
Abstract
One of the central questions in the characterization of enzyme inhibitors is determining the mode of inhibition (MOI). Classically, this is done with a number of low-throughput methods in which inhibition models are fitted to the data. The ability to rapidly characterize the MOI for inhibitors arising from high-throughput screening in which hundreds to thousands of primary inhibitors may need to be characterized would greatly help in lead selection efforts. Here we describe a novel method for determining the MOI of a compound without the need for curve fitting of the enzyme inhibition data. We provide experimental data to demonstrate the utility of this new high-throughput MOI classification method based on nonparametric analysis of the activity derived from a small matrix of substrate and inhibitor concentrations (e.g., from a 4S × 4I matrix). Lists of inhibitors from four different enzyme assays are studied, and the results are compared with the previously described IC50-shift method for MOI classification. The MOI results from this method are in good agreement with the known MOI and compare favorably with those from the IC50-shift method. In addition, we discuss some advantages and limitations of the method and provide recommendations for utilization of this MOI classification method.
Introduction
Currently, many drug discovery programs rely on screening massive chemical libraries against molecular targets with a biologically relevant assay in an effort to find novel chemotypes as entry points.1,2 As the scale of such high-throughput screening (HTS) is generally very large (hundreds of thousands to millions of chemical substances screened), the number of active compounds (“hits”) resulting from screening efforts is also quite large, and therefore effective filtering of the large primary hit lists in a systematic manner to provide a “clean” and information-rich structure-activity profile is critical for effective hit-to-lead selection. In addition to routine confirmation testing and counterscreens to weed unwanted hits that occur for various reasons, including data variability (false-positives), assay technology interferences, and/or interaction with biological off-targets, early assessment of hits with the desired mode of action (MOA) and pharmacological properties is among the most important parameters in selecting chemical starting points for hit-to-lead optimization.
Enzymes and receptors still comprise the majority of biochemical and cellular targets in current lead discovery efforts. There are more than 300 marketed drugs that work by modulating the activity of an enzyme and many others by modulating the activity of a receptor. 3 For instance, inhibitors with a novel MOA for protein kinases were initially discovered through screening efforts.4-6 Understanding the mode of inhibition (MOI) of enzyme targets enables one to prioritize chemotypes for further optimization of inhibition specificity and potency. 7 Because of the complexity and low throughput of the conventional MOI determination methodologies, for any large collection of enzymatic inhibitors identified from a screening campaign, the MOI information for these hits is not readily obtainable. To remove or alleviate such bottlenecks downstream of HTS (sometimes referred to as the postscreening dilemma) in the drug discovery process, a number of approaches and methodologies have been explored in recent years, including the use of high-throughput infrastructure (e.g., automation) to increase the scale of conventional MOI determination. For example, using robotics and instrumentation, a fully automated steady-state enzyme kinetic assay in compound profiling has been described for dipeptidyl peptidase IV, and the setup was capable of producing full mechanistic inhibition analysis in a timely fashion to support drug discovery. 8 Chemoinformatic and computational tools are employed to prioritize the hit scaffolds based on in silico models and analysis.9,10 In the case of assessing the MOI of CK2 kinase inhibitors identified from a screening effort, the binding mode of the inhibitors was determined via combining structure-activity relationships with a molecular docking procedure. 5 There are other novel strategies and methodologies for enabling enzyme mechanism and MOA studies, such as combining molecular docking with in vitro studies to elucidate the inhibitory effects of herbal compounds on human cytochrome P450 1A 11 and a method in which the MOI can be determined using a fixed inhibitor concentration over a range of substrate concentrations. 12
To increase the throughput in MOI characterization, a few high-throughput approaches have been explored recently.12,13 Based on the conventional IC50 shift under different substrate concentrations and the relationship of IC50, substrate concentration, Km and Ki, which has been described by in the Cheng and Prusoff equation, 14 Wei et al. 13 described a high-throughput MOI classification approach for evaluating hits from HTS. The MOI of large number of hits can be classified as competitive, uncompetitive, noncompetitive, or mixed mode by measuring the ratio of IC50 values (or percentage inhibition) obtained at two separated substrate concentrations. In this method, the ratio (R) of two IC50 values for an inhibitor under two selected substrate concentrations is calculated (where [S1] < [S2]; concentrations are chosen to bracket the known Km value). In this case, R = IC50S2/IC50S1 = (1 + [S2]/Km)/(1 + [S1]/Km) for competitive inhibitors and R = IC50S2/IC50S1 = (1 + Km/[S2])/(1 + Km/[S1]) for uncompetitive inhibitors, and so forth. Using these R values and the standard deviation of their measurements as a guide, this higher throughput method was applied to analyze the MOI for more than 900 screening hits to distinguish compounds between competitive, noncompetitive, or mixed MOIs. 12 Such large-scale MOI profiling would be difficult to implement with conventional methods.
Although simple to apply, the IC50-shift (R-value) method for MOI classification of a large number of hits also has some practical limitations. First, the approach is based on the IC50 ratio under the two different substrate concentrations. To distinguish among all types of MOI reliably, the two substrate concentrations need to be sufficiently below and above the known Km value so that the separation of R value (IC50 shift) for the inhibitors is sufficiently large. In practice, this demands wide separation between high and low substrate concentrations, which sometimes is practically unattainable because of substrate solubility, signal strength, and linearity at these substrate concentrations. The assay signal at high and low substrate concentrations needs to be strong and sensitive enough to yield reliable IC50 values. As well, individual curve fitting to the data at both testing substrate concentrations needs to be performed, which is generally time-consuming. Error accumulation both from measurement and curve fitting can sometimes be significantly large in the IC50 ratio R. Lastly, and maybe more practically important, hits from screening tend to have a wide range of activity; some compounds have strong activity, but many have weak activity, and both very strong and weak hits often do not generate full concentration-response curves from which reliable IC50 values can be determined in the chosen compound titration range. If no IC50 value or only one IC50 value is obtained from the concentration-response curves at the two selected substrate levels, it is not possible to calculate the R ratio and this limits the classification of their MOI.
In this article, we describe in detail a new approach for high-throughput MOI assessment for rapid classification of screening hits against targeted enzymes. The method is based on pattern classification of the inhibition activity surface from testing a small matrix covering different substrate and inhibitor concentrations. Our method neither involves curve fitting nor determination of IC50 values. To demonstrate the practical value of this approach as a rapid and effective method with experimental data sets, we selected four enzymatic inhibitor case studies. In all cases, the compounds and enzyme assays were originally derived from screening efforts. In a few cases, we had some prior knowledge of the MOI profile of the testing compounds, but in other cases, we had no MOI information available at the time of testing. Those cases with prior MOI knowledge serve to compare and validate the results of our new MOI classification method, whereas those that do not have any prior knowledge act as a blinded study in which we can compare results from matrices with different dimensions (small and large matrix testing) as well as to the IC50-shift method. 13 We expand the study in one case to a large hit list containing 1363 compounds. The enzymes varied in class ranging from firefly luciferase (FLuc), 15 an NS5 RNA dependent RNA polymerase (RdRp), 16 as well as protein methyl transferases. The assay formats and readouts also varied and included both direct and indirect detection methods including fluorometric and matrix-assisted laser desorption/ionization (MALDI) mass spectrometry (MS) detection. After testing various (substrate x inhibitor) matrices, we applied the MOI classification algorithm based on the S-gradient signature (J value) to assign the MOI. The data illustrate the advantages and limitations of this new method, and we provide several recommendations for performing the analysis. The outcome of these case studies clearly demonstrates that this simple MOI classification scheme is effective for the rapid profiling and characterization of a large number of enzyme inhibitors derived from HTS. This enables prioritization of enzyme inhibitors for lead development based on the MOI.
Materials and Methods
Matrix-Based MOI Classification Method
The activity of an enzymatic reaction can be described by Michaelis-Menton reaction kinetics. 17 The inhibition of an enzymatic reaction at different substrate and inhibitor conditions can be derived from the Cheng and Prusoff equation. 14 Based on these fundamental enzymatic equations, the following formulae for the percentage inhibition by an inhibitor (Inh%) were derived for each of the MOIs for reversible inhibitors.
