
Abstract
Malignant melanomas often arise from nevi, which result from initial oncogene-induced hyperproliferation of melanocytes that are maintained in a CDKN2A/p16-mediated senescent state. Thus, genes that can bypass this senescence barrier are likely to contribute to melanoma development. We have performed a gain-of-function screen of 17,030 lentivirally expressed human open reading frames (ORFs) in a melanoma cell line containing an inducible p16 construct to identify such genes. Genes known to bypass p16-induced senescence arrest, including the human papilloma virus 18 E7 gene (HPV18E7), and genes such as the p16-binding CDK6 with expected functions, as well as panel of novel genes, were identified, including high-mobility group box (HMGB) proteins. A number of these were further validated in two other models of p16-induced senescence. Tissue immunohistochemistry demonstrated higher levels of CDK6 in primary melanomas compared with normal skin and nevi. Reduction of CDK6 levels drove melanoma cells expressing functional p16 into senescence, demonstrating its contribution to bypass senescence.
Introduction
The CDKN2A/INK4A gene, which encodes the p16 protein, is one of the most commonly mutated tumor suppressors. 1 It is deleted, mutated, or epigenetically silenced in variety of tumor types, including melanomas, and germline CDKN2A mutations are observed in melanoma-prone families. 1 The p16 protein is thought to suppress tumorigenesis through its ability to induce cellular senescence. p16 binds to and inhibits the activity of the CDK4/6–cyclin D complex, which normally phosphorylates and inactivates the retinoblastoma tumor suppressor (RB) protein. Thus, inactivation of p16 or other components of the p16-CDK4-RB1 pathway allows E2F1-dependent transcription and cell cycle progression. Indeed, dysfunction of this pathway is a common feature in melanomas. 1
Senescence can be triggered by DNA damage, reduction in telomere length during aging, and expression of oncogenes that drive excessive mitogenic signaling. 2 Senescence also plays a role in tissue remodeling during active repair and early development. Senescent cells are commonly characterized by enlarged morphology, absence of the Ki-67 proliferation marker, and presence of senescence-associated β-galactosidase (SA-β-gal) activity. 2 The best described in vivo example of oncogene-induced senescence is the melanocytic nevus or mole. 3 Melanocytic nevi are benign, clonal lesions of cutaneous melanocytes that most often harbor activating mutations in BRAF or NRAS mitogenic signaling proteins. 1 Increased p16 expression, as seen in nevi, is sufficient to drive senescence in melanoma cell lines with functional RB1. 4
Between 20% and 50% melanomas arise from preexisting nevi. 5 Thus, nevi represent a reservoir of premalignant cells, although the transformation rate is low. Early stage melanomas with a high proliferative activity commonly retain p16 expression, which is generally lost only in late-stage disease. 1 This suggests that there must be mechanisms for bypassing the p16-induced senescence arrest during early stages of melanoma development. Several genes have been identified that are capable of bypassing a senescence barrier. Activated Wnt and Hedgehog pathway signaling and overexpressed TBX2 and BMI1 all act by suppressing p16 expression.6,7 Overexpression of ZBTB7A/LRF or MYC 8 upregulates cyclin E expression and consequently cyclin E/CDK2 activity; this, in turn, promotes cell cycle progression by bypassing the p16-CDK4/6-RB arrest point. However, there is little evidence for ZBTB7A or PDK1 overexpression in primary melanoma, and while MYC gene amplification has been found in primary melanomas, MYC expression was elevated in only a small proportion of these. 9 This suggests that there are as yet unidentified factors that can drive melanoma development past the p16-induced senescence seen in a nevus.
High-throughput functional genome-wide screens are a powerful, unbiased approach that can be used to elucidate novel potentially oncogenic mechanisms. Loss-of-function screens have been used to identify additional genes required for maintenance of cellular senescence.7,10 However, potential oncogenic drivers of senescence bypass are more likely to be revealed using gain-of-function genome screening. Therefore, we chose this approach to identify genes that can trigger bypass of p16-induced senescence in a melanoma cell line. Here, we used a recently constructed lentiviral expression library comprising 17, 030 human open reading frames (ORFs) 11 to look for genes that can overcome senescence in a melanoma cell line containing an inducible p16 expression construct. 4
Materials and Methods
Unless otherwise noted, all regents were obtained from Sigma-Aldrich (Sydney, Australia).
