduce the cysteine residue. This peptide was then incubated with a threefold excess of TAMRA C5 maleimide at room temperature overnight. The labeling reaction was quenched with 10-fold excess of DTT. The labeled peptide was purified by reverse phase high-performance liquid chromatography (HPLC) and its identify confirmed by mass spectrometry. The Alexa Fluor 488–labeled 2pY peptide used in the TR-FRET assay consists of the following sequence: XGNQLpYNELNLGRREEpYDVLD, where X is propargyl-glycine. Alexa Fluor 488-azide (Life Technologies, Grand Island, NY) was incubated in a threefold molar excess over 2pY peptide with the addition of ascorbic acid and copper sulfate at room temperature overnight. The labeled peptide was then purified by reverse phase HPLC and its identity was confirmed by mass spectrometry.
Fluorescence Polarization Binding Assays
ZAP-70:2pY FP binding reactions were carried out in 20 mM Tris pH 8.0, 150 mM NaCl, 2% glycerol, 1 mM TCEP, 0.01% Tween-20, 0.01% Triton X-100, 2 nM TAMRA-2pY, and 100 nM ZAP-70. This reaction mixture was prepared in bulk and dispensed as 20 µL volumes with a liquid handling instrument (Matrix WellMate Bulk Dispenser, Thermo Scientific, Waltham, MA) into Corning 3820 384-well plates for the high-throughput screen. Percent inhibition is calculated relative to free labeled 2pY peptide in the assay buffer as the positive control for 100% inhibition of the assay and ZAP-70:2pY treated with DMSO as a control for 0% inhibition. Concentration–response curve fits are calculated with Prism GraphPad (GraphPad Software, La Jolla, CA) four-parameter fit for inhibition.
High-Throughput Screen
Fifty nanoliters of compound in DMSO was transferred by pin tool to 20 µL of the FP reaction mixture (25 µM final compound concentration, 0.25% DMSO) with shaking (Beckman Biomek FXP, Brea, CA). The ZAP-70:2pY binding mixture was incubated with compound for 20 min before reading on an EnVision plate reader (PerkinElmer, Waltham, MA; flash lamp, 531/25 excitation filter, 579/25s and 579/25p emission filters, and BODIPY TMR FP DUAL mirror).
TR-FRET Assay
The binding reaction mixture consisted of 2 nM ZAP-70, 100 nM Alexa Fluor 488-2pY, 20 mM Tris pH 8.0, 150 mM NaCl, 1% glycerol, 1 mM TCEP, 0.01% Tween-20, 0.01% Triton X-100, and 2 nM terbium chelate-streptavidin (Cisbio, Bedford, MA). The compounds were dispensed as with the FP assay. Plates were read on the EnVision plate reader with a 340/30 excitation filter and 495/10 and 520/25 emission filters. FRET was calculated as a ratio of 520/495 nm fluorescence. Percent inhibition is calculated relative to free ZAP-70-biotin-streptavidin-terbium in the assay buffer as the positive control for 100% inhibition of the assay and ZAP-70:2pY treated with DMSO as a control for 0% inhibition. Concentration–response curve fits are calculated with Prism GraphPad four-parameter fit for inhibition.
Results and Discussion
Design and Optimization of Fluorescence Polarization-Based Assay
FP is an established biophysical method used to assay interactions between biological macromolecules. It provides a cost-effective means of measuring protein–peptide interactions in solution, making it well suited to high-throughput screening. We have previously reported FP assays for measuring the ZAP-70:ITAM interaction.8,10 Here, we describe the development and optimization of this assay for use in a high-throughput screen.
As a first step toward developing an assay to measure disruption of the ZAP-70:2pY binding, we measured the affinity between ZAP-70 and a fluorophore-labeled peptide derived from the first (most N-terminal) ITAM motif in the ζ-chain. 11 This measurement had been performed previously with fluorescein (excitation peak at 494 nm and emission peak at 521 nm), a green fluorescent dye. 10 We replaced fluorescein with TAMRA, a red-shifted dye (excitation peak of 542 nm and emission peak of 568 nm), to reduce the potential for fluorescence interference from the compound library. 12 Additionally, this assay was miniaturized from a 200 µL volume performed in cuvettes to a 20 µL microplate well volume for use in 384-well plates, to facilitate high-throughput screening.
