
Abstract
Four inhibitory compounds were identified using a poly-uridylic acid (polyU) mRNA-directed aminoacylation/translation (A/T) protein synthesis system composed of phenylalanyl-tRNA synthetases (PheRS), ribosomes, and ribosomal factors from Pseudomonas aeruginosa in an in vitro screen of a synthetic compound library. The compounds were specific for inhibition of bacterial protein synthesis. In enzymatic assays, the compounds inhibited protein synthesis with IC50 values ranging from 20 to 60 μM. Minimum inhibitory concentrations (MICs) were determined in cultures for a panel of pathogenic organisms, including Enterococcus faecalis, Escherichia coli, Haemophilus influenzae, P. aeruginosa, Staphylococcus aureus, and Streptococcus pneumoniae. All the compounds were observed to have broad-spectrum activity and inhibited an efflux pump mutant strain of P. aeruginosa with MICs of 0.5–16 μg/mL. The molecular target of two compounds was determined to be PheRS. These two compounds were bacteriostatic against both Gram-positive and Gram-negative pathogens. In competition assays, they were not observed to compete with the natural substrates ATP or phenylalanine for active site binding. The other two compounds directly inhibited the ribosome and were bactericidal against both Gram-positive and Gram-negative pathogens. In cytotoxicity MTT testing in human cell lines, the compounds were shown to be from 2500- to 30,000-fold less active than the control staurosporine.
Introduction
A Gram-negative microorganism, Pseudomonas aeruginosa, is an opportunistic bacterial pathogen and a causative agent in a wide range of infections. Infections range from bacteremia and urinary tract infections to burn wound infections and pulmonary infections. In the hospital setting, P. aeruginosa is responsible for approximately one-seventh of all infections, and has been identified as the most common Gram-negative pathogen located in the intensive care unit. 1 Clinical isolates of antibiotic resistance strains of P. aeruginosa are significant and growing. In recent reports, the Infectious Disease Society of America (IDSA) has summarized the current and future public health burden resulting from drug-resistant bacteria. According to these reports, the current annual cost to the U.S. health care system for antibiotic-resistant infections is as much as $34 billion and growing. 2 On a global scale, antibiotic resistance has become a serious problem due to an increase in incidences, as well as the lack of innovative antibiotics available. The number of new antibacterial agents approved by the Food and Drug Administration (FDA) has dramatically decreased since 1983, and of the new antibiotics that have been approved for use by the FDA, most are modified forms of previously marketed antibiotics. 3 Although this has been a successful short-term strategy, it is no fix for a growing worldwide resistance issue.
The focus of this study was to use an optimized in vitro protein synthesis system to screen a synthetic chemical compound library and analyze the antibacterial activity of the inhibitors identified. Previously, we developed poly-uridylic acid (polyU) mRNA-directed aminoacylation/translation (A/T) assays and monitored the activity of these systems using scintillation proximity assays (SPAs). These systems have been validated in high-throughput assays.4,5 Using this system from P. aeruginosa as a platform, we have screened a synthetic chemical compound library, and four compounds were identified as inhibitors of protein biosynthesis. These compounds were characterized for activity against bacterial growth, mechanism of action, and toxicity in human cell cultures.
Methods and Materials
Materials
All chemicals were obtained from either Sigma Aldrich (St. Louis, MO) or Fisher Scientific (Pittsburgh, PA). Radioactive isotopes, SPA beads, and 96-well screening plates were from PerkinElmer (Waltham, MA). Escherichia coli tolC mutant, P. aeruginosa PAO200 (efflux pump mutant), and P. aeruginosa hypersensitive (ATCC 35151) strains were a kind gift from Urs Ochsner (Crestone Pharma, Boulder, CO). All other bacteria were from the American Type Culture Collection (ATCC) (Manassas, VA). Penicillin-streptomycin solution was from Mediatech, Inc. (Centerville, IA). The synthetic Anti-Infectives Library was from TimTec LLC (Newark, DE) and included 890 low-molecular-weight, druglike molecules with scaffolds found in antiseptic agents with antibacterial, antifungoid, and antimicrobial activities. Compounds were supplied as 10 mM stocks dissolved in DMSO, stored at −20 °C, and thawed immediately before analysis. The compounds have an average purity of 95%, and the minimum purity is at least 90%.
Chemical Compound Screening
An SPA was developed for the P. aeruginosa A/T assay, and the concentrations of biological and nonbiological components were as previously described. 5 The screening reactions were carried out in 96-well microtiter plates (Costar). The final assay (50 μL) contained 50 mM Tris-HCl (pH 7.5), 25 mM KCl, 10 mM MgCl2, 0.03 mM spermine, 1.5 mM ATP, 0.5 mM GTP, 40 μM [3H]phenylalanine (Phe), 0.3 mg/mL polyU mRNA, and 20 μM E. coli tRNA. Constant levels of ATP and GTP were maintained in the assay using a nucleotide regeneration system composed of 4 mM phosphoenolpyruvate (PEP) and 0.025 units/μL pyruvate kinase (PK). P. aeruginosa biological components were in the following concentrations: ribosome (0.2 μM), PheRS (0.1 μM), EF-Tu (1 μM), EF-Ts (0.05 μM), and EF-G (0.2 μM).