Cells and Culture Conditions
WMM1175 isopropyl β-D-1-thiogalactopyranoside (IPTG)–inducible p16 wild-type (WT) clone B17 cells 4 were a kind gift from Dr. Therese Becker (Westmead Millennium Institute, Westmead, Australia) and cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% Serum Supreme calf serum (Lonza, Waverley, Australia), 4 mM L-glutamine, 1 mM sodium pyruvate, and 10 mM HEPES. To induce p16 expression, IPTG (4 mM) (Amresco, Solon, OH) was added to culture medium. Additional melanoma-derived cell lines were cultured as described previously. 12
Viral Supernatant Library and Plasmids
Lentiviral ORF expression library consisting of viral supernatants arrayed in 96-well plates (n = 205) was obtained from and screened at the ARVEC facility at UQ Diamantina Institute. Lentiviral expression constructs were generated in pLV411G (synonym pLVEIG, accession KF486506.1), a Gateway destination vector that allows EF1α promoter-driven coexpression of an upstream ORF and a downstream green fluorescent protein (GFP), separated by an intervening IRES sequence. Control wells on each plate were four wells containing negative control supernatant derived from plv411 (empty expression plasmid, no ORF and no ccdB gene), two “mock” wells containing viral particles without the expression plasmids, and two positive control wells expressing mutant CDK4R24C. The positive control expression constructs were derived from pcDNA3-HA-Cdk4R24C (kind gift from Dr. William Hahn, Dana Farber Cancer Institute, Boston, MA). The CDK4R24C fragment with a C-terminal FLAG tag was PCR amplified and cloned into pENTR/D-TOPO according to the manufacturer’s directions (Life Technologies, Mulgrave, Australia). The resultant pENTR-CDK4R24C-FLAG entry clone was sequence verified and then recombined with pLV411G as described previously. 11
To generate additional plates for hit validation, expression clones were rearrayed from bacterial glycerol stocks, DNA isolated, and fresh virus generated by packaging in HEK293T cells as described previously. 11
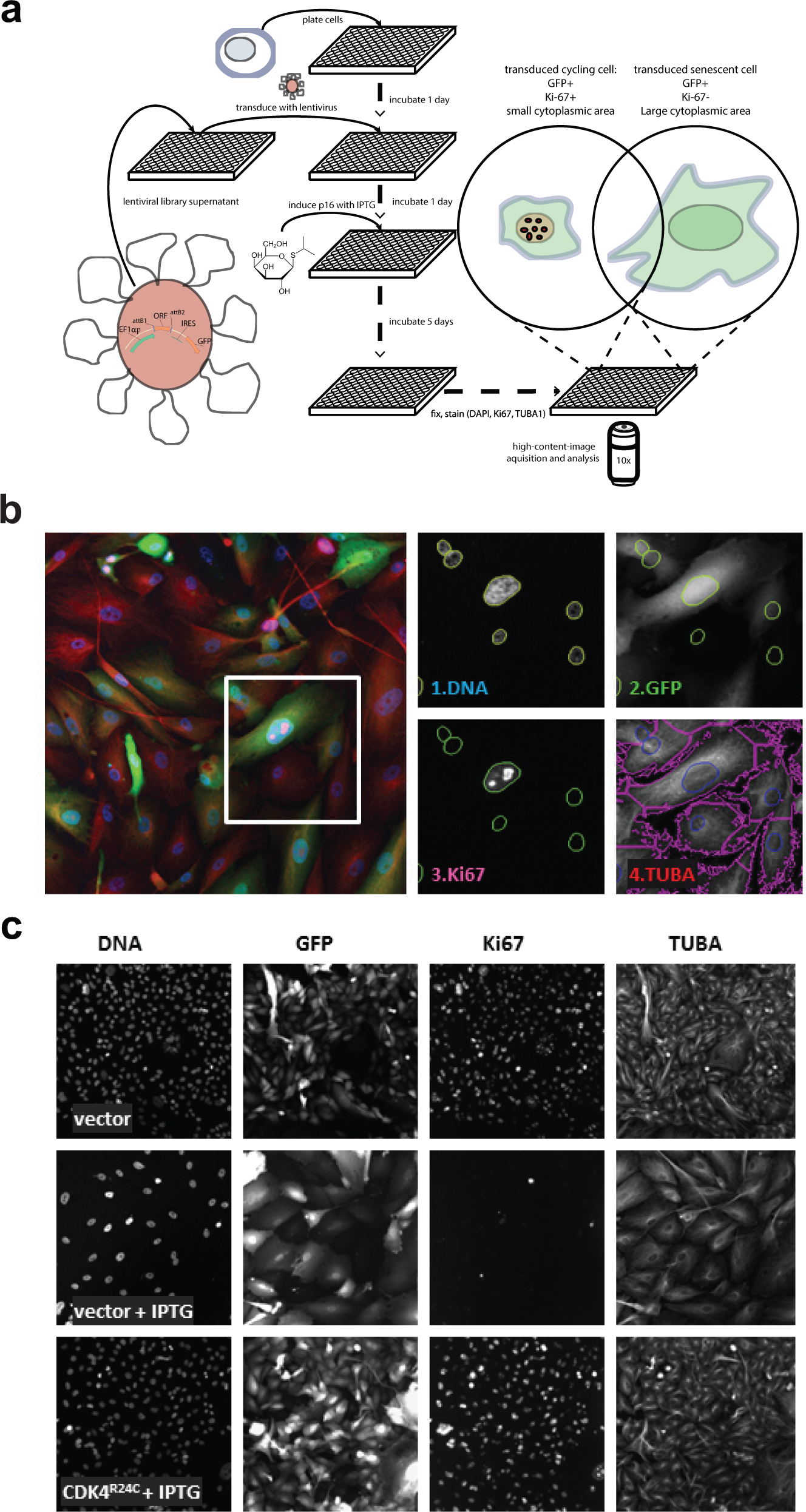
High-Throughput Transductions and Plate Processing for Imaging
Cell seeding and medium and fixative bulk dispensing were performed using a WellMate microplate dispenser (Thermo Fisher Scientific, Hudson, NH). An ELx405
WMM1175 p16 WT clone B17 cells were seeded into ViewPlate96-Black plates (PerkinElmer, Waltham, MA) at 1000 cells per well in 120 µL/well and incubated in the Liconic (Liechtenstein) robotic multiwell plate tissue culture incubator. The following day, media were removed. For this and all subsequent aspirate steps, aspirate tip height was adjusted so that there was a residual volume of 20 µL/well. Plates containing lentiviral supernatants were thawed and hexadimethrine bromide (polybrene) added at 12 µg/mL. This supernatant was then added to cells (30 µL/well), incubated for 2 h, and then topped up with 100 µL per well of medium. The next day medium was aspirated and replaced with 180 µL per well medium with or without 4.4 mM IPTG (Amresco). After an additional 5-day incubation, media were aspirated and replaced with 180 µL/well 3.7% formaldehyde in PBS. After a 15-min incubation at room temperature (RT), wells were washed by streaming 700 µL/well PBS. Unless they were processed immediately, fixed cells were stored at 4 °C in 75 µL/well PBS.