Using the TAMRA-labeled 2pY peptide, we measured the affinity between ZAP-70 and the 2pY peptide and determined the value of the dissociation constant, KD, to be 45.7 ± 4.7 nM. This is comparable to the previously measured KD value of 76.6 nM 10 ( Fig. 1A ). To further examine the sensitivity of this assay, we measured the KD value for 2pY peptide binding to ZAP-70 Y315F/Y319F, a construct in which two tyrosine residues crucial to the autoinhibited conformation of ZAP-70 are mutated to phenylalanine. These mutations have been previously shown to stabilize ZAP-70 in the autoinhibited conformation and reduce the affinity of the protein for ITAM. 10 We verified this result and determined a KD value of 82.2 ± 5.6 nM ( Fig. 1B ) for the ZAP-70 Y315F/Y319F mutant. In a final verification step, we measured the affinity between the TAMRA-2pY peptide and a chimeric protein that replaced the kinase domain of ZAP-70 with that of Src kinase ( Fig. 1C ). Because this chimeric protein should not be able to adopt a compact inhibited tertiary structure, the ZAP-Src chimera should exhibit an increased affinity for TAMRA-2pY and therefore a lower KD value. The experimental results were consistent with our predictions (measured binding constant for 2pY peptide and the ZAP-Src chimera is 15.8 ± 1.3 nM). To maximize both the signal window and sensitivity for screening purposes, a ZAP-70 concentration of 100 nM was used in the assay, which corresponds to ~80% of the maximum binding value (EC80).
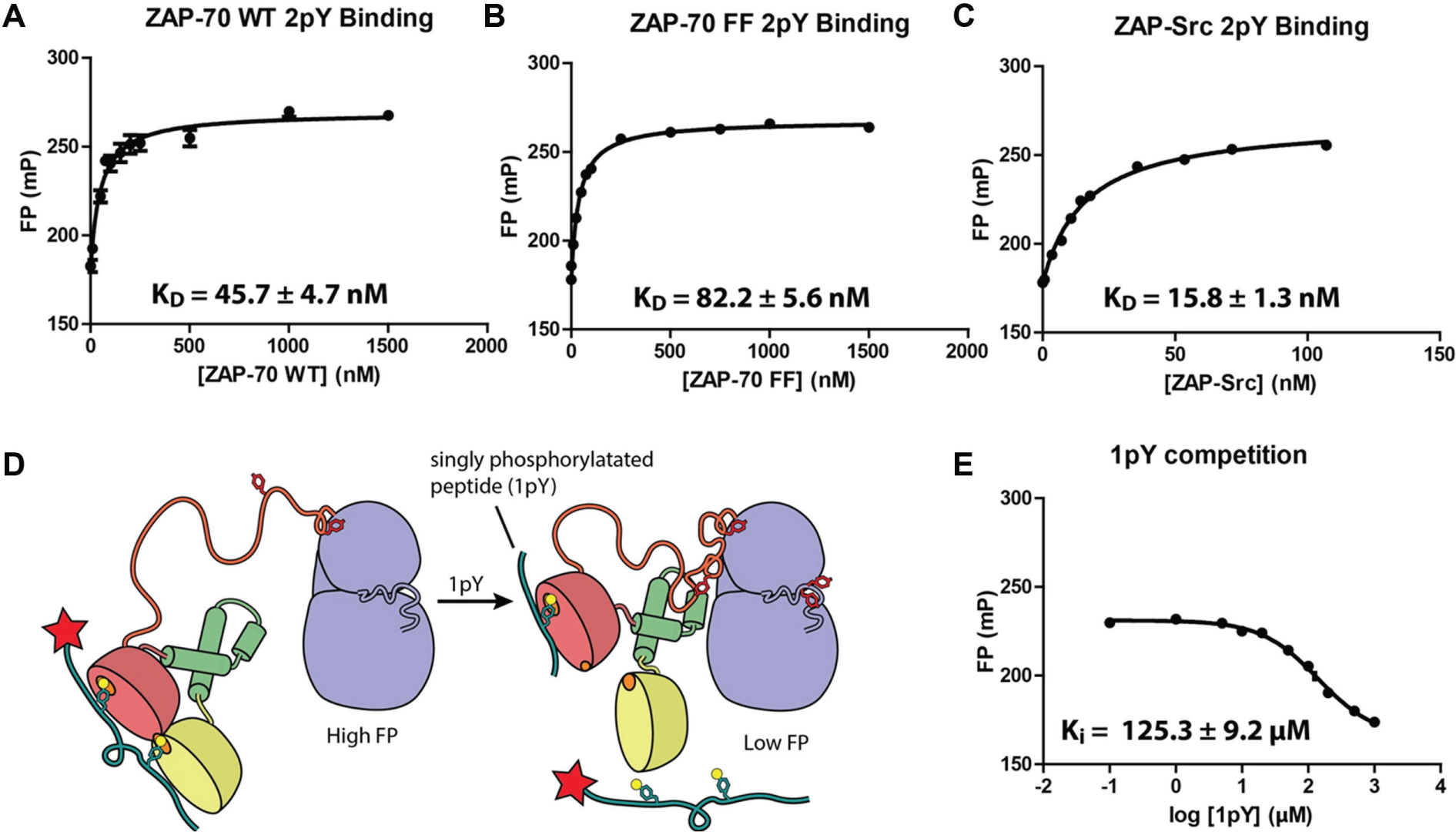
Binding curves of ZAP-70 and TAMRA-2pY measured by FP. (
Attempts have been made to develop phosphotyrosine-competitive SH2 inhibitors of ZAP-70.13,14 Similar to previous inhibitors of SH2 domains, these compounds are primarily phosphotyrosine mimetics and demonstrated moderate specificity and low affinity. In order to select against phosphotyrosine competitors in our screen, we supplemented the FP assay with a truncated ITAM-based peptide (sequence: CGNQLpYNELNLGRREE), denoted 1pY. The residues preceding the second phosphotyrosine in the ITAM motif were excluded in this peptide.
