
Abstract
In the present study, we sought to define genes associated with immune thrombocytopenia (ITP). Microarray analysis revealed that of 1002 genes associated with ITP, 309 genes had downregulated expression and 693 genes had upregulated expression in patients with ITP. Gene set enrichment analysis revealed that 11 pathways were positively correlated to ITP, such as type I diabetes mellitus, intestinal immune network for IgA production, and oxidative phosphorylation. The messenger RNA expression levels of the indicated genes, including HLA-DRB5, IGHV3-66, IFI27, FAM212A, PLD5, tumor necrosis factor (TNF)–α, interferon-γ, interleukin (IL)–1β, and IL-4, were significantly increased in patients with ITP compared with healthy humans, while MMP8, SLC1A3, CRISP3, THBS1, FMN1, and IL-10 were decreased. In conclusion, the gene expression profile of patients with ITP has established a foundation to study the gene mechanism of ITP progression.
Introduction
Immune thrombocytopenia (ITP) is an acquired immune-mediated autoimmune disease characterized by low peripheral blood platelet counts (<100 × 109/L), which are considered thrombocytopenia, and increased risk of bleeding. 1 The mechanism of ITP was first identified in 1951, in which both impaired platelet production and T-cell-mediated effects played a critical role.2,3 Recently, ITP has been classified by duration into acute (<3 months), persistent (3–12 months), and chronic (>12 months). 1 ITP in adults usually follows a chronic course that has an onset with a preceding viral or other illness. However, ITP in children is usually short-lived, occurring a few days or weeks after viral infection and more than half recovering within 6 months. 4 It has been indicated that the development of chronic ITP in adults is associated with advanced age, sex, less mucosal bleedings, and an insidious onset. 5 This is in contrast to children with ITP, in whom a low proportion has chronic disease, and incidence between sexes is similar. 6
Blood leukocytes constitute an accessible source of clinically relevant information, and a comprehensive molecular phenotype of these cells can be obtained using gene expression microarray. 7 Genome-wide gene expression profiling with microarrays is a power emerging technology that has permitted development of classification and prediction of cancer outcomes, including lymphoma, leukemia, and colon and breast cancer, and identification of molecular markers and pathways dysregulated in diseases.8,9
RNA-sequencing (RNA-seq) is a relatively new method for analyzing gene expression; it provides digital readouts for mapping and quantifying transcriptomes. 10 RNA-seq studies of model organisms have revealed unknown aspects of transcriptomes through refinement of transcriptional start sites and identification of novel upstream open reading frames. 11 Global surveys of alternative splicing show that nearly 95% of all multiexon genes in humans undergo alternative splicing events. 12 Gene set enrichment analysis (GSEA) evaluates microarray data at the level of gene sets. Mootha et al. 13 performed GSEA to analyzed data from diabetics versus healthy controls and revealed that genes involved in oxidative phosphorylation show reduced expression in diabetics. However, up until now, investigation of the gene expression pattern and signature in ITP has remained unexplored.
At present, it is clear that its pathogenesis depends on both environmental and genetic factors. The most important are the cytokines or growth factors such as interleukin (IL)–2, IL-10, interferon (IFN)–γ, vascular endothelial growth factor (VEGF), and transforming growth factor (TGF)–β, which have been implicated in the pathophysiology of ITP.14–16 However, it is clear that many of the genes involved have still not been identified. In the present study, we describe a gene expression pattern and signature study on a panel of three patients with ITP for the identification of genes using the microarray, with validation in a larger sample of the patients and controls using quantitative real-time PCR (qRT-PCR) and enzyme-linked immunosorbent assay (ELISA). Hence, we could identify several important molecular alternations during the ITP development.
Materials and Methods
Ethics Statements
The study protocol was proved by the ethics committee of Shanghai Children’s Hospital. Written informed consent was obtained from all participants in this study.