Initially, 33 μL of the protein/substrate mix (without tRNA) was added to 2 μL of chemical compound (3.3 mM) dissolved in 100% DMSO. This mixture was allowed to incubate at ambient temperature for 15 min, and then reactions were initiated by addition of 15 μL of E. coli tRNA, followed by a 2 h incubation at room temperature (comparable to 1 h at 37 °C). Reactions were stopped by the addition of 5 μL of 0.5 M ethylenediaminetetraacetic acid (EDTA). A total of 200 μg of SPA beads (RNA Binding Beads [YSI], PerkinElmer) in 150 μL of 300 mM citrate buffer (pH 6.2) was added. The plates were analyzed using a 1450 Microbeta (Jet) liquid scintillation and luminescence counter (Wallac). Assays to determine IC50 values were as described above with the test compounds serially diluted from 200 to 0.4 μM. To determine the mechanism of action of BT04E11 and BT06G06, with respect to PheRS substrates ATP and Phe, competition assays were carried out as previously described. 6
Microbiological Assays
Broth microdilution MIC testing was performed in 96-well microtiter plates according to Clinical and Laboratory Standards Institute (CLSI) guidelines. 7 MIC values were determined for E. coli (ATCC 25922), E. coli tolC efflux pump mutant, Enterococcus faecalis (ATCC 29212), Haemophilus influenzae (ATCC 49766), P. aeruginosa (ATCC 47085), P. aeruginosa PAO200 (efflux pump mutant), P. aeruginosa hypersensitive strain (ATCC 35151), Staphylococcus aureus (ATCC 29213), and Streptococcus pneumoniae (ATCC 49619).
Time-kill experiments were performed according to CLSI guidelines. 8 Growth media was Brain Heart Infusion or Trypticase Soy Broth from Remel (Lenexa, KS). For the experiments, 10 mL of broth medium was inoculated with 0.1 mL of a fresh overnight culture and grown at 37 °C with shaking (200 rpm) for 2–3 h. Prewarmed flasks containing 10 mL of medium alone or 10 mL of medium containing a test compound at 4× MIC were then inoculated with 0.1 mL of the exponentially growing cultures. Samples were removed at 0, 2, 4, 6, and 24 h, and serial dilutions were plated on blood agar to allow for colony enumeration and calculation of live cell density.
Eukaryotic Protein Synthesis Assays
Reactions to determine the inhibitory effect of compounds on eukaryotic protein synthesis were carried out using wheat germ cell extracts as described. 4 The concentrations of the compounds in these assays ranged from 0.4 to 200 µM, and the concentration of the cycloheximide in the control reactions was from 0.3 to 300 µM.
Assays were carried out to test the inhibitory effect of compounds on the activity of human mitochondrial PheRS (hmPheRS). hmPheRS was prepared as described. 9 The final assay (50 µL) contained 50 mM Tris-HCl (pH 7.5), 1 mM spermine, 10 mM MgOAc, 2.5 mM ATP, 1 mM 1,4-dithiothreitol (DTT), 75 μM [3H] Phe, 2 μM E. coli tRNAPhe, 0.5 μM hmPheRS, and 2 µL of compound. 6 A total of 33 µL of the mix (minus tRNA) was incubated for 15 min at room temperature, with 2 µL of compound serially diluted to result in concentrations from 200 to 0.04 μM in the final reactions. The reaction was then initiated by addition of 15 µL tRNA. The reactions were stopped by diluting into 3 mL of ice-cold 5% trichloroacetic acid (TCA) and filtered through glass fiber filters as described. 4
Cytotoxicity Test against a Human Cell Line
To determine the effect of hit compounds on the growth of human cell cultures, in vitro cytotoxicity testing was carried out as described using human embryonic kidney 293 cells (HEK-293). 10 The Trevigen TACS MTT Cell Proliferation Assay Kit (Gaithersburg, MD) was utilized to assess impacts on human cell proliferation and/or viability. The control staurosporine was serially diluted in assays from 1 to 0.001 μg/mL, and the compounds tested were serially diluted in assays from 120 to 3.75 μg/mL or from 400 to 25 μg/mL.