The rationale for the use of this peptide is that if the screen identified an allosteric inhibitor of ZAP-70, which stabilized the protein in the autoinhibited conformation, then the affinity between ZAP-70 and the doubly phosphorylated peptide (2pY) would be reduced to a value closer to that for a singly phosphorylated peptide. This is because we expect the autoinhibited conformation to have only one intact phosphorylation binding site in the tandem SH2 domain. The excess of 1pY peptide would then outcompete any residual 2pY binding and further increase the signal window and assay sensitivity. This is possible because of the ~1000-fold difference in affinity between the 1pY and 2pY peptides for ZAP-70. 15 An additional function of the excess of 1pY peptide would be to outcompete any compounds that bind to the phosphotyrosine pocket binding compounds.
To determine the appropriate concentration of the singly-phosphorylated peptide that would outcompete fluorescent peptide in the presence of an inhibitor, we titrated 1pY peptide into the assay to disrupt the ZAP-70:2pY binding interaction. We determined the Ki value of 1pY to be 125.3 ± 9.2 µM ( Fig. 1D , E ). A 1pY concentration of 30 µM, corresponding roughly to the IC20 value, was then used for future assays. This concentration of 1pY optimizes the assay’s sensitivity by facilitating a rapid change in response to an inhibitor through dynamically displacing 2pY.
Because FP values are dependent on the tumbling rate of the fluorophore in solution, we adjusted the viscosity of the buffer by varying the amount of glycerol. A range of glycerol concentrations (0%–10% v/v) was examined to determine their impact on FP signal window. Reducing the glycerol concentration increased the signal window by further decreasing the lower-boundary FP value (TAMRA-2pY alone). We chose a 2% glycerol concentration, which is sufficient to reduce protein instability, but which afforded an enlarged signal window (data not shown).
DMSO can interfere with assay readouts through its action as a protein denaturant. Assay tolerance up to 5% (v/v) was established (data not shown). This concentration is well in excess of the 0.25% DMSO concentration that is used in the high-throughput screen.