Patient Specimens
Peripheral leukocytes from healthy controls or ITP patients in Shanghai Children’s Hospital were included for RNA-seq and GSEA analysis, including three healthy controls (two females and one male) and three patients with ITP (two males and one female), ages 2 to 4 years. Peripheral leukocytes from healthy controls or ITP patients in Shanghai Children’s Hospital were also included for qRT-PCR and ELISA analysis, including 34 females and 41 males, ages 3 months to 12 years (median, 4.5 years); these patients were healthy (n = 24), had chronic ITP (CITP, n = 20), or had acute ITP (AITP, n = 31). None of these patients had received radiotherapy or chemotherapy.
RNA-seq and GSEA
RNA-seq was performed as previously described. 10 Affymetrix (Santa Clara, CA, USA) Human Genome U133 plus2 Array for peripheral leukocytes was performed on ITP patients and healthy controls. RNA-seq data were log2-normalized and gene expression changes were considered. RNA-seq peripheral leukocyte samples from ITP patients and controls have been deposited in NCBI (http://www.ncbi.nlm.nih.gov/sra, AC: SRP065146). To gain further insight into the biological pathways involved in ITP pathogenesis, GSEA was performed on the KEGG (Kyoto Encyclopedia of Genes and Genomes) pathway. The pathways used in this analysis were chosen according to the one in which the selected genes belonged. The gene sets showing p < 0.05 and q value false discovery rate (FDR) ≤0.05 were considered enriched between ITP patients and healthy humans.
Validation of Differential Expression by qRT-PCR