Subsequently cells were permeabilized in 0.05% (w/v) Triton-X-100 in PBS for 15 min, washed in PBS, and then incubated in blocking buffer (1.5% bovine serum albumin [Amresco] and 0.1% Saponin in PBS) for at least 1 h. Blocking buffer was aspirated (residual volume 20 µL) and primary antibodies added in 10 µL/well at 3× concentration in blocking buffer. Plates were incubated for minimum 1 h at RT or overnight at 4 °C. Wells were washed in PBS and then incubated in secondary fluorescently tagged antibodies as above. After another PBS wash, plates were stained with 300 nM 4′,6-diamidino-2-phenylindole (DAPI) in PBS for 1.5 h. Wells were washed and the cells imaged in 75 µL/well PBS.
High-Content Imaging
All antibodies were diluted in blocking buffer as indicated. For the primary screen and initial validation experiment, primary antibodies used were mouse monoclonal anti–Ki-67 (1:600, clone MIB-1; Dako, Campbellfield, Australia) and rabbit polyclonal anti–α-tubulin (1:1500, ab18251; Abcam, Cambridge, MA). Secondary probe-conjugated antibodies used in the screen were goat Cy3-anti-mouse-IgG (1:1500) and goat Cy5-anti-rabbit-IgG (1:1500) (Life Technologies). For subsequent validation experiments, additional antibodies used were as follows: rabbit anti-pRB Ser780 antibody (1:1000; Cell Signaling), rabbit polyclonal anti–Ki-67 (1:1000, ab15580; Abcam), and mouse monoclonal anti-p16 (1:800, clone G175-405; BD Biosciences, San Jose, CA) for primary and goat Cy3-anti-rabbit-IgG (1:1500; Life Technologies) and goat Alexa Fluor 647–anti-mouse-IgG (1:1500; Life Techno-logies) for secondary, as indicated in the figure legends. Plates were scanned using the ArrayScan VTI HCS Reader (Thermo Scientific, Rockford, IL) coupled to a Twister II Plate Handler (PerkinElmer, Waltham, MA). Fluorescent images were captured using a 10× objective and the XF2046 (400-485-558-640QBDR) quadband dichroic and excitation/emission filter set (Omega, Brattleboro, VT). Images were acquired and processed using the Cellomics CellHealthProfiling.v3 algorithm (Thermo Scientific, Rockford, IL, USA) to obtain numerical parameters in four filter channels: blue, green, red, and far red. For the primary screen and initial validation experiments, channels were assigned as follows: (1) blue, DAPI (for setting nuclear mask and collecting cell counts and nuclear DNA content); (2) green, GFP (for transduced cell selection); (3) red, Cy3 (for Ki-67 detection within the nuclear mask and proliferating cell selection); and (4) far red, Cy5 (for α-tubulin detection and setting the cytoplasmic area mask). For the subsequent validation experiments, channels 3 and/or 4 were reassigned to alternative antibodies and/or quantification area masks (e.g., nuclear, cytoplasmic), as indicated in the results.
Data Analysis and Hit Selection
Obtained object parameters were processed using the R system for statistical computation and graphics (http://www.r-project.org/). Data from all four imaging channels for each cell were linked to each object (i.e., nucleus identified by setting a threshold-based mask in the DAPI channel). The screen was performed in five batches of plates, which were stained and imaged separately so that there was considerable variation in signal intensity. To correct for batch-wise differences in signal intensity and thus apply common thresholds for assigning object GFP and Ki-67 status and compare relative DNA content across the whole primary screen, intensity values from these three channels were adjusted with a batch-specific correction factor such that the modes of the density histograms for each of these parameters from all five batches were aligned (
Each of the 205 screened plates contained four negative control wells, two positive control wells, and two mock-transduced wells (treated with supernatant with empty viral particles), one of which was not stained with primary antibodies. The two mock wells were used to set fluorescence intensity thresholds for assigning GFP and Ki-67 status, respectively, to individual nuclei. For quantitation of GFP expression and Ki-67 staining, average nuclear fluorescence intensities were used. For determination of relative DNA content and assigning cell cycle phase, total integrated intensities from staining with DAPI were used. The thresholds were batch-adjusted postscreening using density plots (
Details of the other methods used are provided in the Supplementary Material.
Results
Immunohistochemical staining of a panel of 57 nevi and 34 primary melanomas showed that 51 of 57 nevi and 29 of 34 melanomas were positive for p16 staining and that overall staining for p16 was very similar in both groups (
To identify such genes, an overexpression screening approach was adopted, using a clonal isolate of the melanoma-derived WMM1175 cell line (which has homozygous deletion of endogenous CDKN2A) containing an IPTG-inducible wild-type p16 construct. These cells have functional RB and undergo senescence upon IPTG-induced expression of p16. This is independent of p53 and does not involve the DNA damage response often associated with senescence.