Z′ and Pilot Screen
We next assessed the performance of the FP assay in a high-throughput format by first testing the assay in an automated screening format. We began by measuring the Z′ value of the assay with automated instrumentation. We divided a 384-well plate into two parts and measured the FP values of all the assay components (ZAP-70, 1pY, and TAMRA-2pY) as the top value or negative control of the assay. The second half of the plate contained only TAMRA-2pY. We used this as a positive control and minimum FP value for the assay. Our Z′ plate reported a Z′ value of 0.78, which is an excellent value to proceed with the assay.
We next assessed the robustness of the assay by performing a pilot screen against a collection of 2000 molecules curated by the UCSF SMDC as bioactive. After filtering out for molecules that exhibited total fluorescence greater than 1σ above the mean fluorescence, there were 12 hit compounds that showed inhibition greater than 3σ above the mean. A single molecule, pyrithione zinc, showed concentration-responsive inhibition. We went on to characterize the mode of inhibition of this molecule and found that it is a reversible precipitator of the ZAP-70 tandem SH2 domains (data not shown).
HTS and Concentration–Response
We performed the high-throughput screen at UCSF’s SMDC, and screened against the SMDC’s diversity library, a collection of 132,842 compounds. The assay performed well, and an overall Z′ score of 0.70 was achieved. We first removed fluorescence artifacts by removing from consideration all compounds that exhibited fluorescence values that were greater than 1σ above or below the mean fluorescence value of the library compound wells ( Fig. 2A ). Comparison of the correlation between perpendicular and parallel channels for all compounds provides one way to check for fluorescence anomalies, but this was not done. We further filtered the screening hits by selecting those compounds that showed inhibition greater than or equal to 3σ above the mean (~20% inhibition) ( Fig. 2B ). The resulting 428 compounds corresponded to a ~0.3% hit rate. We then took the most potent 320 compounds from the primary screen and tested