For competitive inhibitors:
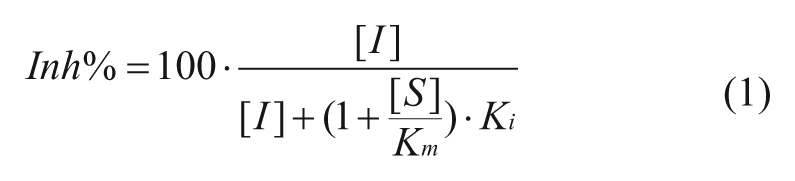
For uncompetitive inhibitors:
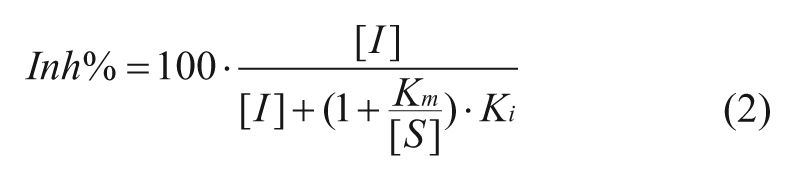
For noncompetitive inhibitors:
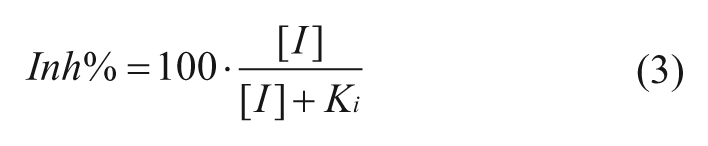
For mixed inhibitors:
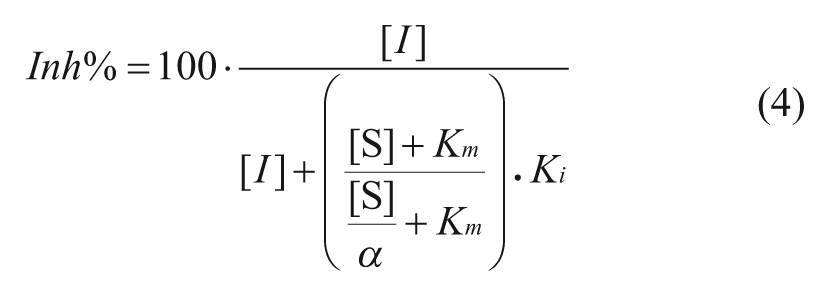
In the above formulae, the Inh% in top three cases can be determined by the substrate concentration [S], inhibitor concentration [I], and the corresponding constants for Km and Ki. The Km is the Michaelis-Menten constant for the substrate. Competitive inhibitors bind exclusively to the free enzyme with a dissociation constant Ki. Uncompetitive inhibitors bind exclusively to the enzyme-substrate complex with a dissociation constant Ki. Noncompetitive inhibition refers to an inhibitor that binds to the free enzyme and enzyme-substrate complex with equal potency. Equations (1), (2), and (3) illustrate the relationship between Inh% and the substrate and inhibitor presented ([S] and [I]) for inhibitors in an enzymatic reaction (with characteristic constants Km, and Ki). Equation 4 describes mixed inhibition, which is modulated by the ratio of the Ki values for both the free enzyme and the enzyme-substrate complex (designated as α). Equation 4 reduces to pure inhibition modes at certain values of α: noncompetitive when α = 1, competitive when α >>1, and uncompetitive when α << 1. From these equations, the theoretical Inh% can be calculated at different inhibitor concentrations ([I]) at any given substrate concentration ([S]). Analyzing the inhibition at various [I] and [S] levels should allow not only the determination of the MOI (competitive, noncompetitive, uncompetitive, and mixed) but also, when the Km is known, the Ki values for inhibitors.
Based on these equations, we propose a simple way to classify a large number of inhibitors and assign the MOI via the activity surface pattern of a small substrate x inhibitor matrix test at several selected concentrations. The approach is described as follows. Suppose an enzymatic assay is tested at m substrate levels (denoted S1, S2, . . . , Sm) and n inhibitor levels (denoted I1, I2, . . . , In), and the test conditions compose a m × n matrix. The corresponding Inh% values also make a m × n matrix. In the following description of this nonparametric approach, we use a 4S × 4I matrix to illustrate the method and a two-step procedure for a real test.
Step 1: Determine the Normalized S Gradient at Each I Level
In a 4S × 4I matrix test, at each I level, there are four levels of S (S1 to S4) and four corresponding Inh% values (obtained from test). This gives four Inh% activity gradients (one at each I) along the substrate levels (referred to as the S gradient). We considered the parameter to evaluate the S gradient at each I level.
Local S-Gradient Average ( Jl)
In this evaluation, the slope using every point-to-point activity data was used to estimate the activity surface gradient along the S dimension (in a logarithmic scale). We use the formula (Xi – Xj)/(logSi – logSj) to calculate the normalized activity change (i.e., slope) between data point Xi and Xj (where i, j are two data points in the four S levels test at the same I level, and X is Inh% value). The four S levels would generate a total of six local gradient values. The average gradient is obtained from simply taking the average of the six local gradient values, as shown below (n = 6 in this case). The average gradient (denoted J) is used because of its greater resistance to data outliers in a real matrix test.
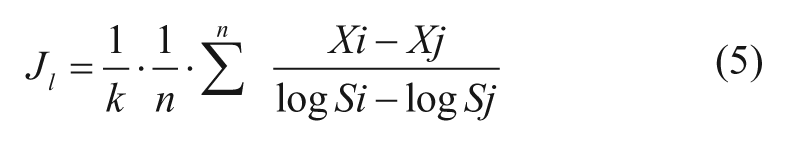
Global S-Slope (Jg)
Alternatively, a slope function is used to estimate the S gradient at each I level.
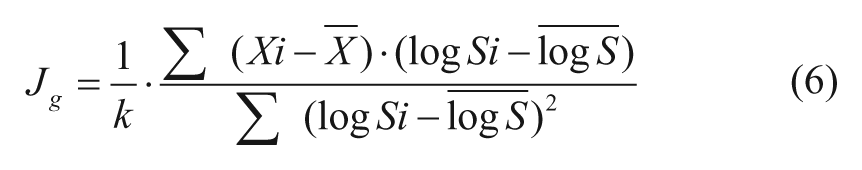
where X and logS with top bar notation are the corresponding mean values for the four data points along S at each I. In both Equation 5 and 6, the term 1/k in the J formulae is a constant modifier of J value for convenience of classification (see next section).
Step 2: Apply S-Gradient Signature for MOI Classification
From step 1, a small m × n matrix testing (such as 4S × 4I) would generate a set of J values (J1, J2, . . . , Jn), with each Ji being the S-gradient vales at inhibitor concentration Ii of the matrix testing. This set of J values is referred to as the S-gradient signature of the matrix test. Key features of this signature are used to classify the MOI of the inhibitor (more details are given in the next section).
Assays for the Lysine Methyl Transferase EZH2
EZH2 HTRF assay
Europium conjugated anti-histone H3K27Me2 antibody was obtained from Cisbio (No. 61KC2KAE). Compounds were tested in eight concentrations in a twofold dilution series (20 µM to 0.15 µM final concentration in a 4 µL reaction). Assay-ready plates were prepared by dispensing 40 nL compound into each well of 384 Proxiplates (6008289, PerkinElmer, Waltham, MA) using Echo Liquid Handler (Labcyte Inc., Sunnyvale, CA). The substrate mixtures were prepared by mixing biotinylated H3K27Me0 peptide (21–44 aa on H3, from AnaSpec No. 64440-1, final reaction concentration 0.1 µM) and each of the four different S-adenosyl methionine (SAM; No. S018, AK Scientific, Union City, CA) concentrations (20 µM, 5.7 µM, 1.6 µM, 0.5 µM final concentration) in final 4 µL reaction volume). To set up the assay, 2 µL substrate mixture and 2 µL of PRC2 enzyme (50 nM final concentration, produced in house) were incubated in 1x assay buffer containing 20 mM Tris buffer, pH 8, 0.005% Triton X-100, and 0.5 mM DTT for 3 h at ambient temperature. Two microliters of detection reagent (1 nM anti-dimethyl-H3K27-Eu and 40 nM SA-APC) was added to each well to a final reaction volume 6 µL. The plates were then incubated for an additional 2.5 h at ambient temperature. The fluorescent outputs were detected using PHERAstar microplate reader (BMG LABTECH, Ortenberg, Germany) with a homogeneous time-resolved fluorescence filter (excitation 337 nm, emission at 665 nm and 620 nm).