Results and Discussion
We previously developed a screening platform using an A/T protein synthesis assay from P. aeruginosa to detect inhibitors of protein biosynthesis. 5 The biological components of the A/T system include ribosomes, elongation factor Tu (EF-Tu), elongation factor Ts (EF-Ts), elongation factor G (EF-G), and phenylalanyl-tRNA synthetase (PheRS) from P. aeruginosa. The concentration of all components of the system was optimized by multiple titrations of each component into the system to determine the inflection point of saturation on a titration curve. Concentrations were set just below the saturation points to facilitate maximum sensitivity to inhibition of each and every enzymatic component of the system. 5 In the SPA assay, the RNA portion of ribosomes was used to localize the ribosome to scintillation beads, enabling detection of the nascent radiolabeled poly-phenylalanine (poly-Phe) peptide still attached to the ribosome. The optimal pH for ribosome/bead binding was determined to be pH 6.2, and at this pH, in the absence of ribosomes, negligible amounts of tRNA charged with [3H]Phe or free [3H]Phe bound to the beads.
Screening of a Chemical Compound Library against the P. aeruginosa A/T System
A synthetic chemical compound library containing 890 compounds was tested in a medium-throughput format. The screening reactions were very robust, with an average signal-to-background ratio of 54:1 and a percent coefficient of variation (%CV) of 0.31. The Z′ and Z factors across all plates averaged 0.6 and 0.1, respectively. The initial screen contained single-point assays with chemical compounds at 132 μM; compounds that inhibited more than 50% of poly-Phe synthesis were defined as hits. The hit compounds were retested in triplicate for confirmation of inhibitory activity.