them in a concentration–response FP assay. One hundred two of these compounds exhibited concentration-responsive inhibition, minimal cooperativity (Hill slope, nH ≤ 2; note that the Hill slopes are not well determined for compounds where the titration is not complete), and at least 25% inhibition at 33.3 μM compound concentration. The IC50 values calculated ranged from >33 to 3.7 µM. Representative data are shown ( Fig. 2C , D ). Those compounds that failed to show concentration-responsive inhibition were possibly fluorescent artifacts, protein destabilizing compounds, or errors in the single-point measurements from the primary screen. Further analysis showed these hit compounds to be cysteine-dependent covalent modifiers of ZAP-70, as described. 8
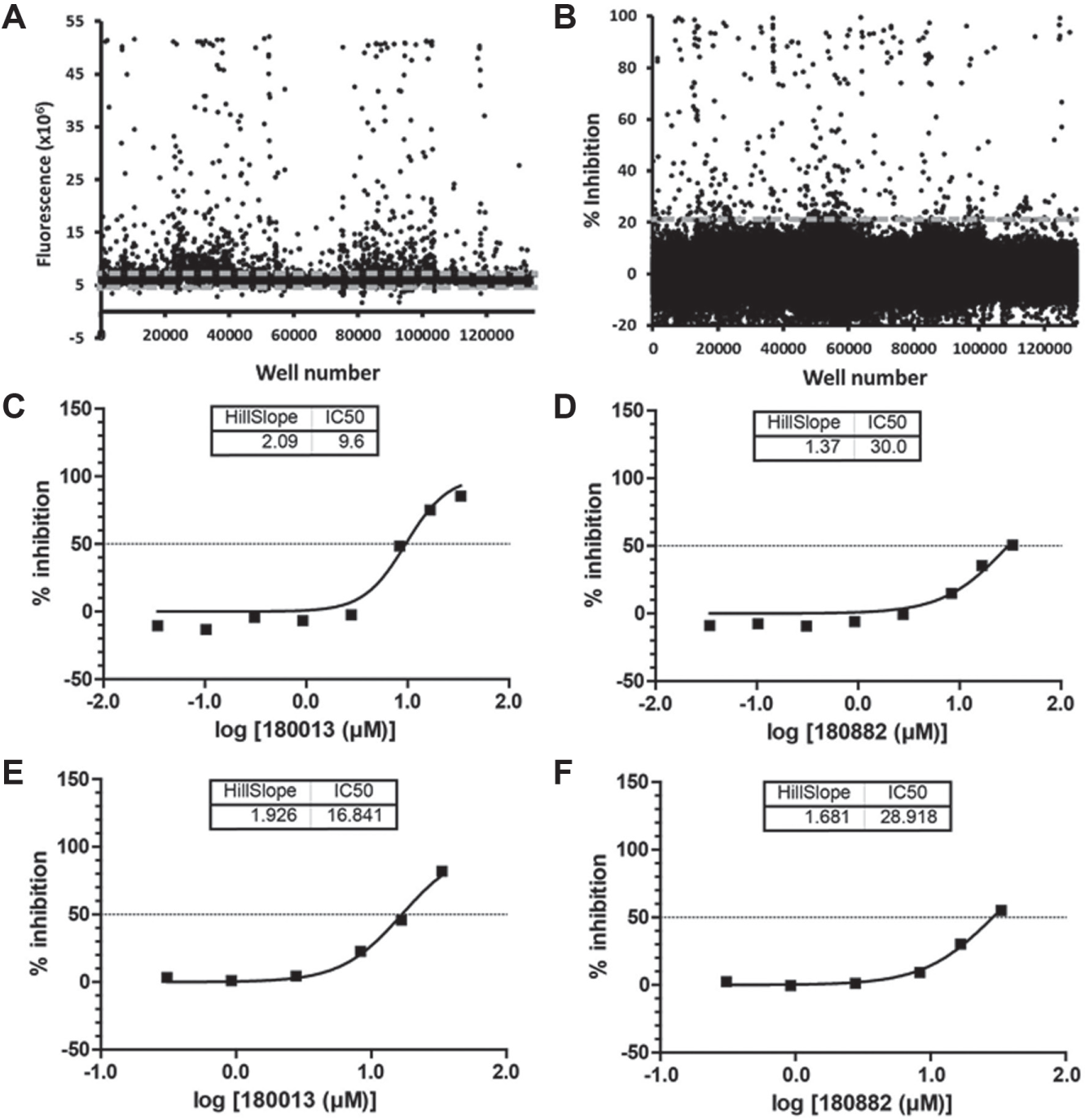
(
Development of ZAP-70:2pY TR-FRET Assay
In order to verify the results of the primary screen and FP concentration–response assay, we developed an orthogonal screen using TR-FRET as a measurement of ZAP-70:2pY binding, which was performed in a concentration–response format. This assay utilizes a streptavidin-bound terbium as the FRET donor. This assay has the advantage of using a far red-shifted excitation wavelength that should avoid fluorescence artifacts from the library compounds. Furthermore, the fluorescence lifetime of the terbium excitation exceeds that typically seen for organic molecules. These two aspects of the TR-FRET assay make it an ideal method for screening small-molecule libraries. For this assay, we engineered a ZAP-70 construct that could be specifically biotinylated on a lysine residue on a genetically encoded tag sequence known as an Avitag (Avidity). The biotin ligase, BirA, then adds biotin to the Avitag lysine residue. Biotinylated ZAP-70 was then labeled by the addition of streptavidin labeled with a terbium cryptate (Cisbio, excitation peak at 340 nm, emission peak of 495 nm) to produce a fluorescent ZAP-70 FRET donor. The addition of an Alexa Fluor 488–labeled ITAM-derived peptide (excitation peak of 495, emission peak at 519 nm), denoted AlexaFluor488-2pY, acts as a FRET acceptor upon binding to terbium-conjugated ZAP-70 ( Fig. 3A ).