4
The induction of p16 was verified by immunoblotting (
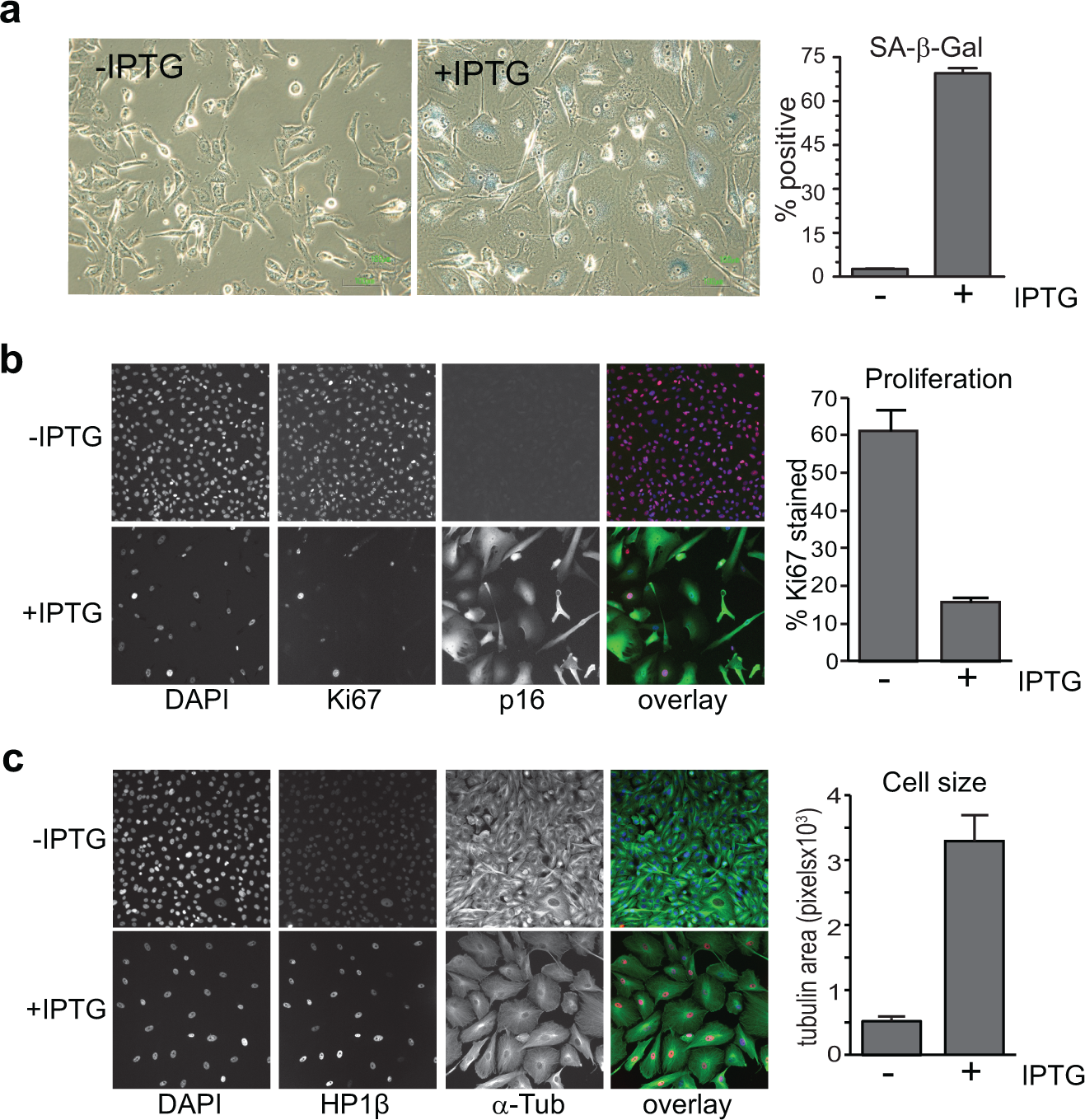
(
(
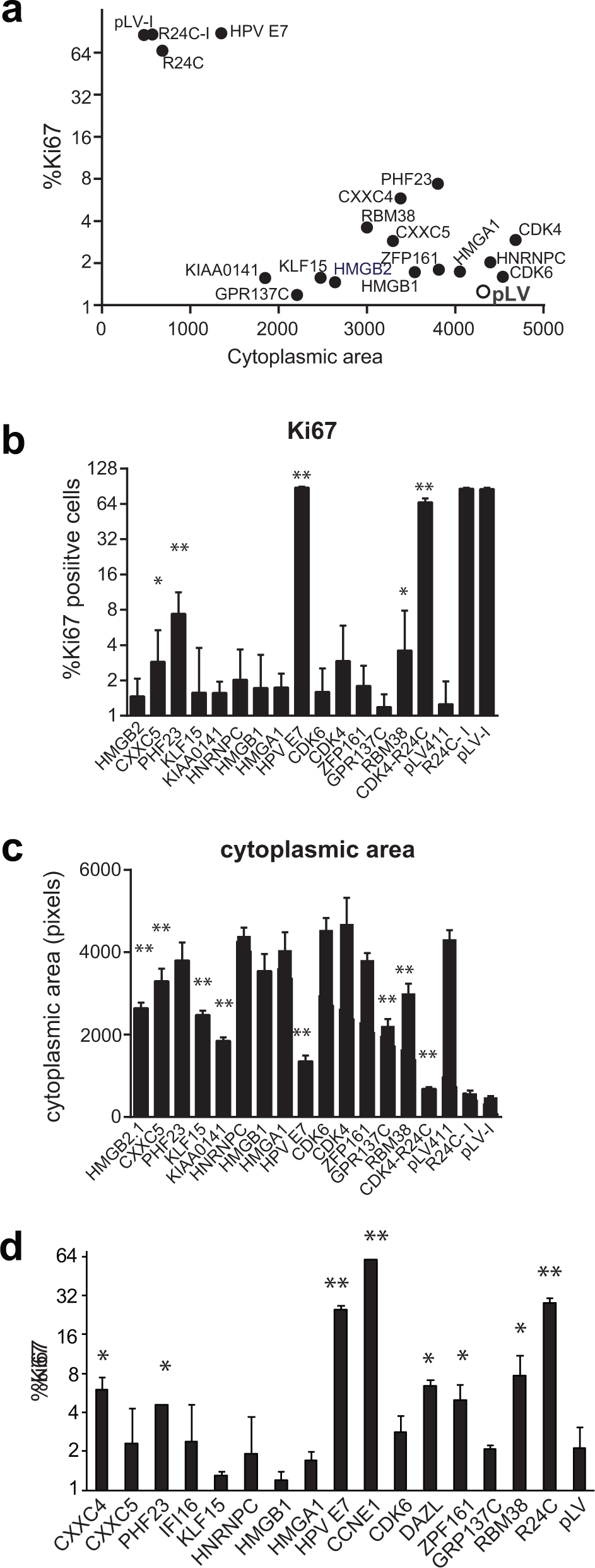
Data for valid wells (defined as having >100 GFP-positive cells) from the 202 plates that passed technical quality control are presented in
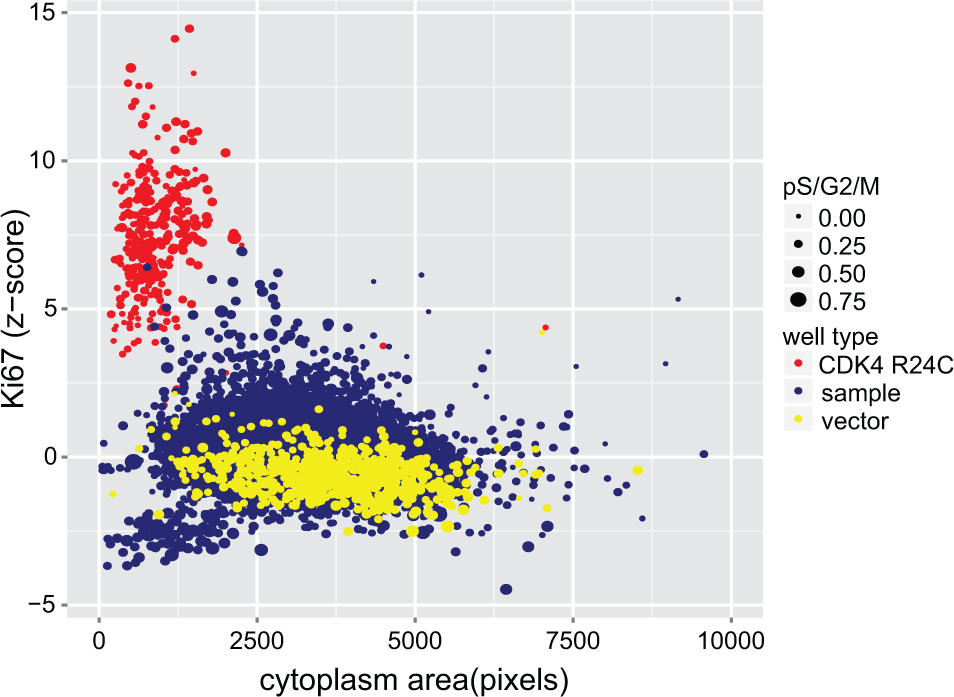
Scatterplot of variables used for hit selection; Ki-67 robust z score (y-axis), cytoplasm area (pixels, x-axis), and combined proportion of nuclei in S, G2, and M phases of the cell cycle (point size), representing data for all wells analyzed in the primary screen. Sample wells (blue) were considered putative hits if they grouped with CDK4R24C (red) and away from vector (yellow) wells.
To confirm these candidate hits, we performed a secondary screen in triplicate. Sequence-verified hit clones were rearrayed from bacterial glycerol stocks and fresh virus generated. Validated hits were selected by comparison of Ki-67 scores (
Confirmed Hits Grouped by Functional Category.