The confirmed hit compounds were next tested in specificity assays against each of the four accessory proteins (EF-Tu, EF-Ts, EF-G, and PheRS) as described. 5 Four compounds (BT04E11, BT06D07, BT06G06, and BT08F11) were found to be specific inhibitors of a single component of the A/T assay ( Fig. 1 ). Two of the hit compounds (BT06D07 and BT08F11) did not inhibit an accessory protein but did inhibit poly-Phe synthesis in the biochemical assay, indicating the mechanism of action of these compounds was direct inhibition of the ribosome. The other two compounds (BT04E11 and BT06G06) were found to specifically inhibit PheRS. The IC50 values for these four compounds were determined by serially diluting the compounds from 200 to 0.4 µM in A/T assays or tRNA aminoacylation assays. The two compounds BT04E11 and BT06G06 inhibited the activity of PheRS in the A/T assay with IC50 values of 48.4 and 63.3 µM, respectively ( Fig. 2A , B ). When these two compounds were retested using the aminoacylation assay, similar IC50 values were observed ( Fig. 2 E , F ). BT06D07 and BT08F11 inhibited the function of the ribosome with IC50 values of 23.4 and 29.1 μM, respectively ( Fig. 2 C , D ).
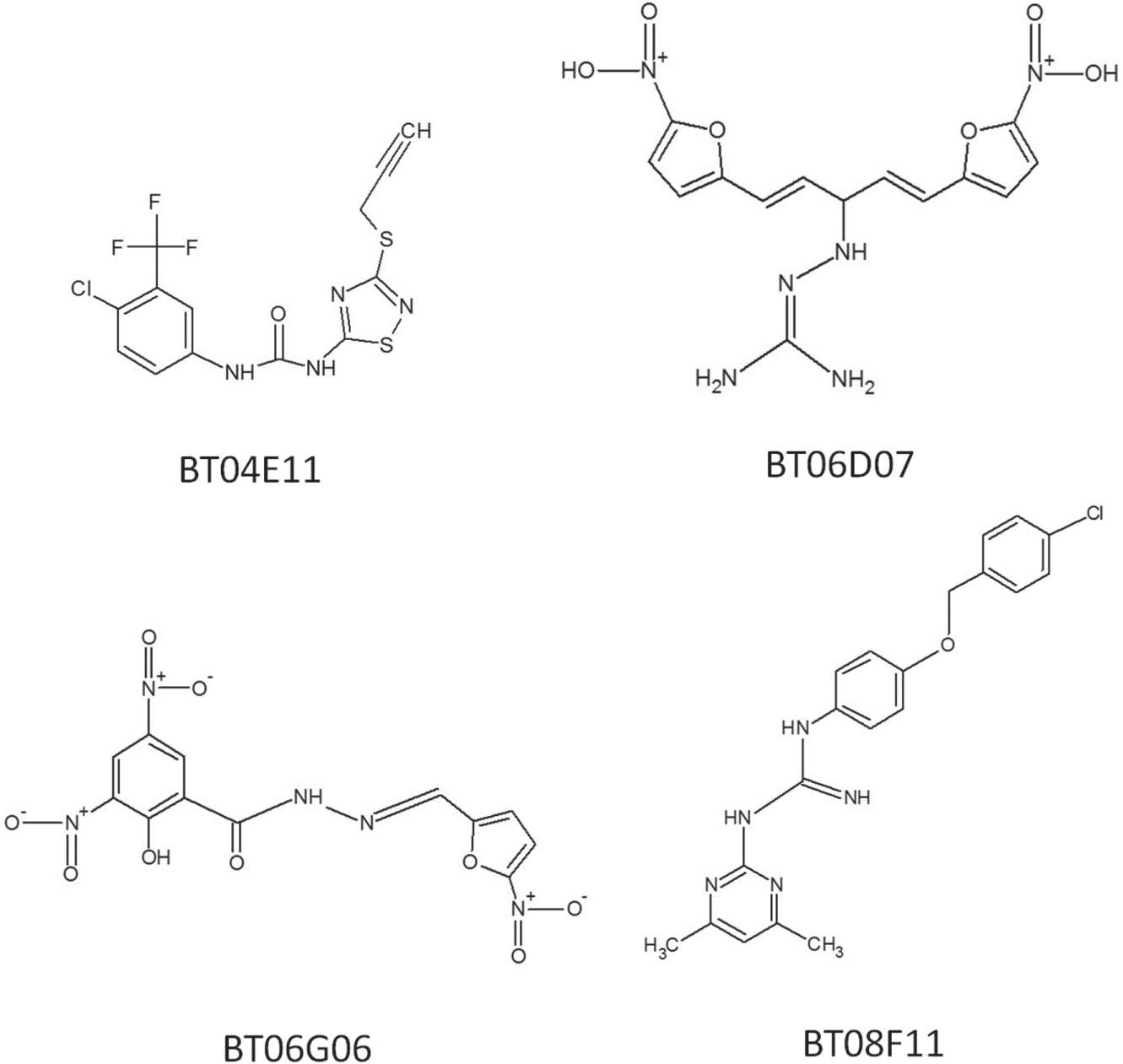
The chemical structure of the four hit compounds: BT04E11, BT06D07, BT06G06, and BT08F11.
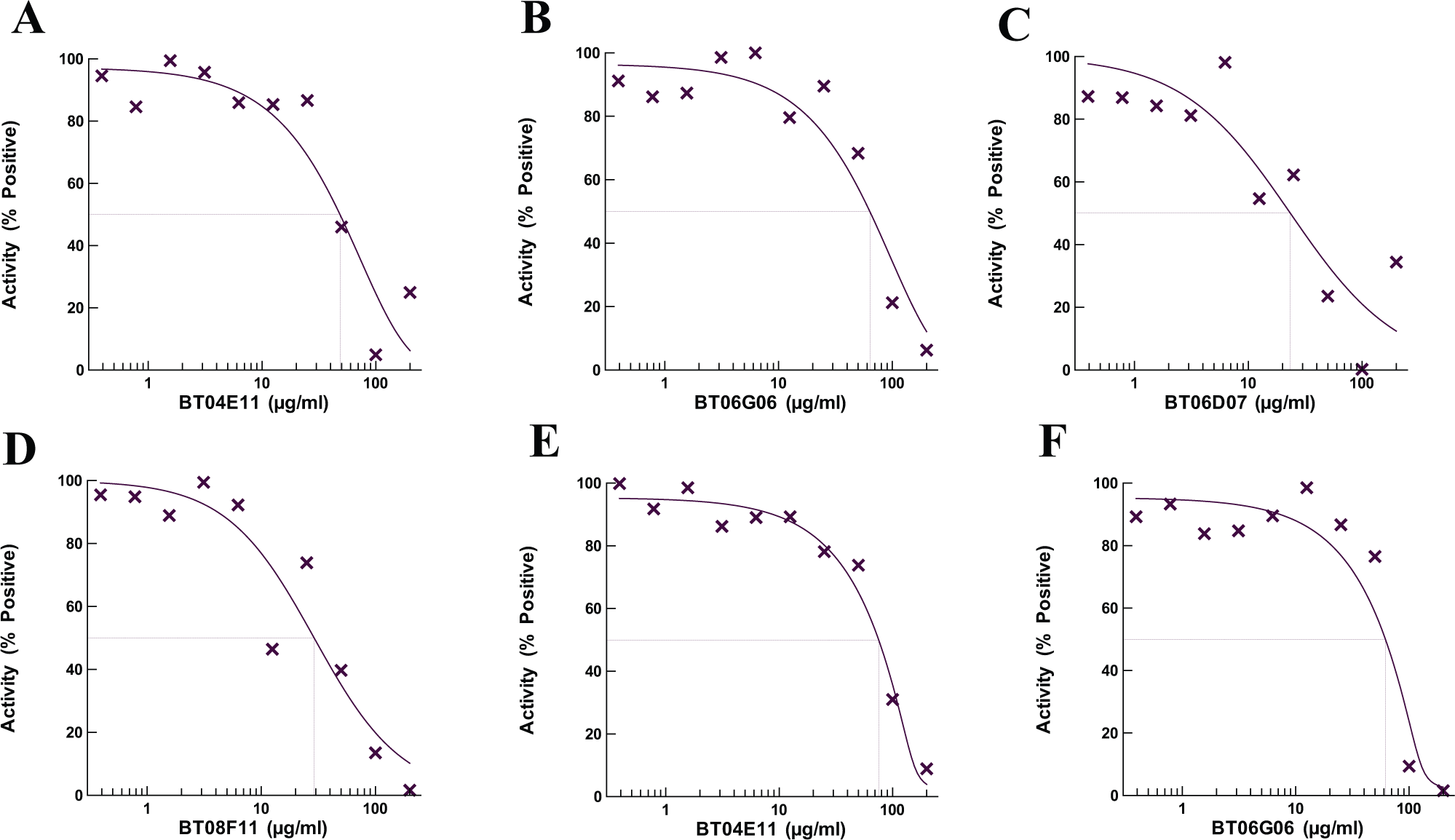
Determination of IC50 values in enzymatic assays. The test compounds were serially diluted from 200 to 0.4 μM in assays to determine the IC50 values. The IC50 values were initially determined using A/T assays (
Excluding tRNA, PheRS has two molecular substrates, ATP and phenylalanine. To determine if BT04E11 and BT06G06 inhibited the activity of PheRS by competing with one or both of these substrates for binding in the active site, competition assays were carried out. The IC50 values were monitored to detect a possible increase or decrease as each substrate was titrated into the assays. The mechanism of inhibition with respect to ATP was determined at various ATP concentrations (25, 50, 100, 250, 500, and 1000 µM) while holding the Phe concentration constant. To determine the mechanism of inhibition with respect to the amino acid, the same assay was used, except ATP was held constant and the IC50 was determined at different concentrations of Phe (25, 50, 100, 200, and 300 µM). There was no shift in the IC50 observed in any of the assays, indicating that both compounds were noncompetitive with both of the natural substrates (data not shown).