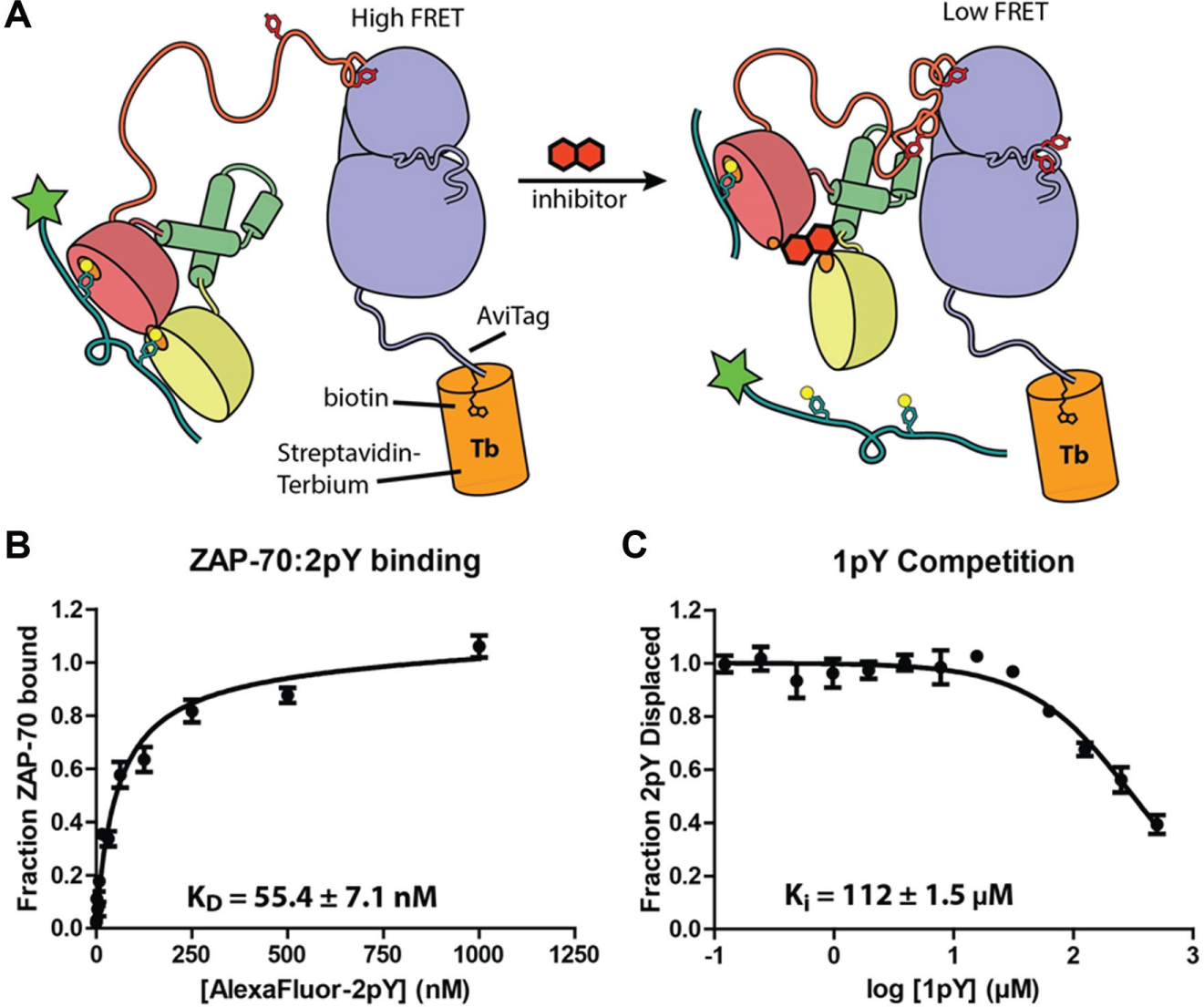
(
Using this method, we determined the binding constant between ZAP-70 and phosphorylated 2pY to be 55.4 ± 7.1 nM; this is very similar to the value measured in the FP assay (45.7 nM) ( Fig. 3B ). We further characterized the assay by competing off the doubly phosphorylated 2pY peptide with singly phosphorylated 1pY and obtained a Ki value of 112 ± 1.5 µM ( Fig. 3C ), which was also comparable to the Ki value of 1pY measured in the FP assay (125.3 µM) ( Fig. 1E ).