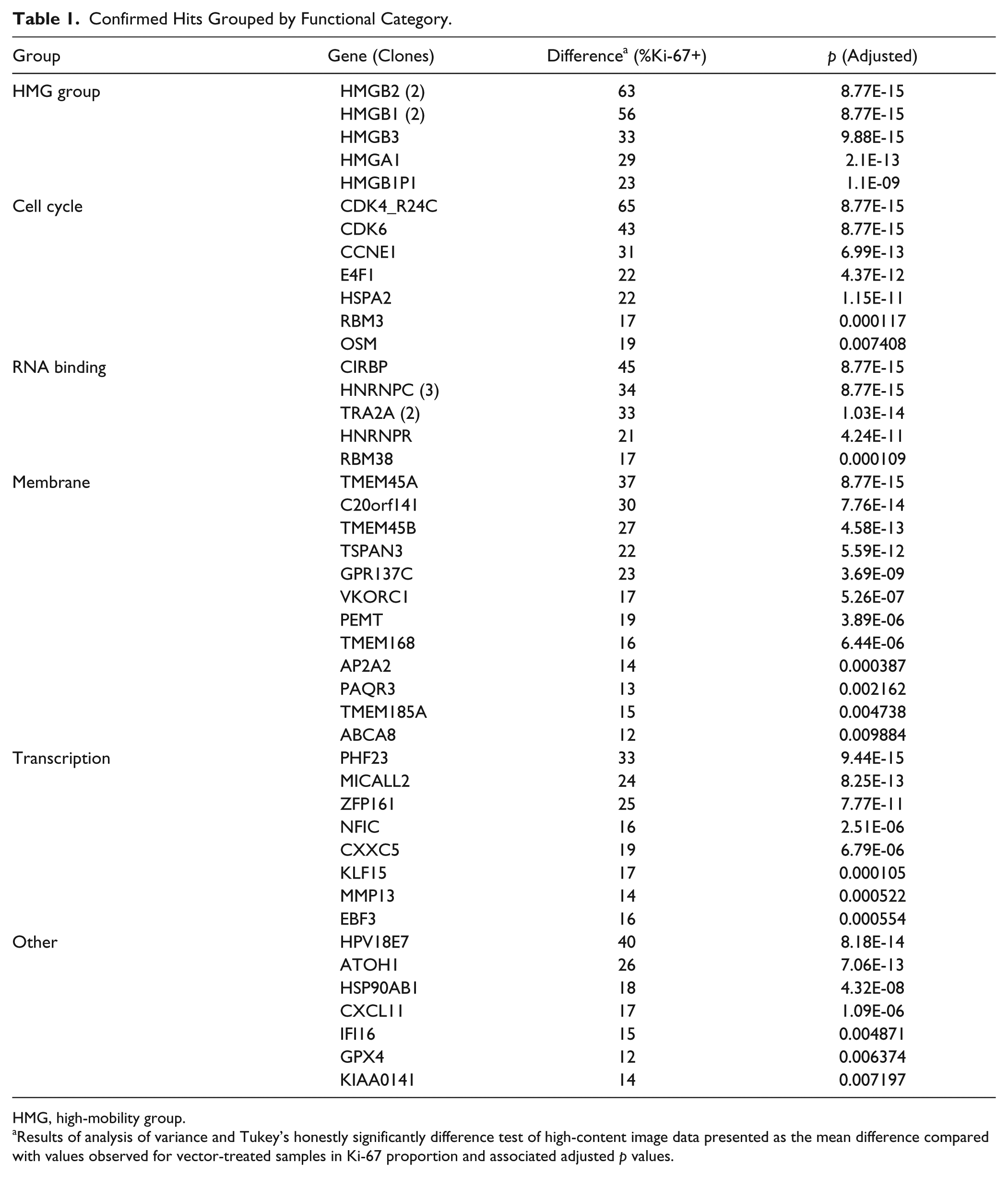
HMG, high-mobility group.
Results of analysis of variance and Tukey’s honestly significantly difference test of high-content image data presented as the mean difference compared with values observed for vector-treated samples in Ki-67 proportion and associated adjusted p values.
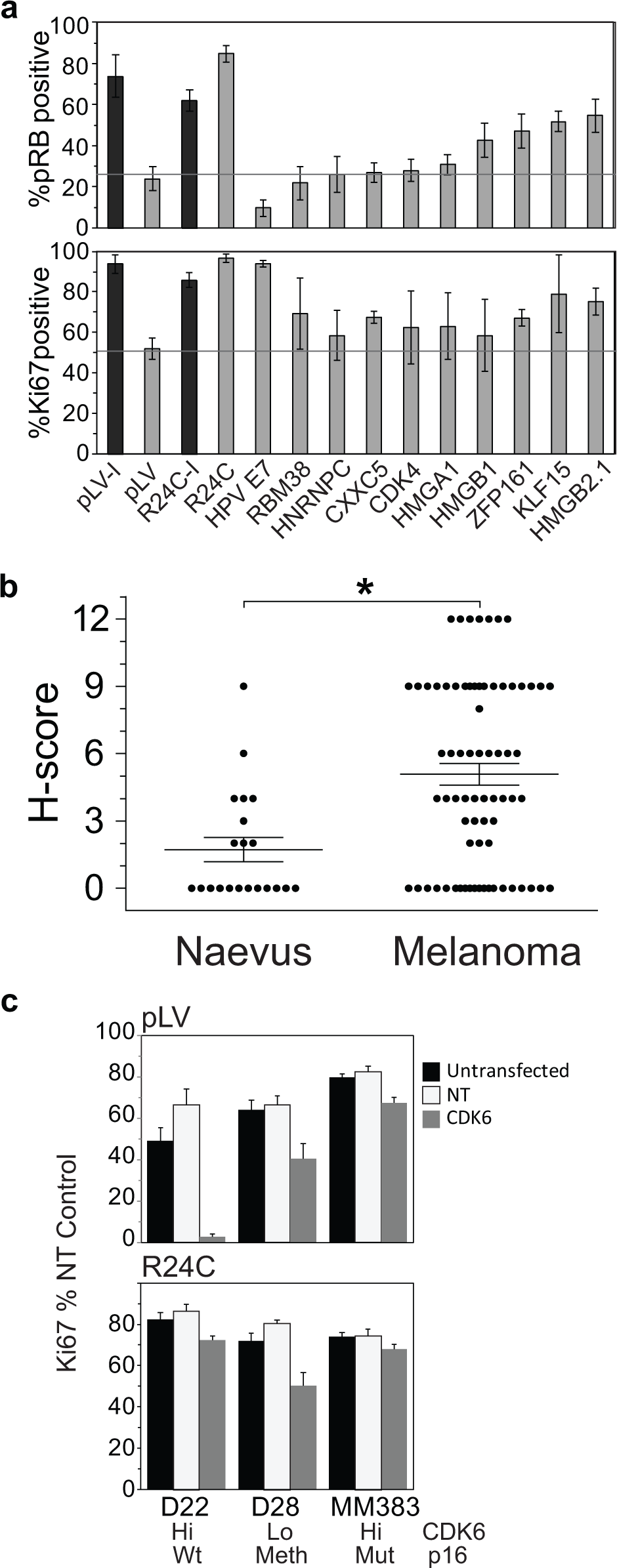
Hit genes were also assessed in two other p16-inducible models of senescence derived from breast cancer–derived MDA-MB-231 cells and melanoma-derived SKMel13, 12 respectively. In each of these cases, overexpression of the control genes CDK4R24C and our hit HPV E7 drove a strong bypass of the p16-induced proliferative arrest ( Fig. 4 ). Cyclin E overexpression also strongly drove senescence bypass in the SKMel13 model ( Fig. 4d ), as in the WMM1175 model. Two genes (PHF23, RBM38) consistently increased proliferation in both the MDA-MB-231 and the WMM1175 models, albeit to a lesser degree than the positive controls ( Fig. 4a , d ). CXXC5 and ZPF161 each produced a weaker bypass in one of the model lines, while CDK6 just failed to reach significance in SKMel13. HNRNPC, HMGA1, and HMGB1 had no obvious activity in these models. A subset of genes from the initial hit list, KIAA0141, KLF15, and GPR137C, and the strongest hit from the screen, HMGB2, failed to increase proliferation but significantly (p < 0.001) reversed the increased cell size associated with p16-induced senescence in MDA-MB-231 cells ( Fig. 4c ). KLF15 affected cell size in both uninduced and p16-induced conditions. Thus, at least six hits were validated in at least one other model of p16-induced senescence, and a further four significantly reduced the increased cell size that is a hallmark of senescence. The robust effect of HMGB proteins and CDK6 in bypassing p16-induced senescence arrest in the WMM1175 but weaker effects in the other models suggests that cell line–intrinsic features, such as the relative levels of induced p16 expression and HMGB and CDK6 expression, can influence the outcome.
MDA-MB-231 cells with inducible p16 were transduced with the indicated lentivirus and induced to express p16 by addition of doxycycline the next day. After 5 days, cells were fixed and assessed for Ki-67 (
The primary target of p16 inhibition of CDK4/cyclin D that results in G1 phase arrest and senescence is RB, particularly the phosphorylation of RB on Ser780 (pRB), a CDK4/cyclin D phosphorylation site. We examined the effect of selected hits on RB phosphorylation. Three genes, HMGB2, ZFP161, and KLF15, increased Ki-67 staining and pRB levels, suggesting that they may also operate upstream of CDK4/cyclin D ( Fig. 5a ). Cells expressing HPVE7, which induces RB degradation, 13 had reduced levels of pRB, as expected ( Fig. 5a ). None of the other hit genes analyzed had similarly reduced level of pRB, indicating that degradation of RB was not a mechanism of senescence bypass for these genes.