Microbiological Assays
The four hit compounds were tested in broth microdilution assays to determine minimum inhibitory concentrations (MICs) against a panel of nine pathogenic bacteria, including efflux pump mutants of E. coli and P. aeruginosa and a hypersensitive strain of P. aeruginosa ( Suppl. Table S1 ). None of the compounds had good inhibitory activity against the native strains of either E. coli or P. aeruginosa; however, all the compounds did inhibit the efflux pump mutants of both E. coli and P. aeruginosa and the hypersensitive strain of P. aeruginosa, indicating that the likely reason for the lack of activity against the wild-type bacteria was efflux. The compounds all had good activity against H. influenzae, the other Gram-negative bacteria in the panel, and modest activity against the Gram-positive bacteria.
Next, time-kill analysis was used to determine if the compounds were bacteriostatic or bactericidal. The compounds were tested against H. influenzae and S. aureus in time-kill studies at four times the MIC between 0 and 24 h. BT04E11 and BT06G06 inhibited the function of PheRS, and both were shown to be bacteriostatic against both bacteria. Both compounds were observed to have constant growth but a decrease in colony-forming units (CFU) of 2 to 4 log10 compared with the control during the first 6 h, and then an increase in growth at 24 h ( Fig. 3A , C ). BT06D07 and BT08F11 were shown to be bactericidal against both pathogens ( Fig. 3B , D ). BT06D07 inhibited the growth of both bacteria completely during all times tested, but at 24 h there was some regrowth observed in cultures containing BT08F11. This may be due to the inability to completely kill all the bacteria present at 4× MIC.