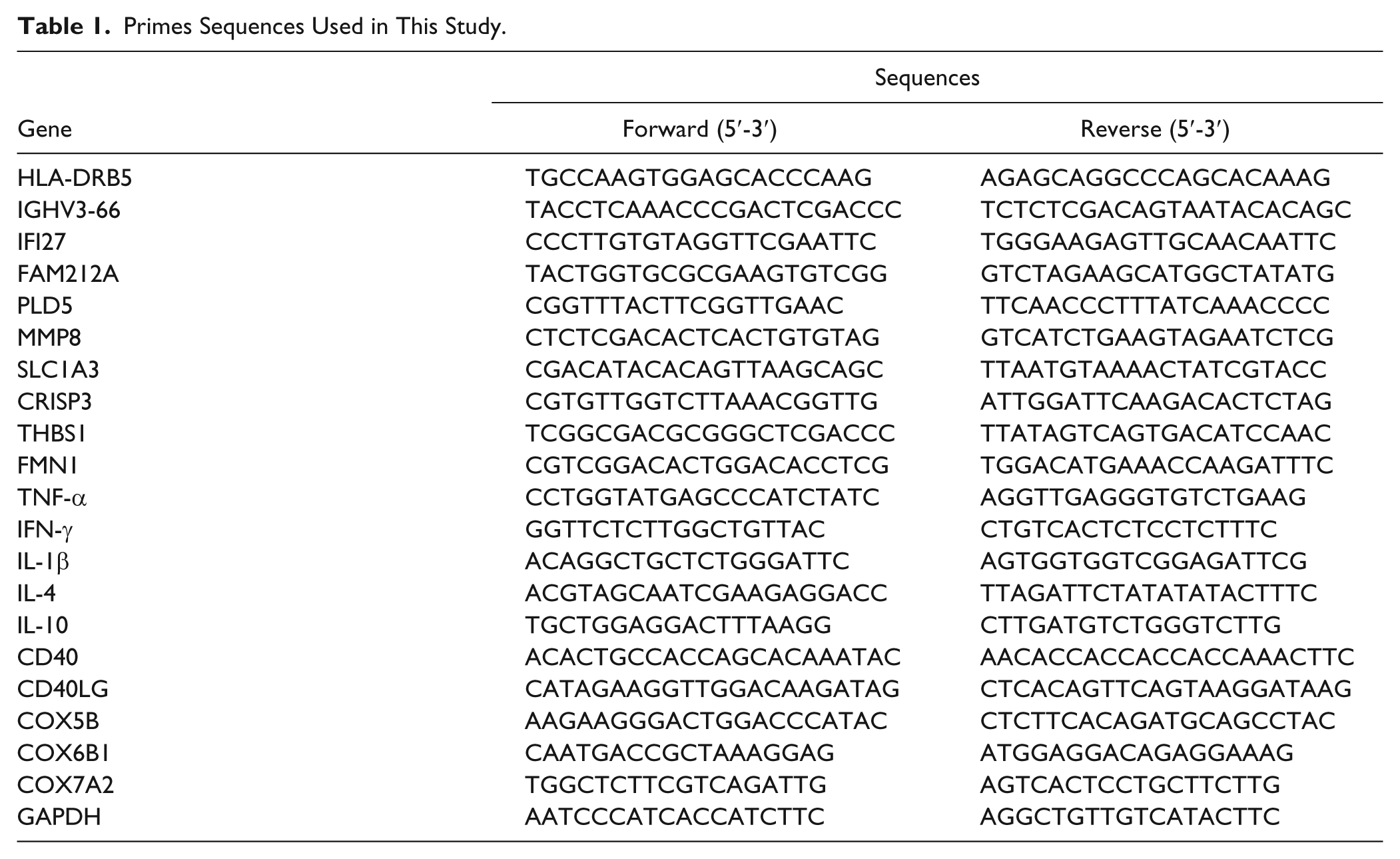
Total RNA was extracted from peripheral leukocytes using the TRIzol reagent (Invitrogen Life Technologies, Carlsbad, CA), and reverse transcription PCR was done using the RNA-cDNA synthesis kit (Thermo Fisher Scientific, Rockford, IL) according to the manufacturer’s protocol. qRT-PCR analysis was performed using the Maxima SYBR Green qPCR Master Mix (Thermo Fisher Scientific) and the ABI 7500 sequence detection system (Applied Biosystems, Foster City, CA). GAPDH level was used as an internal control, and fold changes were calculated using the 2–ΔΔCt method. The primers used are listed in Table 1 .
Primes Sequences Used in This Study.
ELISA
Secretions of tumor necrosis factor (TNF)–α, IFN-γ, IL-1β, IL-4, and IL-10 were determined by ELISA. After the 8 × 109/L peripheral leukocytes were collected from healthy controls or ITP patients in Shanghai Children’s Hospital, the relative content of TNF-α, IFN-γ, IL-1β, IL-4, and IL-10 was measured by ELISA according to the manufacturer’s protocol (Boster BioTech, Wuhan, China).
Statistical Analysis
All experiments were performed in triplicate unless otherwise stated. Data are presented as the mean ± SD. Statistical analyses were performed with GraphPad Prism 5 software (GraphPad Software, La Jolla, CA). Significance analysis of microarray (SAM) method was used to find mostly significant gene expression changes between patients and controls. Student’s t test was used for comparison between the healthy controls and CITP or AITP patients. A p value of 0.05 or less was considered to indicate statistical significance.