Compounds Inhibit 2pY Binding to ZAP-70 in an Orthogonal TR-FRET Assay
The TR-FRET concentration–response assay was performed similarly to the FP concentration–response assay. This assay also proved to be extremely robust, and we calculated a Z′ value of 0.8 for this TR-FRET concentration–response assay. The most potent 320 compounds from the FP-based primary screen were delivered in DMSO in a range of 33.33 µM to 0.102 µM in a 12-point concentration–response format. One hundred twenty-five compounds showed total fluorescence within 1σ of the mean fluorescence at the highest compound concentration of 33.33 µM. These 125 compounds also exhibit at least 30% inhibition at 33 μM compound concentration in the TR-FRET assay. Eighty-six hit compounds scored positively in both the FP and TR-FRET concentration–response assays. The majority of the IC50 values generated in the TR-FRET concentration–response assay were very similar to those obtained in the FP concentration–response assay. Representative data are shown ( Fig. 2E , F ). The IC50 values calculated ranged from >33 to 0.80 µM. A possible difference in compounds that inhibited in the FP assay but not in the TR-FRET assay could have been fluorescent artifacts specific to each assay format. Alternatively, the two different assay formats may have resulted in distinct sets of hit compounds. The difference in the ratio of ZAP-70:2pY in the two assays may also contribute toward the differing results. By excluding compounds that scored positively in one assay, but not the other, it is possible that our analysis excluded false negatives. However, we did not explore this further.
Hit Compounds Can Act Directly on the Tandem SH2 Unit
To further characterize the mechanism of inhibition of these compounds, we developed an assay to measure the binding between 2pY peptide and the isolated tandem SH2 domains of ZAP-70. The tandem SH2 unit of ZAP-70 bound to the 2pY peptide with a dissociation constant nearly identical to the Src-ZAP fusion protein (data not shown). This is expected as the fusion protein is not predicted to form any interactions that should hinder the tandem SH2 unit from binding the 2pY peptide. We chose a concentration of tandem SH2 that corresponded to the EC80 value on the tandem SH2:2pY-2pY binding isotherm. We then tested the hit compounds against this assay to probe for compounds that directly competed with 2pY for binding to the tandem SH2 unit ( Fig. 4 ). We performed this experiment with a fresh stock of compound, which may account for the increased potency of the compounds for the tandem SH2 unit compared with those experiments performed with full-length ZAP-70. All the hit compounds from the primary screen also inhibited the isolated tandem SH2 unit. This finding indicates that none of the hit molecules stabilize the autoinhibited conformation formed by full-length ZAP-70. Rather, the inhibitors of 2pY peptide binding act directly on the tandem SH2 domains of ZAP-70.