EZH2-MALDI assay
Compounds were tested in eight concentrations with a twofold dilution series (20 µM to 0.78 µM final reaction concentration in 10 µL reaction). Five microliters of PRC2 enzyme (10 nM final concentration) and 5 µL of substrate (20 µM of SAM and seven doses of H3K27Me0 peptide starting from 20 µM to 300 nM in a 1:2 titration series) were incubated in 1x assay buffer (20 mM Tris, pH 8, 0.01% Tween, 0.5 mM DTT) for 4 h at ambient temperature. A total of 2.5 µL of stop reagent containing trifluoroacetic acid at a final 0.1% and H3K27Me3 (AnaSpec No. 64367-1), as positive internal control, at final concentration 0.5 µM) was added to the plate. The plates were stored at −20 °C before MALDI MS analysis. MALDI MS analysis was performed similarly as described below for a protein methyl transferase.
MALDI MS Analysis of a Protein Methyl Transferase (PMT) with Peptide Variation
For PMT assays, 6 nM enzyme was used in five separate reactions with variable amounts of substrate for the MOI study. SAM substrate concentrations were fixed at 20 µM for each of these reactions, and substrate was used in the following concentrations (4.5 µM, 1.5 µM, 0.5 µM, 0.167 µM, and 0.056 µM). Each reaction was performed in 20 mM HEPES, pH 8, with 0.002% CHAPS, 0.5 mM NaCl. These variable substrate reactions were used to generate concentration-response curves (CRCs) for 26 putative PMT inhibitors of various MOIs. To generate the CRCs for each of the five reactions, assay-ready plates were generated by depositing 100 nL of compound into a 384-well small-volume Greiner plate with concentrations ranging from 20 µM to 0.150 µM. The peptide substrates from the five distinct reactions were then plated into each of the different assay-ready plates, and the reaction was performed at room temperature for 2 h. To quench the reactions, trifluoroacetic acid at a final concentration of 0.1% was added to stop the enzyme and then the reaction plates were stored at −80 °C until read by the MALDI MS. To read the reaction plates, each plate was thawed, 300 nL of each reaction was spotted onto a stainless steel MALDI MS plate and dried, and then 300 nL of the MALDI matrix alpha-cyanohydroxycinnamic acid (CHCA) was added to the dried spots and allowed to dry. The dried reaction spots from each 384-well plate were analyzed by a Bruker Autoflex MALDI MS. Substrate and product peptides were detected by measuring the intensity of the two ions for every spot (1084.5 Da and 1098.5 Da, respectively) representing the Me0 and Me1 peptides after data were acquired using 1000 shots/spot. Ratios of total product intensity divided by the sum of the substrate and product intensities were calculated for all doses of compound as well as the neutral control (DMSO only) and the active control (pre-quenched enzyme) reactions. The percentage activity of the enzyme in each well was determined by taking the product/substrate ratios for each reaction (R) and comparing them to the ratios of the neutral controls (NC) and active controls (AC) in the following equation: percentage activity = (R – (NC – AC))/(NC + AC) × 100%.
MALDI mass spectrometry analysis of PMT with SAM variation
For PMT assays, 6 nM enzyme was used in five separate reactions with variable amounts of substrate for the MOI study. Peptide concentrations were fixed at 0.5 µM for each of these reactions, and SAM was varied in the following concentrations (8.3 µM, 2.8 µM, 0.92 µM, 0.31 µM, and 0.1 µM). Each reaction was performed in 20 mM HEPES, pH 8, with 0.002% CHAPS, 0.5 mM NaCl. These variable substrate reactions were used to generate concentration-response curves for 26 putative enzyme inhibitors of various MOIs. To generate the CRCs for each of the five reactions, assay-ready plates were generated by depositing 100 nL of compound into a 384-well small-volume Greiner plates with concentrations ranging from 20 µM to 0.150 µM. The five distinct reactions were then plated into each of the different assay-ready plates, and the reaction was performed at room temperature for 2 h. To quench the reactions, trifluoroacetic acid at a final concentration of 0.1% was added to stop the enzyme, and then the reaction plates were stored at −80 °C until read by the MALDI MS. To read the reaction plates, each plate was thawed, 300 nL of each reaction was spotted onto a stainless steel MALDI MS plate and dried, and then 300 nL of the MALDI matrix CHCA was added to the dried spots and allowed to dry. The dried reaction spots from each 384-well plate were analyzed by a Bruker Autoflex MALDI MS. PMT substrate and product peptides were detected by measuring the intensity of the two ions for every spot (1084.5 Da and 1098.5 Da) representing the Me0 and Me1 peptides after data were acquired using 1000 shots/spot. Ratios of total product intensity divided by the sum of the substrate and product intensities were calculated for all doses of compound as well as the neutral control (DMSO only) and the active control (prequenched enzyme) reactions. The percentage activity of the enzyme in each well was determined by taking the product/substrate ratios for each reaction (R) and comparing them to the ratios of the NC and AC in the following equation: percent activity = (R –(NC – AC))/(NC + AC) × 100%.
Assay for NS5 RNA-Dependent RNA Polymerase (RdRp) from Dengue Virus
The fluorescent alkaline phosphatase-coupled polymerase assay was performed as described by Smith et al. 16 Briefly, the fluorescent-based assay consisted of two steps: a polymerase initiation/elongation reaction and a signal generation reaction. The nucleoside triphosphates (GTP, ATP, UTP) and ATTO-CTP are the substrates used by NS5 polymerase for incorporation into the growing RNA chain. Release of the by-product ATTO-pyrophosphate during the polymerization reaction is catalyzed by NS5 RdRp. 18 The ATTO-PPi is subsequently hydrolyzed by calf intestinal alkaline phosphatase (CIP) to liberate free product whose fluorescence signal can be detected by excitation wavelength at 420 nm and emission wavelength at 560 nm. The assay used viral genome-derived RNA (576-nt in length, derived by in vitro transcription [IVT]) 16 as an RNA substrate template to enable DENV NS5 de novo initiation activity. The assay was set in a 4 uL reaction volume in 1536-well plate (Greiner BioOne, Kremsmünster, Austria) containing 100 nM RdRp enzyme, 20 µM ATP, 20 µM GTP, 20 µM UTP, and 5 µM ATTO-CTP with IVT RNA at 0.156-10x of KM value, respectively. The assay buffer contained 20 mM Tris-HCl, pH 7.5, 10 mM KCl, 1 mM MgCl2, 0.3 mM MnCl2, 0.02% CHAPS, and 2 mM TCEP. Both enzyme and substrate mixtures were dissolved in assay buffer. Fluorescence from the coupled reaction using ATTO-CTP was used in the assay to track polymerase elongation. The RdRp enzyme was dispensed at 2 µL volume into each well, and then 100 nL inhibitors were acoustically added for preincubation for 15 min at 22 °C. The substrate mixture at 2 µL was dispensed to initiate the reactions. The incubation time for each IVT RNA concentration varied as follows: 120 min for 0.156x Km, 90 min for 0.625x Km, 60 min for 2.5x Km, 30 min for 10x Km. The apparent Km for RNA in the assay was determined to be ~125 nM. The negative control consisted of RNA and nucleotides without the enzyme. The stop solution contained 20 nM CIP in AttoPhos buffer supplemented with 160 mM NaCl and 20 mM MgCl2. The BioRaptr liquid dispenser (Beckman Coulter, Brea, CA) was used for all enzyme, substrate, and stop solution addition. The Echo liquid handler (Labcyte, Sunnyvale, CA) was used for compound addition. After stop solution addition, the plates were incubated at 22 °C for 60 min prior to fluorescence measurement on PHERAstar microplate reader (BMG Labtech). A total of 165 compounds were tested in duplicates as eight-point twofold serial dilution in the concentration range of 0.39 to 50 µM. The percentage inhibition of the compounds was calculated using the positive and negative controls. ATTO-CTP was custom made by Trilink; nucleotides ATP, GTP, and UTP were purchased from Sigma (St. Louis, MO); CIP was obtained from New England Biolab (Ipswich, MA), AttoPhos buffer was purchased from Promega (Madison, WI).