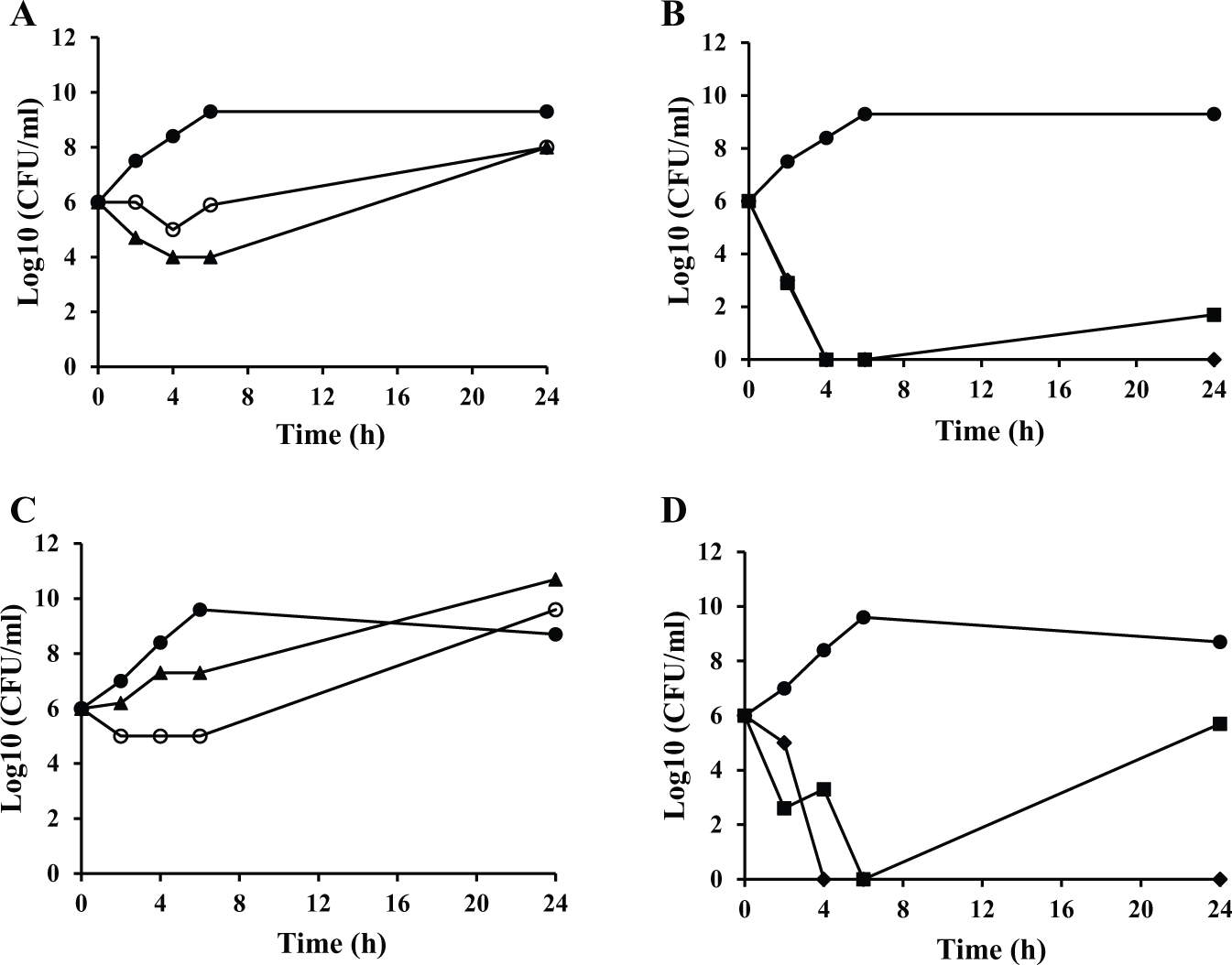
Time-kill kinetics against selected bacteria. The activity of the hit compounds against growths of (
Compound Inhibition of Eukaryotic Protein Synthesis
Mitochondria, the organelle present in eukaryotic cells, contains its own protein synthesis system, and even though the proteins involved in mitochondrial translation are encoded by genes contained within the cellular genome, they are strictly required for eukaryotic cell viability. Two of the hit compounds targeted PheRS; therefore, we tested the ability of these compounds to inhibit the activity of hmPheRS. The compounds were assayed at concentrations from 0.4 to 200 µM in hmPheRS aminoacylation assays. 6 In these assays, the activity of hmPheRS was observed to be only modestly affected by both BT04E11 and BT06G06 at high concentrations ( Suppl. Fig. S1A,B ).
To determine the effect of the hit compounds on cellular eukaryotic protein synthesis, the compounds were next tested for the ability to inhibit protein biosynthesis using wheat germ cell extract assays. Previously, poly(U) messenger RNA, yeast tRNAPhe, [3H]Phe, and Mg2+ concentrations were optimized for poly-Phe synthesis in wheat germ cell extract assays. 4 The hit compounds were compared with a known inhibitor of protein synthesis, cycloheximide, which inhibits greater than 80% of protein synthesis at 30 µM. The test compounds were not observed to significantly inhibit protein synthesis in wheat germ cell extract at concentrations up to 200 µM ( Suppl. Fig. S1C–F ).
Cytotoxicity Effects
To test for cytotoxicity in human cell lines, HEK-293 cells were dosed with each of the hit compounds or with staurosporine, a potent protein kinase inhibitor that is characteristically used as a control inhibitor of cell cultures 11 ( Fig. 4A ). The control staurosporine was serially diluted in assays from 1 to 0.001 μg/mL, and the IC50 (or cytotoxicity data, CC50) was determined to be 0.003 μg/mL. When dosed in these cell cultures, BT06D07 was not observed to inhibit cell proliferation at concentrations up to 400 μg/mL. The IC50 for inhibition of cell growth by BT04E11 was 90 μg/mL ( Fig. 4B ), while both BT06G06 and BT08F11 inhibited cell cultures with lower IC50 values of 18.5 and 8.3 μg/mL, respectively ( Fig. 4C , D ).