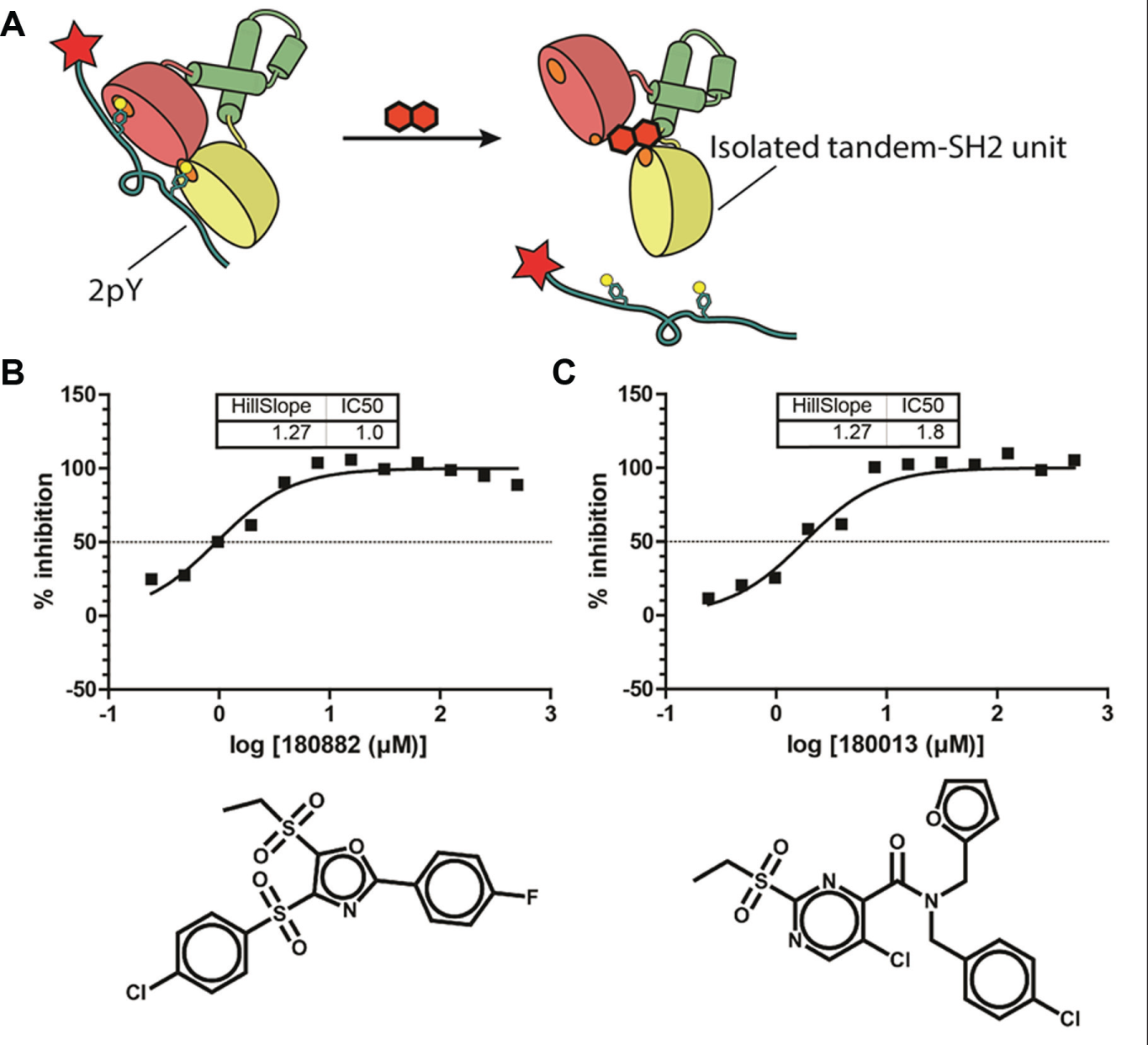
(
Further investigation revealed that the mechanism of action involved covalent modification of the tandem SH2 domains. 8 This covalent modification occurred in the presence of 1 mM TCEP, the reducing agent used in the screening assay. We found that replacement of TCEP with DTT prevented the covalent modification, as described elsewhere. 8
The remaining 108 compounds filtered through the primary screen that were not characterized were picked for further investigation. None of these compounds showed a concentration-responsive inhibition in the presence of DTT (data not shown). This set of 108 compounds was not characterized further. The sensitivity of ZAP-70 to inhibition by thiol-modifying compounds skewed the results of the screen such that the most potent compounds were all thiol-modifying compounds. Examination of the chemical composition of the 86 final hits revealed a diverse group of thiol-sensitive inhibitors across 20 unique scaffolds. Sulfone and sulfonamide compounds were prevalent chemical groups in the hits; the abundance of these groups was not representative of the screening library. The time during which ZAP-70 equilibrated with compounds selected for compounds with a higher rate of reactivity. A shorter equilibration time may reduce the number of covalent hits from the primary screen. Additionally, modification of the protocol described here to use DTT instead of TCEP might provide an opportunity to discover compounds that bind noncovalently.
Footnotes
Acknowledgements
The authors are very grateful to David King (HHMI) for peptide synthesis and mass spectrometry, Tony Iavarone (UC Berkeley, QB3) for mass spectrometry, and Kyle Simonetta, Terri Kadlecek, and Keen-Hoon Ang for invaluable discussions.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by National Institute of Arthritis and Musculoskeletal and Skin Diseases/National Institutes of Health American Recovery and Reinvest Act grant to A.W. and J.K. A Rogers Family Foundation Grant awarded to J.K. and A.W. also supported this work. P.R.V. was supported by a National Institutes of Health/National Cancer Institute UC Berkeley Cancer Lab training grant. K.L was supported by a Berkeley SURF/Rose Hills Undergraduate Fellowship.