Assay for FLuc Enzyme Activity
The FLuc enzyme assay has been previously described in Thorne et al. 15 The assay was performed in 1536-well white Greiner MB plates (Greiner BioOne). Three microliters of substrates (ATP kept at 0.01 mM and D-luciferin was varied) was dispensed, and then compounds were added using a 20 nL pintool. The reaction was then started by the addition of FLuc enzyme (Sigma Aldrich, cat. No. L9506) at a final concentration of 10 nM. The assay buffer contained 50 mM Tris acetate (pH 7.5), 13.3 mM Mg-acetate, 0.01 mM D-luciferin (Sigma Aldrich, cat. No. L9504), 0.01 mM ATP, 0.01% Tween-20, and 0.05% bovine serum albumin. Following incubation for 10 min at room temperature, the plates were read on a PerkinElmer Viewlux using 1x binning and 30 s of exposure. The percentage inhibition of the compounds was calculated using the full activity (positive) and no enzyme (negative, as 100% inhibited) controls.
Results and Discussion
For a matrix activity pattern based classification for MOI types, both parametric and nonparametric parameters can be considered. In theory, an algorithm to match the theoretical inhibition surface models provided with Equations 1 to 3 to a parametrically modeled inhibition surface of the testing matrix can be developed and then used to predict the MOI type by the best fit of the surface models. However, similar to the IC50 ratio approach, individual surface-fitting routines (vs. curve-fitting routines in the IC50 ratio approach) is needed for each matrix test. To develop surface models would require significant computing power and detailed information about the enzymatic reaction such as the Michaelis-Menten constant Km. As well, in the IC50 ratio method, two IC50 values must be calculated at high and low [S]/Km values, and oftentimes, one of these values is not determinable, for example, a competitive inhibitor at high [S]/Km conditions may show only weak inhibition. Alternatively, a nonparametric classification approach could be simpler to perform and less sensitive to error propagation. Because of these potential merits, we chose to further explore the nonparametric approach for MOI classification trials.
Figure 1 shows Inh% surface pattern from the theoretical inhibition ( Fig. 1A ) and a more discrete Inh% plot of a 4S × 4I matrix ( Fig. 1B ), both derived directly from Equations 1 to 3. From Figure 1 , it can be seen that, even for a small m × n matrix testing (such as 4S × 4I), the Inh% surface pattern is very distinctive for each inhibition mode and a good representative of the theoretical surface pattern. It should be noted that for most enzymatic assays, the enzyme concentration present in the reaction compared with substrate and inhibitor concentrations is very low, and the product conversion is usually also low, so one can use the initial substrate and inhibitor concentrations to approximate the free concentration. Also we note here that -100% activity is defined as full inhibition, 100 Inh%.
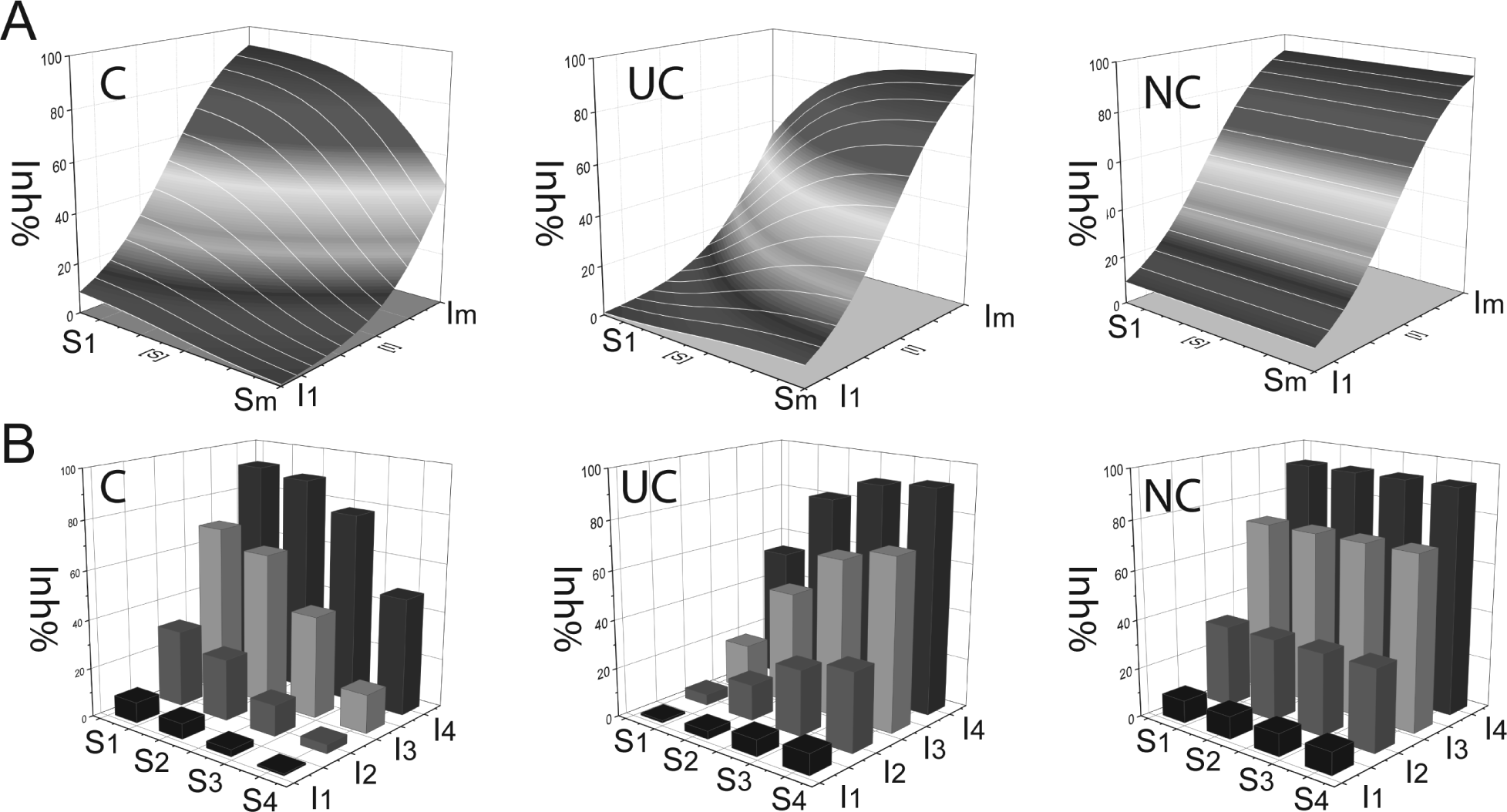
Dependence of percentage inhibition on substrate and inhibitor concentration. Plots representing a S versus I matrix test and the corresponding Inh% values. (
For this approach, we evaluated two strategies to calculate the normalized Inh% activity gradient along the substrate levels (i.e., S gradient), as described in the Materials and Methods section. We compared both the local gradient average (Jl) and global S-slope (Jg) in our approach. For all the cases tested, Jl and Jg always yielded very similar or almost identical values, with a Pearson correlation coefficient in the range of r = 0.999 (data not shown). Therefore, the two gradient methods should almost always give the same MOI classification results in practical applications. The global S-slope in reality is easier to calculate than local S-gradient average, and we therefore recommend using the global S-slope (Jg) for J calculation.
Key Features of S-Gradient Signature
From step 1 of the Materials and Methods section, the S gradient (J: Jl ≈ Jg) at any one specific I level is obtained. For a 4S × 4I matrix test, the four S-gradient values at the ordered four I levels (e.g., I level from high to low) is referred here to a S-gradient signature ( Fig. 2a ). This S-gradient signature contains the critical Inh% surface pattern information of the matrix test and forms the basis for MOI classification. Three features of the signature are used for the nonparametric MOI classification.
Sign of J: As shown in Figure 2a , the S-gradient signatures of the three types of MOI have different signs. Competitive inhibition (C) gives negative J values, and uncompetitive inhibition (UC) gives positive J values for the signature components. For noncompetitive inhibition (NC), the theoretical J values of its signature components are all zero. In practical cases, NC may likely give J ≠ 0 but instead will have very small positive or negative values, so the J value amplitude needs to be considered as well.