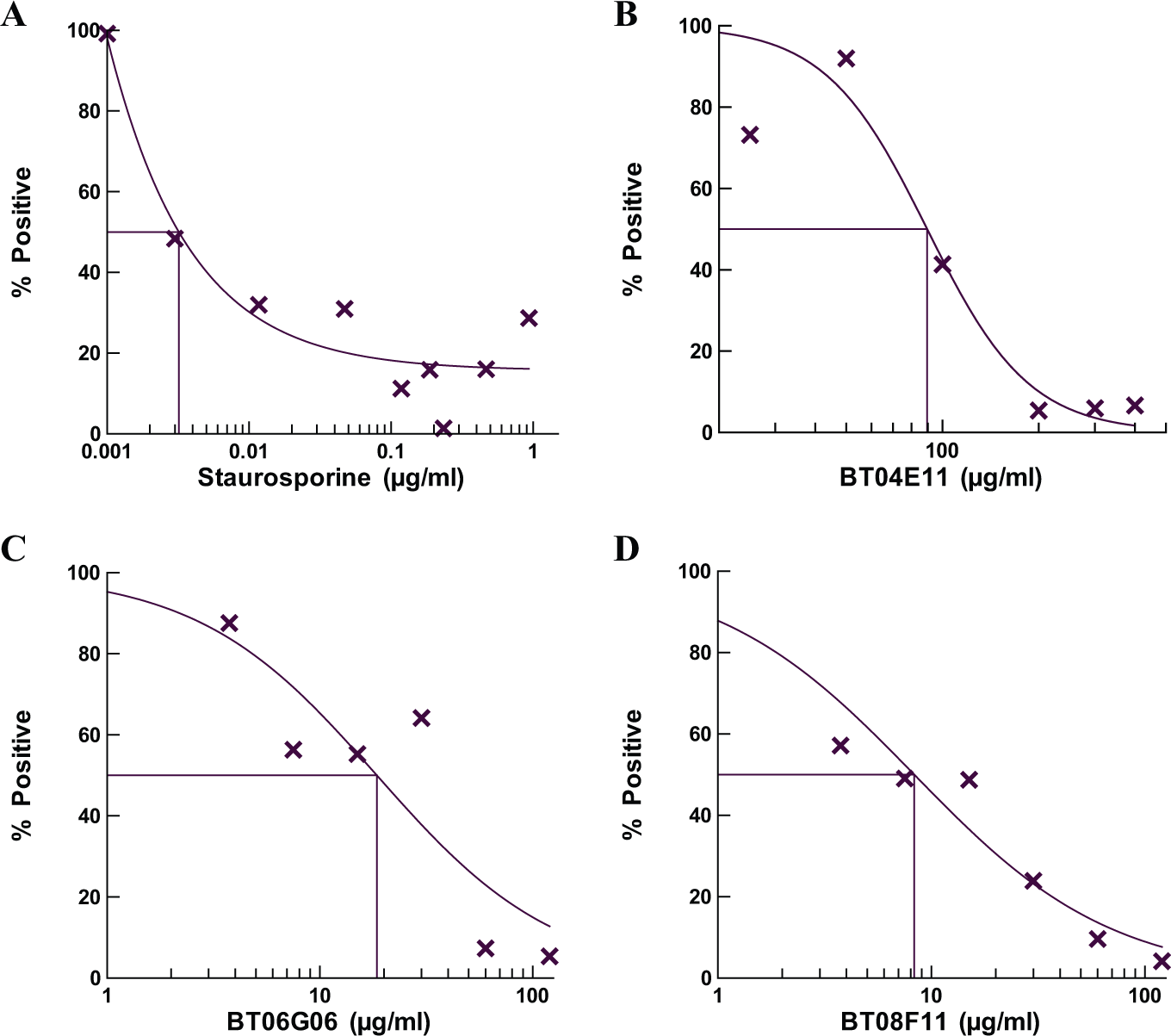
Determination of the toxicity of the hit compounds relative to the control staurosporine in human cell cultures. MTT assays were performed with the indicated dose of drug for 24 h under standard tissue culture conditions as described in Materials and Methods. The control (
The results presented here show the use of an in vitro poly(U) mRNA directed protein synthesis system to screen a synthetic compound library containing 890 compounds and the identification of four compounds that are bacterial protein synthesis inhibitors. Specificity screening identified PheRS as the molecular target in the A/T assay of two of the compounds, while two of the compounds targeted the ribosome. The compounds identified in the A/T screen were subjected to microbiological assays in which they displayed varying levels of activity against a broad spectrum of pathogenic organisms. MIC determination indicated that the compounds had little inhibitory effect against E. coli and P. aeruginosa activity, but good activity was observed against the efflux mutant forms of these bacteria, indicating that efflux was the probable cause of the low activity against the wild-type strains. The MIC assays confirmed the broad-spectrum activity of these compounds against both Gram-positive and Gram-negative bacteria. In particular, they were active against the respiratory pathogens, S. pneumoniae and H. influenzae, as well as S. aureus.