(
CDK6 was one of the top hits in the screen. The level of CDK6 protein was assessed on the set of lesions as in
Figure 1
. A subset of melanomas with strongly increased CDK6 levels compared with normal skin or nevi (
Fig. 5b
). Immunoblot analysis of cell extracts from a panel of melanoma cell lines revealed MM383, SKMel28, and D22 had elevated levels of CDK6 (
Discussion
Here we have reported a genome-wide overexpression screen to identify novel drivers of bypass of p16-induced senescence. The identification of HPV18 E7 validated the screen, while the ability of cyclin E overexpression to overcome the p16-induced arrest in multiple models is further evidence that the screen was capable of identifying potential bypass genes. Cyclin E was identified as an essential target of both ZBTB7A and MYC to overcome oncogene-induced senescence arrest. 8
Among the novel hits, the incidence of HMG family members was striking and is discussed further below. Other strong hit genes have previously been reported to either interfere with senescence or at least be involved in regulation of a G1 phase cell cycle arrest. Cold-inducible RNA-binding protein (CIRBP) can bypass replicative senescence without affecting p16 expression and is upregulated in a range of cancers but not in melanoma. 14 TMEM45A has been reported to be overexpressed in ovarian cancers, and its depletion resulted in G1 phase accumulation. 15 HSPA2, a member of HSP70 family, is overexpressed in breast cancers although rarely in melanoma, and its depletion drives a senescence-like phenotype. 16 ZNF161 (ZBTB14) is related to ZBTB7A and may act in a similar manner to bypass senescence arrest by upregulating cyclin E expression. 8 Oncostatin M (OSM) has a range of effects, dependent on the cellular context. It can drive a senescence phenotype by upregulating STAT3 activity in the context of a normal cell, but in MYC-overexpressing cells, OSM promotes a transformed phenotype. 17 TRA2A is a nuclear RNA binding protein phosphorylated by CDK4/6, suggesting a role in regulating G1 phase progression. 18 In addition to these genes, we have identified four novel genes with modest senescence bypass activity in all three models assessed here. PHF23 contains a plant homeodomain (PHD) motif that binds H3K4me3 residues and is present in an oncogenic fusion with NPU98 in leukemia. 19 RBM38 is an RNA binding protein that regulates the translation of p53. 20 CXXC4 and CXXC5 are inhibitors of WNT signaling through direct binding to the Disheveled protein, which in turn drives degradation of β-catenin. 21 Little is known about how these genes contribute to senescence bypass during transformation from nevus to melanoma.