J value amplitude (gradient strength): In any practical test, as mentioned above, the three MOI types (i.e., C, UC, and NC) probably cannot be classified unambiguously by the sign of J alone, particularly for NC, which gives a very small J value (J ≈ 0), and its sign will be easily affected by any measurement variation. In the same vein, experimental data variation will always obscure the border lines for C or UC types of MOI from NC. In addition, there are likely many inhibitors that do not belong to any one pure type of MOI classification, for they behave as mixed-mode inhibitors. For these reasons, the strength of the gradient signature (J value amplitude) needs to be combined with its sign for the MOI classification. We defined a gradient strength indicator (Jmax) for this purpose.
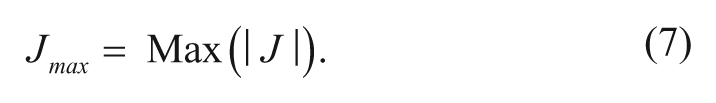
Jmax has the maximum amplitude of the S-gradient signature components. Jmax is a key indicator in this MOI classification approach. A large Jmax with a negative sign is indicative of substrate competitive MOI, whereas a large Jmax with a positive sign is indicative of uncompetitive MOI. Very small Jmax values may suggest either a noncompetitive MOI or in special cases other inhibition types (see next section and Table 1 ). The only challenge is when the matrix testing data contain severe outliers. To minimize the outlier interference, we have also developed an algorithm to extract Jmax effectively in data sets with outliers.
Jmax position (pattern of the signature and the corresponding Inh%): Even though the combination of Jmax and sign can usually give a good indication of the MOI type, confusion may arise in certain special occasions. For example, when the Jmax value is close to zero, this can indicate an inhibitor with either a noncompetitive MOI (NC) or simply superpotent (or very weak) inhibition behavior over the testing range. The clue to distinguish these more intriguing situations lies in the pattern of the signature components. An NC inhibitor will produce not only a very small Jmax (i.e., Jmax ≈ 0) but also corresponding X values (Inh%) that are well spread between high and low values in the signature at different I concentrations. In contrast, a very small Jmax value with the corresponding X values (Inh%) that show either constantly high or constantly low values in the signature at different I concentrations may simply mean either a superpotent or very weak inhibitor. More intriguingly, when Jmax resides at the highest (or lowest) I level, it also suggests competitive or uncompetitive MOIs (based on the sign), although Jmax has a moderate value. Altogether, these S-gradient signature patterns, combined with the corresponding Inh% values in the series, can distinguish the MOI type reliably from the matrix test. For example, for a 4S × 4I matrix test four S-gradient values are calculated (one for each inhibitor concentration based on all four substrate levels), and the sign and maximum S-gradient value (Jmax) are used to classify the MOI provided that the Jmax value taken meets certain additional criteria aimed at flagging very weak or strong inhibitors, as illustrated in the details of Table 1 . However, as will be shown in the section below, ambiguity in MOI classification does sometimes occur when the Ji and X values change is not covered well by the test matrix.
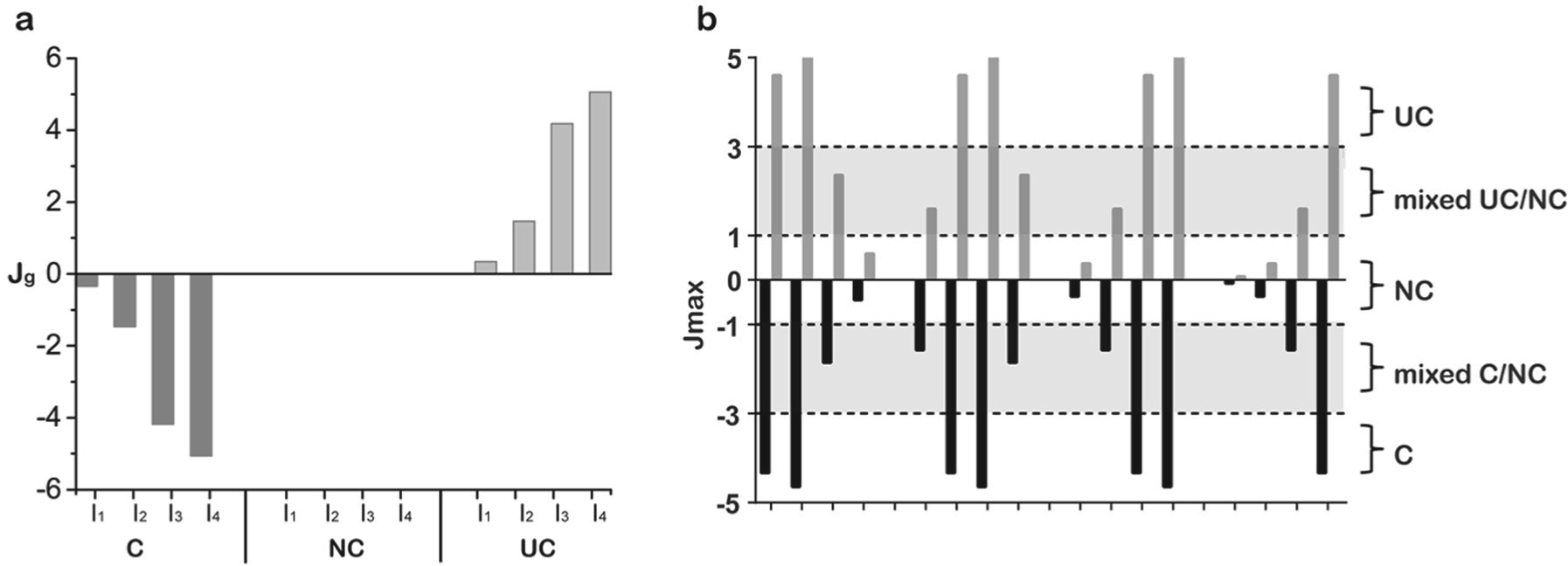
Diagrams of mode of inhibition (MOI) categorization based on the S-gradient signature (
MOI Nonparametric Classification Scheme.
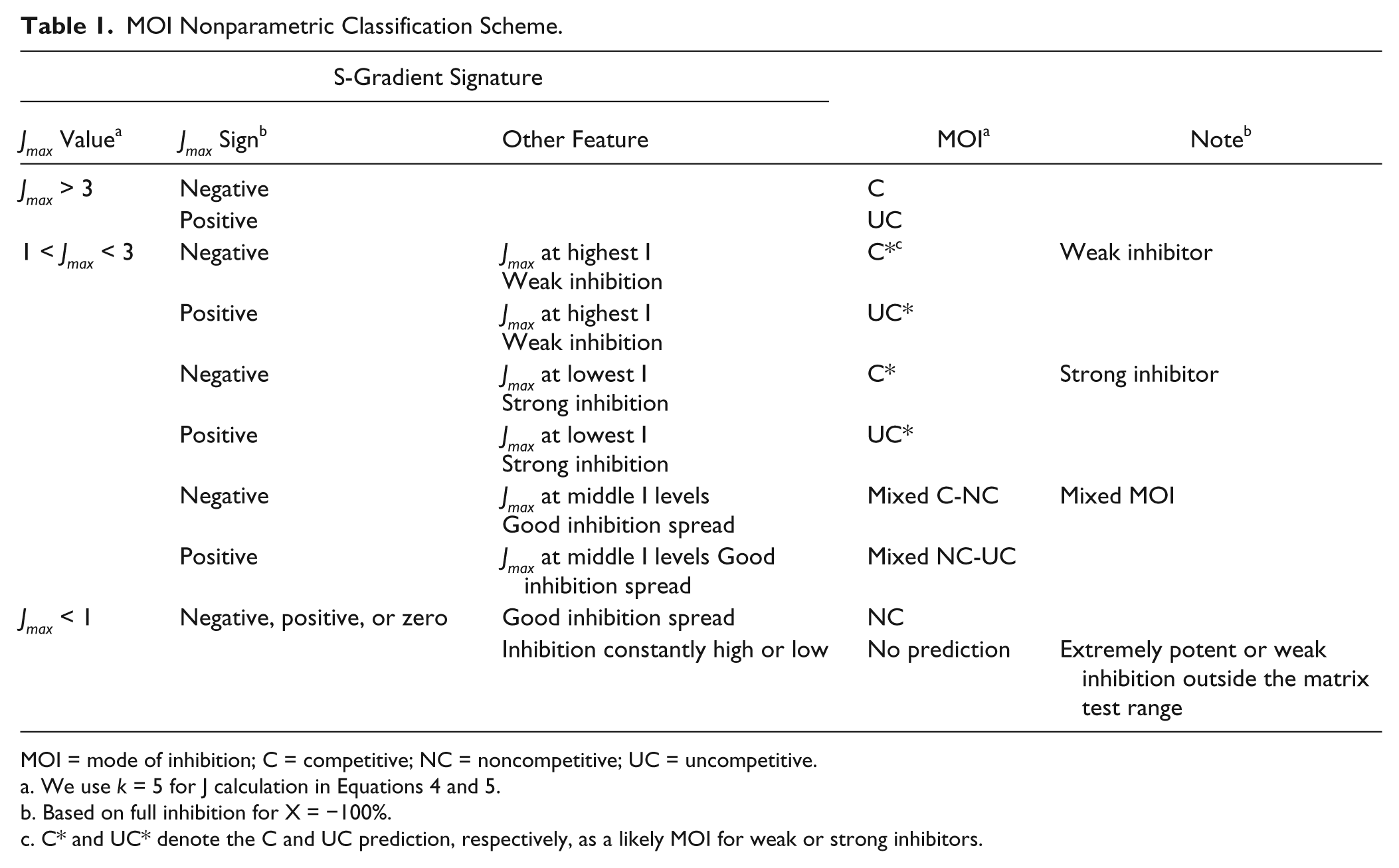
MOI = mode of inhibition; C = competitive; NC = noncompetitive; UC = uncompetitive.