Results
Analysis of Gene Expression Pattern in Patients with ITP
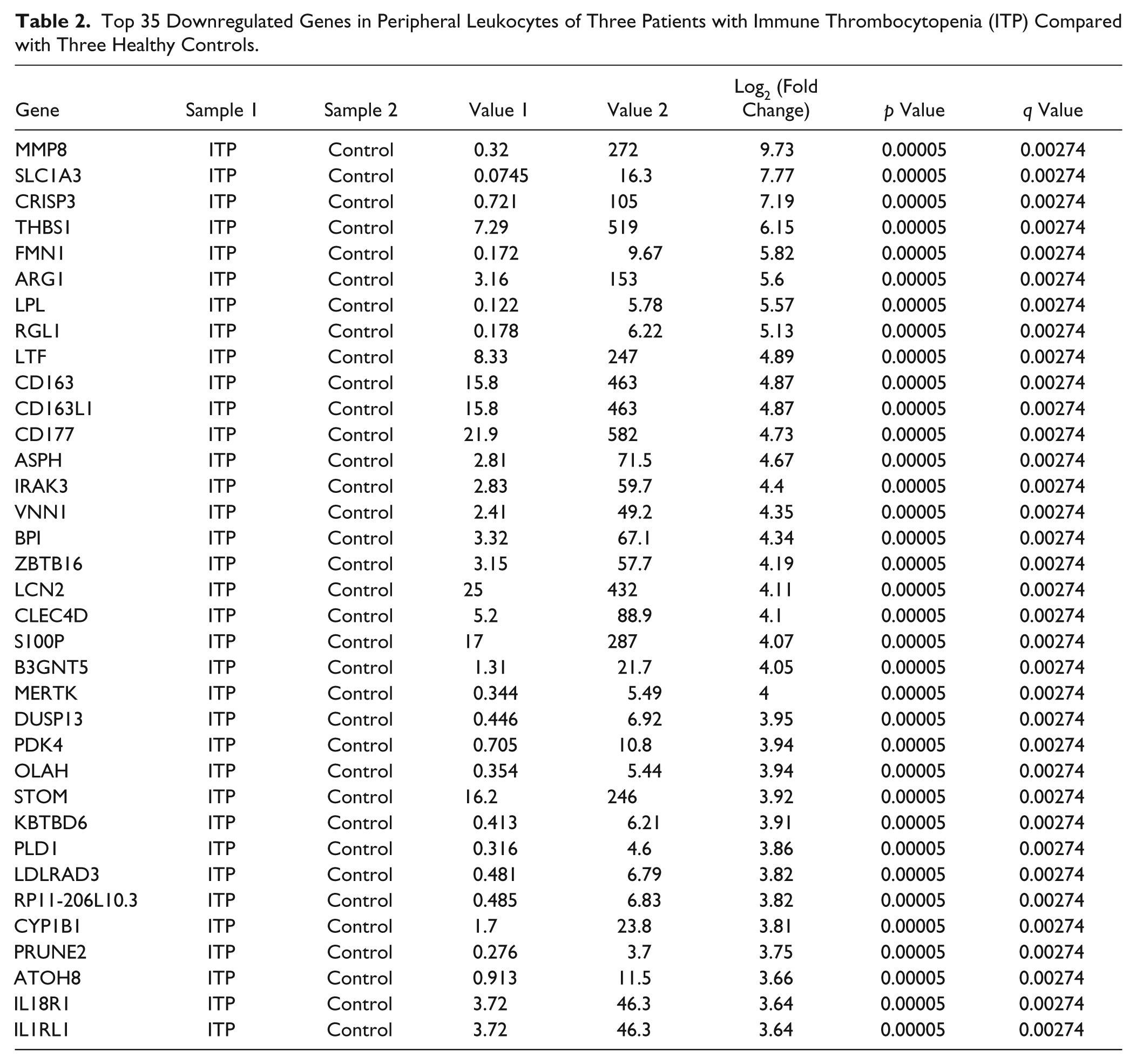
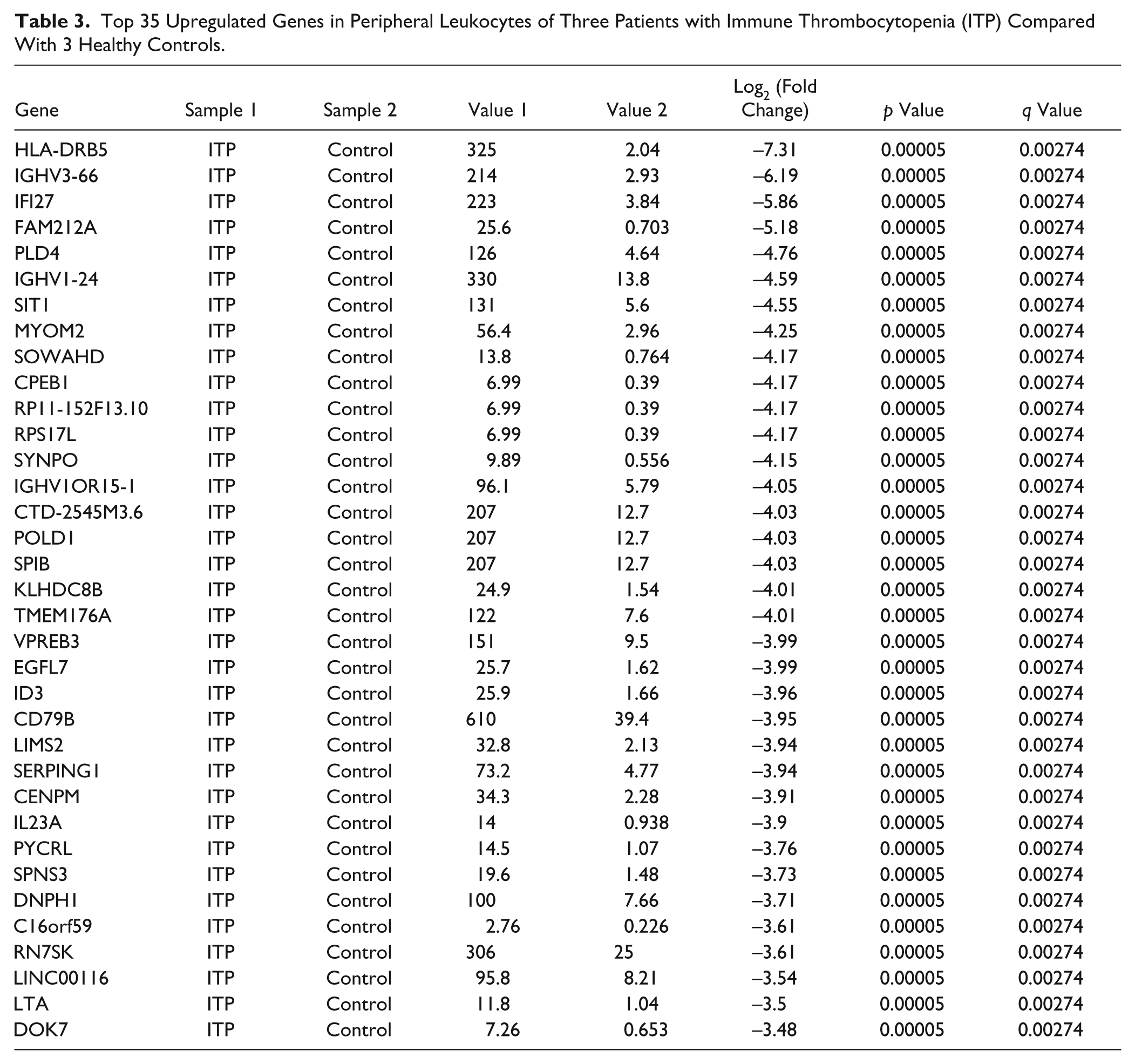
The gene expression pattern was investigated in six independent specimens from peripheral leukocyte messenger RNA (mRNA) expression profiles comprising three healthy controls and three ITP patients, with normal leukocyte numbers (1–10 × 109/L). In total, 1002 ITP-specific genes with significant fold change were identified (p < 0.05 and q value FDR ≤0.05). Of 1002 genes, 309 were downregulated in ITP patients compared with healthy controls ( Table 2 shows the top 35 downregulated genes), whereas 693 of 1002 genes were upregulated ( Table 3 shows the top 35 upregulated genes).
Top 35 Downregulated Genes in Peripheral Leukocytes of Three Patients with Immune Thrombocytopenia (ITP) Compared with Three Healthy Controls.