In time-kill assays, BT04E11 and BT06G06 were shown to have bacteriostatic activity against both H. influenzae and S. aureus. This would be the expected mode of action for compounds that inhibit the activity of PheRS since inhibition of the aminoacylation activity of an aminoacyl tRNA synthetase (aaRS) mimics starvation for amino acids by lowering the ratio of charged to uncharged tRNA, and induces the stringent response, resulting in static bacterial growth. 12 Alternatively, BT06D07 and BT08F11 were observed to be bactericidal, which is in keeping with the inhibition of the function of the ribosome, thereby disrupting protein synthesis and resulting in cell death.
Many compounds that inhibit the activity of an aaRS interfere with binding of ATP and/or the amino acid in the active site. A compound that specifically inhibits ATP binding may be a poor candidate as an antibacterial agent since ATPase enzymes are common in bacterial as well as in eukaryotic cells. To determine the mechanism of action relative to substrate binding of the two compounds targeting PheRS, they were tested in competition assay with varying amounts of ATP or Phe. Neither of the compounds was observed to compete with either of the substrates, indicating that the binding region of both compounds is likely outside the active site for aminoacylation. Additional work will be required to determine the exact mechanism of action of these two compounds.
The use of cell systems in drug discovery as an alternative to whole animal systems for predicting toxic potentials of chemicals is a viable first step in drug development and selection of lead compounds. The hit compounds identified here were tested for cytotoxicity in HEK-293 cell cultures. The compound BT06D07 failed to exhibit any toxic effects at all concentrations tested. BT04E11 exhibited an IC50 of 90 μg/mL, which is 30,000-fold higher than that of the control staurosporine and similar to that observed with ciprofloxacin. 13 The other compounds, BT06G06 and BT08F11, were observed to have lower IC50 values; however, they were comparable to other antibiotics, erythromycin and tetracycline, that target protein synthesis in various cell cultures. 14
The structure of the compounds was searched against the PubChem database for bioassay information. Nitrofurans are a class of functional motifs contained in many synthetic broad antimicrobial spectrum antibiotics. 10 The compound BT06D07 contains two nitrofuran groups, one on each side of the compound, and BT06G06 contains one nitrofuran group on the right-hand side of the molecule. When these two compounds were used to search the PubChem database, BTO6D07 was identified as difurazone (nitrovin) and BT06G06 as nifursol. Both these compounds are feed additive used to aid in increasing weight gain and for the prevention of bacterial and parasitic infections in poultry and livestock in Europe. 15 These compounds will not be considered further. Information for BT08F11 is available in the PubChem Substance and Compound database through the unique chemical structure identifier CID: 9586370. In bioassays at the Southern Research Institute (Birmingham, AL), this compound was found to be one of 1594 compounds active in a high-throughput screen to identify inhibitors of Mycobacterium tuberculosis from a 100,000-compound library from ChemBridge Corporation (San Diego, CA). We will also not pursue additional analysis of this compound. Information for BT04E11 is available in the PubChem Substance and Compound database through the unique chemical structure identifier CID: 4990246. There was no bioassay information available on this compound. The fact that this compound exhibited low levels of inhibition against the wild-type strain of P. aeruginosa ( Suppl. Table S1 ) in initial MIC determination and little cytotoxicity in human cell cultures indicates that it potentially can be used in experimental therapy. The IC50 (or CC50) in human cell cultures was well above the MIC observed in all bacterial growths (except for cultures of E. coli), which is a good starting point for antibiotic development. Also, BT04E11 was specific for inhibition of bacterial protein synthesis and had little effect on eukaryotic protein synthesis, including inhibition of hmPheRS, which increases the possibility for development as an antimicrobial agent. The work here will serve as a starting point for the design of derivatives in attempts to increase the inhibitory activity of BT04E11. We will proceed with structure–activity relationship (SAR) studies of BT04E11 to increase potency and improve efficacy against bacteria in culture.
Footnotes
Acknowledgements
The authors are grateful for the financial support provided by the National Institutes of Health (grant no. 1SC3GM098173). The contents of this article, publication, etc., are solely the responsibility of the authors and do not necessarily represent the official views of the National Institutes of Health. A portion of student support was from a departmental grant from the Robert A. Welch Foundation (grant no. BG-0017).
Supplementary material for this article is available online.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
This work was supported by NIH grant 1SC3GM098173 and Robert A. Welch Foundation grant BG-0017.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.