We use k = 5 for J calculation in Equations 4 and 5.
Based on full inhibition for X = −100%.
C* and UC* denote the C and UC prediction, respectively, as a likely MOI for weak or strong inhibitors.
MOI Classification
Table 1 summarizes the MOI nonparametric classification scheme we propose for characterizing the results from small matrix testing. The classification scheme uses the S-gradient features described and discussed above. The Jmax value categorization was based on theoretical case tests and simulations from Equations 1 to 7 at various S, I, and Ki levels most relevant to practical test (vide infra). Figure 2b shows a further simplified diagram to visualize the MOI categorization based only on Jmax and its sign. In general, mixed inhibitors with α values ~2 to 5 yielded intermediate Jmax values yielding mixed NC/C classifications, whereas α values ~10 showed negative Jmax values >3 yielding the classification as competitive. Similarly, uncompetitive inhibitors showed positive Jmax > 3 for α values ~0.1 and mixed NC/UC classifications at α values ~0.2 to 0.5. It is worth noting that, for the convenience of Jmax presentation, we use k = 5 in J calculation from Equations 5 and 6. The k constant is a modifier of the amplitude. The k value should be held constant so it does not affect the relative amplitude of J. However, this may affect the MOI classification stringency and sometimes can be adjusted slightly according to the experimental data variability and the desired stringency for the MOI classification.
Theoretical Simulations
Based on the calculated J and Jmax data from Equations 6 and 7, respectively, we first conducted several theoretical test cases. It is important to demonstrate the effectiveness of the approach under different S and I concentrations as well as Ki ranges. In reality, however, the highest and lowest S and I concentrations can be limited by the substrate and inhibitor availability (e.g., solubility) and assay sensitivity. Sometimes only a certain range of [S] or [I] can be practically achieved. One would like to see if the matrix approach can effectively cover a wide range of inhibitor potencies, as when dealing with a large number of hits from HTS. The choice of [S] and [I] ranges should be driven by the KM and Ki values with sufficient coverage above and below these values. In our first theoretical matrix (4S × 4I) test case, we used both S and I in 1:3, 1:4, and 1:5 dilution series and Ki values from nanomolar to micromolar range.
Matrix Test Data Analysis and MOI Prediction
The matrix test data were analyzed using the following steps. First, the activity data were normalized with a conventional plate-based method using the positive and negative controls on the test plate to convert the activity into percentage inhibition (Inh%) values. Then, the matrix data set was directly plugged into an Excel worksheet calculation containing a calculation template to convert the matrix Inh% values at each and every inhibitor concentration into the S gradient (J value) and simultaneously apply the classification algorithm for MOI prediction of each compound tested. The MOI classification algorithm is based on the calculations for the global J values (Jg) and a set of modified classification criteria. For example, to obtain a more indicative Jmax and to make this value more experimental error resistant (i.e., outlier resistant), Jmax was further defined in the S-gradient signature as the J with maximum absolute value within 20% to 80% average Inh% range. To further reduce outlier interference in the test data, we found it was usually very helpful to first remove any obvious outliers by either an outlier filter or simply having a quick visual inspection of the matrix Inh% data and excluding any data points that were clearly an outlier to the surrounding data sets. This can be done in a program such as Spotfire, and some examples are given in this article. To flag a hit as “too weak” or “too potent” (i.e., outside the matrix test prediction range), we used average Inh% <20 Inh% at the highest inhibitor concentration and average Inh% >80 Inh% (plus with lowest Inh% >90% of average Inh% at the lowest inhibitor concentration) as the criteria, respectively. In addition, a matrix data set was flagged as noisy data if the average Inh% from the inhibitor titration series failed to show a clear decreasing trend with decreasing inhibitor concentrations. For comparison, data analysis with the IC50 shift method (aka R-ratio) was conducted essentially the same way as previously described by Wei et al. 13
Experimental Case 1: Tests of Two Different Methyl Transferase Enzymes with Inhibitors of Known and Unknown MOI
As a first case, we tested two different methyl transferase assays, each with a small number of inhibitors with both known and unknown MOIs. The enzymes have two substrates: a histone-derived peptide substrate providing methylation sites and SAM as the methyl donor. For each methyl transferase assay, we performed a titration series in the matrix test for each of the two substrates. We also used this case for evaluation of different dimensions of the MS × NI matrix in the MOI classification. At first, we tested 42 compounds in the methyl transferase EZH2 biochemical assay using the PRC2 complex.
19
Two of the inhibitors, dubbed here as B-268 and B-920, have their MOI known: B-268 is a proprietary known competitive inhibitor for the histone peptide substrate, whereas B-920 is a known SAM competitive inhibitor. For the peptide substrate titration, we used a 5S × 8I matrix test, and for SAM, we used a 4S × 8I matrix test. After normalization of the raw data to Inh%, the data were subdivided into several smaller matrices and evaluated with this matrix MOI classification method by the J values (we consistently used Jg in the MOI analysis and the same for other case studies in this work). We evaluated one 5S × 8I, two distinct 4S × 8I, and four distinct 4S × 4I matrices from the peptide titration matrix test and one 4S × 8I and two distinct 4S × 4I matrices from the SAM titration matrix test. The results clearly show that for the same inhibitor, different matrices tend to generate similar (many are the same) MOI predictions for each substrate. Occasionally, a different MOI prediction is obtained by using different matrices; in most of these occasions, the discrepancy was caused by data variations and outliers or because the Jmax value was at the borderline of the classification criteria for some of the matrix test results. Importantly, both of the two known inhibitors have their MOI classified correctly by this method (
To expand on this case, we tested a PMT and performed a large 5S × 12I matrix test with 26 inhibitors. Among these inhibitors, nine are previously known to be competitive for a histone peptide substrate and uncompetitive for SAM from previous conventional MOI studies, with the rest of the inhibitors with unknown MOI but suspected to have similar behavior as the nine known inhibitors. Likewise, either the peptide substrate or SAM was varied in the matrix tests. After normalization of the raw data to Inh%, the data were subdivided into several smaller matrices including various iterations of 4S × 6I, 4S × 4I, 3S × 6I, and 3S × 4I combinations, and the J values were used to classify the MOIs (see
Experimental Case 2: Test of High-Throughput MOI Determination for Dengue NS5 RdRp Inhibitors
To test the matrix-based MOI analysis against a list of inhibitors derived directly from an HTS campaign, we performed a 4S × 8I matrix test of 165 inhibitors in a biochemical RdRP assay.