Top 35 Upregulated Genes in Peripheral Leukocytes of Three Patients with Immune Thrombocytopenia (ITP) Compared With 3 Healthy Controls.
Multiple Pathways Were Bioinformatically Predicted to Be Correlated to ITP
To further characterize the enrichment of pathways that correlates to the development and progression of ITP, we carried out the GSEA in healthy controls and ITP patients. GSEA is a bioinformatical analysis that determines whether an a priori defined set of genes shows statistically significant and concordant differences between two biological states.
17
Specimens were divided into two groups, healthy controls and ITP patients. A ranked gene list was generated by comparing the mRNA microarray data of healthy control and ITP patient groups. Then GSEA was performed to evaluate the enrichment of different pathway gene sets in this ranked gene list. We found that genes that were upregulated in patients with ITP were associated with multiple pathways, including type I diabetes mellitus, intestinal immune network for IgA production, oxidative phosphorylation, and other correlated pathways (
Selection of Predictive Genes
Then we asked whether the genes that were significantly changed and involved in the pathways were associated with the pathogenesis of ITP. To test this hypothesis, the top five downregulated genes (MMP8, SLC1A3, CRISP3, THBSA, and FMN1) and top five upregulated genes (HLA-DRB5, IGHV3-66, IFI27, FAM212A, and PLD5) in ITP patients were selected as the predictor. Moreover, 10 genes associated with the intestinal immune network for IgA production (CD40, CD40LG, and IL-4), type I diabetes mellitus (IFN-γ, TNF-α, and IL-1β), oxidative phosphorylation (COX5B, COX6B1, and COX7A2), and clinical course of ITP (IL-10) 14 were also selected as the predictor. The functions of these genes in the pathogenesis of ITP have not been well characterized. For example, Th1 cytokine IFN-γ was positively correlated with IL-6 in ITP patients, and its levels in plasma were marginally higher in males than in females. 18 Serum IL-10 levels were higher in patients with ITP and could predict the clinical course of ITP. 14 However, the correlation between other selected predictors and the pathogenesis of ITP has not been found. Thus, we hypothesized that these selected genes may have altered their expression in ITP patients.
Validation of Predictive Genes
To experimentally validate the alteration of selected genes in ITP patients with a normal leukocyte number (1–10 × 109/L), samples from 24 healthy controls and 51 ITP patients (20 CITP and 31 AITP) were collected, and qRT-PCR and ELISA assays were performed. As shown in Figure 1A–D, the mRNA expression levels of HLA-DRB5, IGHV3-66, IFI27, FAM212A, and PLD5 were significantly increased in the CITP and AITP groups compared with the healthy control group, while the mRNA expression level of MMP8, SLC1A3, CRISP3, THBS1, and FMN1 was significantly decreased (Fig. 1F–J). Importantly, in the mRNA expression levels of genes, there were notable differences between the AITP and CITP groups, with the exception of HLA-DRB5, IGHV3-66, and THBS1, suggesting that these factors may be associated with the pathological progression of ITP. In addition, the mRNA expression levels of genes involved in the signaling pathways associated with ITP were significantly increased in the CITP and AITP groups compared with healthy control groups ( Fig. 2A–I ), with the exception of a decreased mRNA expression level of IL-10 ( Fig. 2J ). Importantly, the mRNA expression levels of IL-1β, IL-4, COX5B, and COX6B1 were notably increased in the AITP group compared with the CITP group, suggesting that these factors may be associated with the pathological progression of ITP. In addition, the concentrations of TNF-α, IFN-γ, IL-1β, IL-4, and IL-10 were determined by ELISA. The content of the indicated proteins was significantly increased in the CITP and AITP groups compared with the healthy control group ( Fig. 3A–D ), with the exception of a decreased content of IL-10 ( Fig. 3E ). Therefore, the expression pattern of these indicated genes that correlated to ITP was similar to those predicted by microarray and GSEA assays.