HMGA and HMGB gene family members are frequently upregulated in cancer, which often correlates with poor prognosis. 22 HMGA proteins have been demonstrated to have an essential role in Ras-induced senescence, with a major role in establishing senescence-associated heterochromatin foci. 23 HMGA1 has been reported to bind RB directly and inhibit binding of HDAC1 to RB, thereby enhancing E2F activity. 24 Surprisingly, despite the reported direct effect of HMGA1 binding to RB on its activity, HMGA1 was a weak bypass gene in the WMM175 model and failed to bypass senescence in either of the other models tested. The remarkable finding that the HMG proteins B1, B2, and B3 were all strong hits in our screen suggests that p16-induced senescence bypass is an activity shared by this family of proteins, at least when overexpressed. HMGB1 is secreted from senescent cells and regulates the senescence-associated secretory phenotype. 25 Evidence from the literature provides support for potential roles for HMGB proteins in senescence. HMGB2 has been linked to senescence through altering the β-catenin pathway activity by enhancing the β-catenin/Lef transcriptional program. 26 HMGB1 and HMGB2 also modify p53- and p73-dependent transcriptional activity. 27 Secreted HMGB1 acts as a cytokine, signaling through RAGE and TLR4 receptors to activate MAPK and NF-κB pathways. 28 However we found that neither culture supernatant from HMGB2-overexpressing WMM1175 cells nor purified HMGB1 were capable of bypassing p16-induced senescence in WMM1175 cells (unpublished observations). This argues that HMGB proteins are unlikely to act as cytokines to bypass senescence.
HMGB2, along with two other hits from our study, HNRNPC and HSP90AB1, were all strongly downregulated in cells driven into senescence. 29 In addition, CXXC4 and CXXC5 are inhibitors of WNT signaling, which act by directly binding to the Disheveled protein and driving degradation of β-catenin. 21 WNT signaling is a common feature of nevi, 30 and thus inhibition of WNT signaling could contribute to overcoming senescence. Thus, it is possible that multiple hits from this screen use similar signaling pathways to bypass senescence.
We have functionally validated one of the hits from the screen, CDK6. Although CDK6 was highly expressed in a subset of melanomas, CDK6 and wild-type p16 are very rarely coexpressed at a high level. This was detected in only one melanoma cell line examined, D22. In MM383, CDK6 is overexpressed, but CDKN2A is methylated and not expressed. Thus, other mechanisms may supplant the requirement for high CDK6 as tumors progress. The finding that CDK6, but not CDK4, overexpression effectively bypassed senescence may reflect the relative level of CDK4 and CDK6 expression achieved in the screen, as previous studies have reported that CDK4 also exhibited this activity. 31 Compared with CDK6, CDK4 is more commonly upregulated in melanoma, although both occur. CDK6 overexpression drove weak bypass of p16-induced senescence arrest in MB-MDA-231 cells and failed in the SK-Mel-13 cell model, possibly reflecting the level of CDK6 overexpression and p16 induction in these models. Nevertheless, the fact that depletion of CDK6 in an overexpressing melanoma cell line restored p16-induced arrest demonstrated that CDK6 overexpression contributes to senescence bypass in a melanoma context.
In summary, we have described here a high-throughput screening approach using 17,030 human open reading frames for identifying genuine genes that can bypass p16-induced senescence in the WMM1175 cell line when overexpressed. The relevance of this screening approach was supported by the detection of HPV18E7, which is known to abolish p16-induced senescence. In addition, CDK6, which is known to be involved in the retinoblastoma pathway, was identified. Knockdown of CDK6 in a p16-wild-type and CDK6-overexpressing melanoma cell line reinstated proliferative arrest. Our screening approach also identified unexpected genes, notably the HMGB family of proteins, as potential p16-induced senescence bypass genes. These results suggest that the ability of overexpressed HMGB proteins to bypass p16-induced senescence is a class effect of this protein family.
Footnotes
Acknowledgements
We thank Dr. Therese Becker and Prof. Helen Rizos for the WMM1175 cell line, Dr. Alexey Bazarov for the MDA-MB-231-TetR-CMV/TO-p16 cell line, Prof. H. Peter Soyer for advice on histology, and Prof. Nick Hayward and Prof. Dorothy Bennett for discussions on this work.
Supplementary material is available online with this article.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
This work was supported by grants from the Worldwide Cancer Research (formerly AICR), the University of Queensland, and NHMRC Australia. W.J.L. was an NHMRC C. J. Martin Fellow, and B.G. was an NHMRC Senior Research Fellow.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.