16
We evaluated the MOI prediction with three matrices—4S × 8I and two distinct 4S × 4I matrices (details in
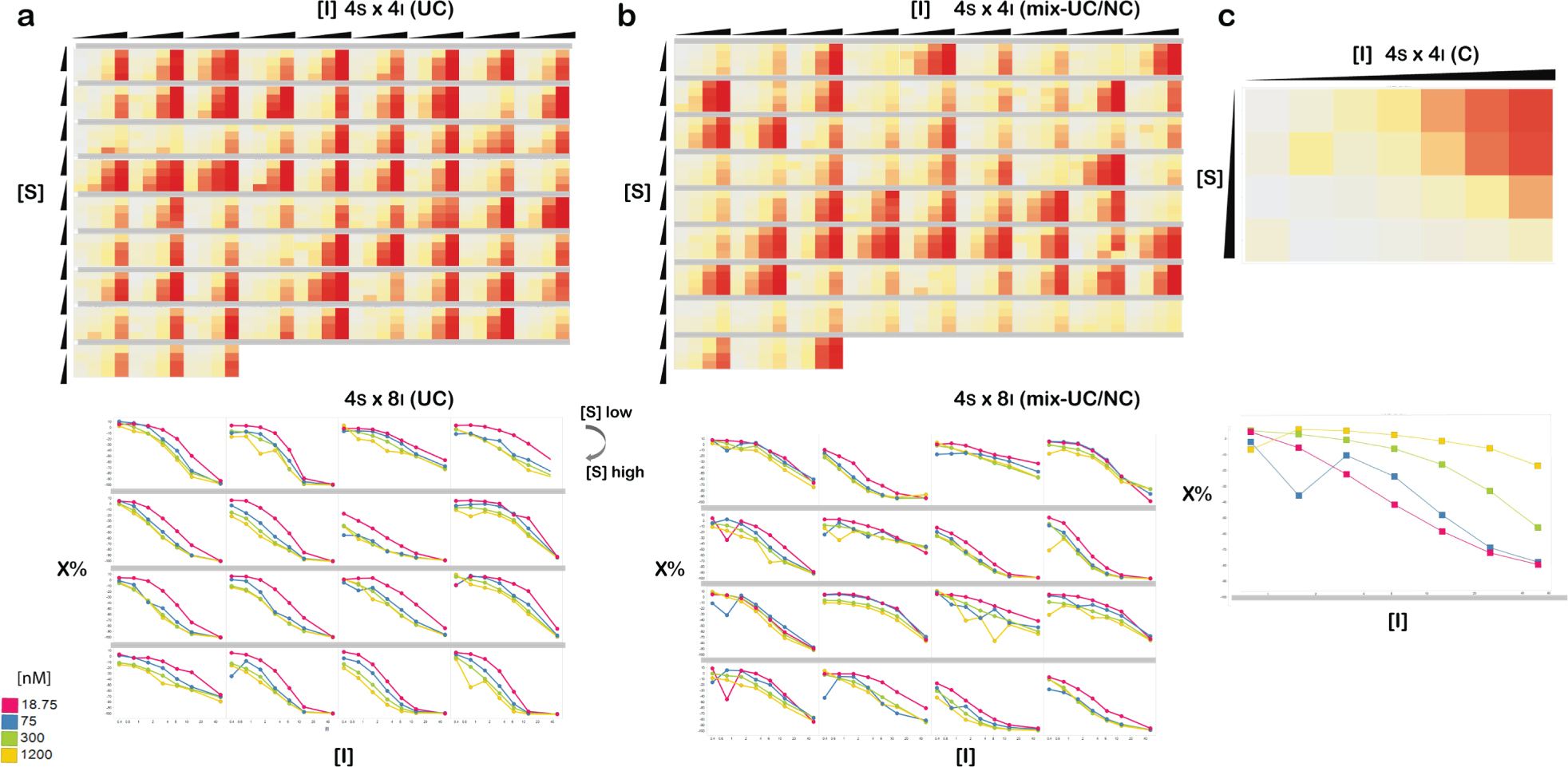
Representative data from the RdDp inhibitor testing. (
Comparison of the IC50 shift method to the matrix S-gradient signature determination for mode of inhibition (MOI) classification for the Dengue RdDp inhibitors. For the IC50 shift method, the data from the 1200 nM and 18.75 nM RNA substrate concentrations were taken. For matrix S-gradient signature determination, 25, 6.25, 1.56, and 0.39 µM of inhibitor and 1200, 300, 75, and 18.75 nM of RNA substrate was used for the matrix. (
Experimental Case 3: Example High-Throughput MOI Determination: Firefly Luciferase Inhibitors
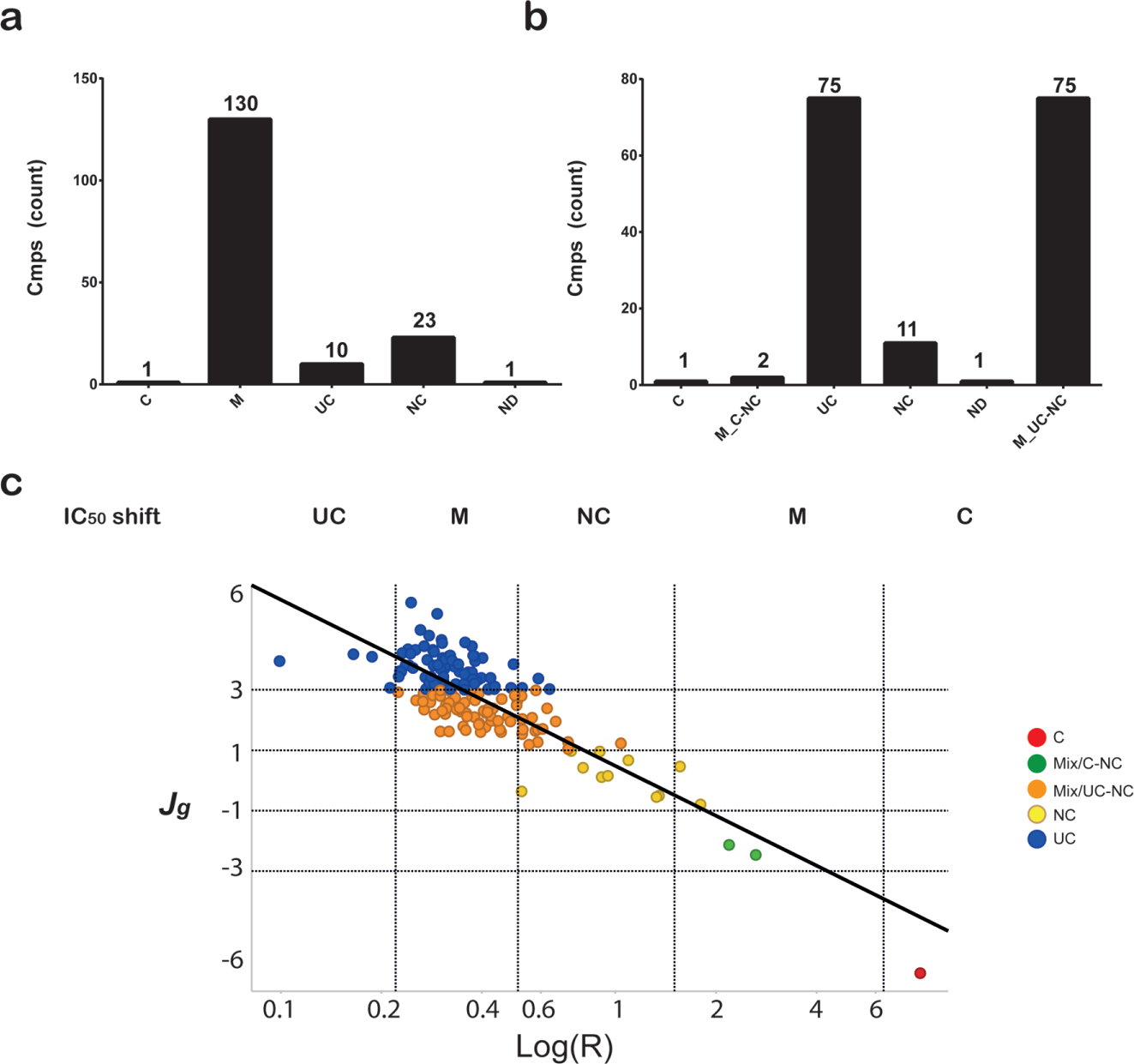
Because the S-gradient matrix method does not require any individual curve fitting as required by the IC50 shift method, one can easily apply this method to any large number of compounds. To examine the MOI classification in a high-throughput mode, we chose to test a series of 1363 FLuc inhibitors that had been previously identified in a screen of the NIBR compound collection aimed at identifying compounds that might interfere with this commonly used reporter enzyme.
22
For MOI determination, these compounds were arrayed as a quantitative HTS (qHTS) format within 1536-well plates and 12 plates were screened, with each plate representing a different inhibitor concentration. The set of qHTS plates was then screened at four different concentrations of the substrate D-luciferin in a biochemical assay measuring FLuc enzyme activity.
22
The data were then broken down to 4S × 8I and two 4S × 4I matrices (
A comparison of the FLuc inhibitor MOIs classified from the IC50 shift ratio (R value) to that derived from Jmax in the current S-gradient matrix method is shown in
Some Important Considerations
This study supports that robust MOI classification can be achieved from nonparametric analysis of activity values derived from a small matrix testing of substrate and inhibitor concentrations. A few recommendations are suggested when implementing this method. For assays with reasonably good data quality and low data variability, the method performs well at smaller test matrices such as 4S × 4I, allowing these to be employed for high-throughput assays. In this case, the number of data points collected is the same as the 2S × 8I employed in the IC50-shift method but without the need for curve fitting. Of course, for assays in which reagents are readily available and low cost, larger 4S × 6I or 4S × 8I matrices could be employed followed by applying the same nonparametric analysis procedure. In every case presented here, because of either data variation or the presence of very strong or weak inhibitors, we found a small subset of compounds whose MOI could not be determined. Therefore, it is important to have checks of the %Inh values at low and high inhibitor concentrations over the tested substrate concentration range. Our analysis takes into consideration both weak or superpotent activity by initially examining the inhibition values at low and high inhibitor concentration to prevent generation of erroneous J values. As well, the Jmax values should be qualified by considering if the S-gradient signature spans a meaningful activity range. For example, we considered Jmax values only if the S-gradient spanned an average of 20% to 80% Inh%. The choice of substrate and inhibitor concentrations is also important, and we found that using a relatively broad range (e.g., covering >100-fold range in concentration) was usually optimal. We used a constant value of k = 5 for the MOI classifications described here, which proved adequate in providing accurate MOI classification based on the amplitude of J values we measured. However, this value can be modified depending on the data variation and assay bias. Overall, one big advantage of the current method is that once a format is judiciously chosen for the matrix testing, the MOI classification could be accurately determined by simply pasting the normalized data into the Excel calculation and classification worksheet. This method provides a rapid and simple procedure for assessing the MOI of large number of compounds arising from screening efforts.
We summarize the overall process of applying the S-gradient signature J with experimental data from matrix testing for MOI classification as illustrated in Figure 5 . The process consists of the following three steps: (1) design of the MS × NI matrix (i.e., selection of the substrate and inhibitor range and dilution series), (2) test of the matrix activity to obtain the corresponding Inh% data matrix after normalization of the activity, and (3) calculation of the S-gradient signature (J) and applying the classification algorithm for MOI prediction of each compound tested. For a 4S × 4I matrix specifically, compounds are arrayed at four concentrations and then tested at four different substrate concentrations. The compound dilution series can be arrayed in four adjacent wells (intraplate dilution series). Alternatively, a qHTS format 24 (i.e., an interplate dilution series) can be applied, and the qHTS set is then screened at four substrate concentrations, as was done for our FLuc case study. Following the assay, the data are normalized by the positive and negative controls on the plates to calculate the Inh% (note in the convention used in this article, complete inhibition is defined as −100%). J values are calculated at the four substrate levels for each compound, and then the MOIs are classified via an algorithm based on the S-gradient signature (J values). This method should also be applicable to binding assays in which ligand and inhibitor concentrations are varied to determine the mechanism of inhibitor binding. In practice, the solubility, background fluorescence, liquid-handling precision, and the possibility of substrate inhibition will affect the test data and thus the classification accuracy. These interfering factors should be taken note of whenever possible no matter which method of MOI determination is employed.
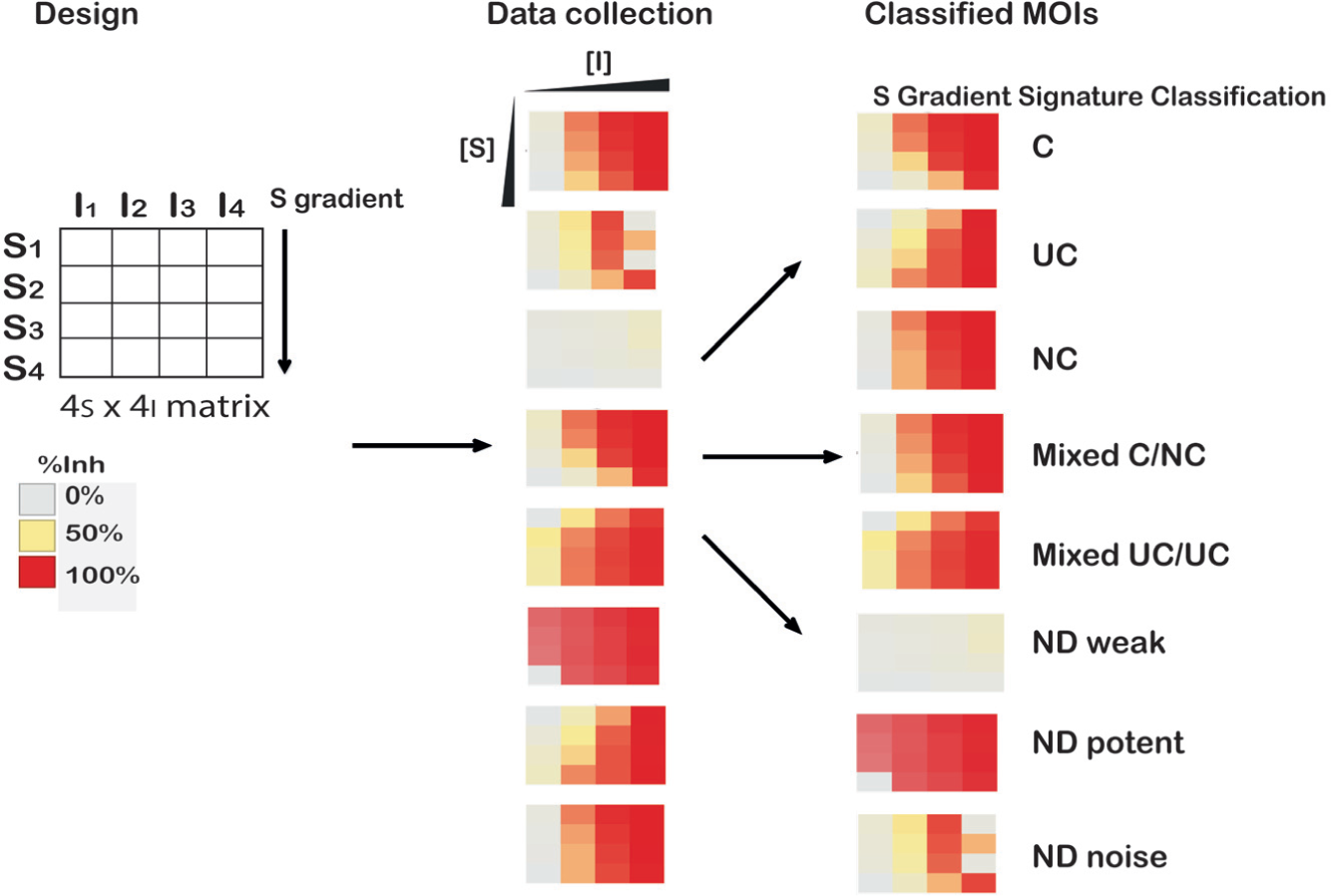
Process for matrix-based experimental testing, data analysis, and S-gradient signature classification for different modes of inhibition (MOIs). An example test of 4S × 4I was performed in the enzyme assays, and example heat maps representing the Inh% values are shown for various patterns. S-gradient signature classification bins the data either as C (competitive), UC (uncompetitive), NC (noncompetitive), or mixed (C/NC or UC/NC). Noisy data as well as very weak or very potent compounds are also correctly identified for which the MOI was not determinable (ND).
Footnotes
Acknowledgements
The authors wish to acknowledge the insightful inputs from Dr. Hanspeter Gubler (Novartis) for this study. We would also like to thank Drs. Guy Herr, Zinger Yang, and John Lin for helpful inputs during the initial discussion. The authors also thank Ms. Pei-I Ho for her technical support in the FLuc assay and testing and Drs. William Hill and Timothy Benson for their support of the study.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.