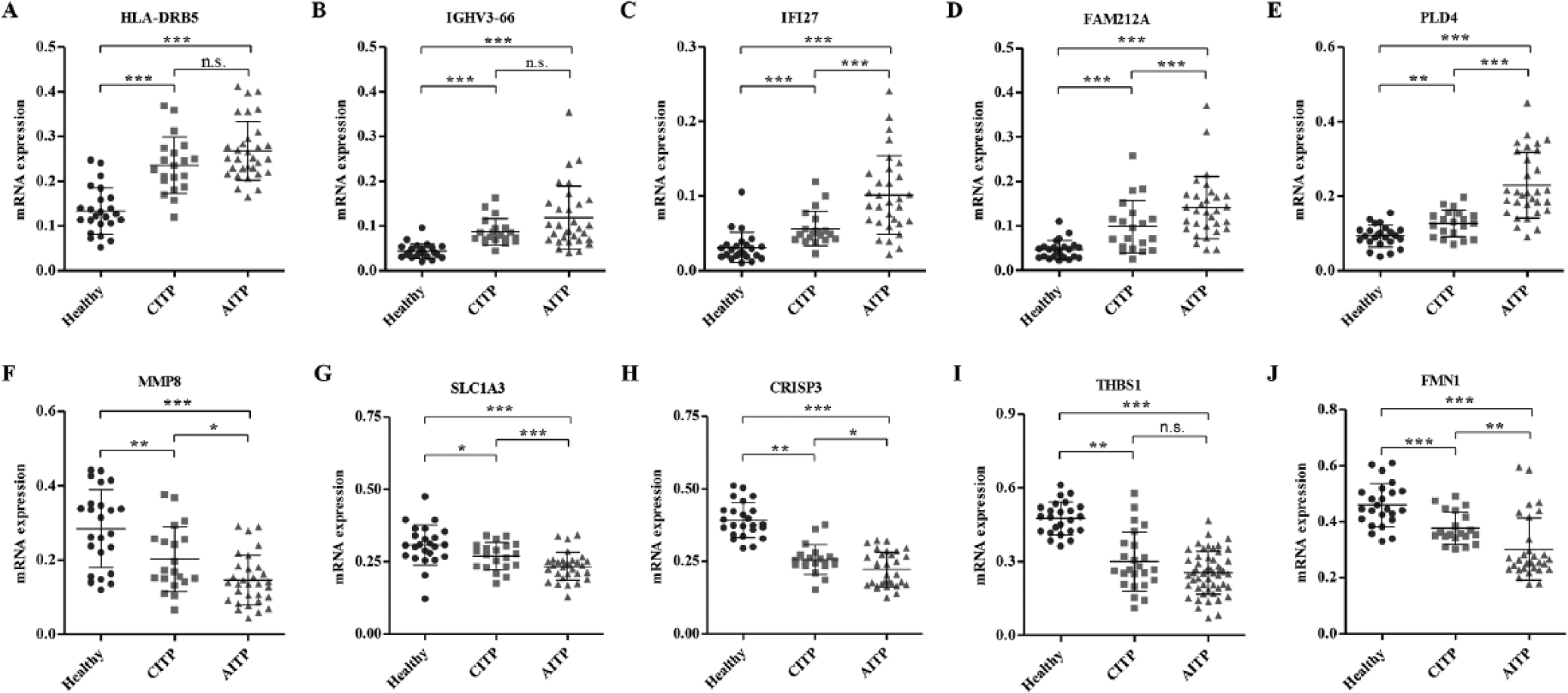
Expression of genes differently expressed between immune thrombocytopenia (ITP) patients and healthy controls. To verify the results from microarray analysis, we analyzed the messenger RNA expression of 10 selected genes in peripheral leukocytes among 20 patients with chronic ITP (CITP), 31 patients with acute ITP (AITP), and 24 healthy controls by quantitative real-time PCR and confirmed the upregulation of HLA-DRB5 (
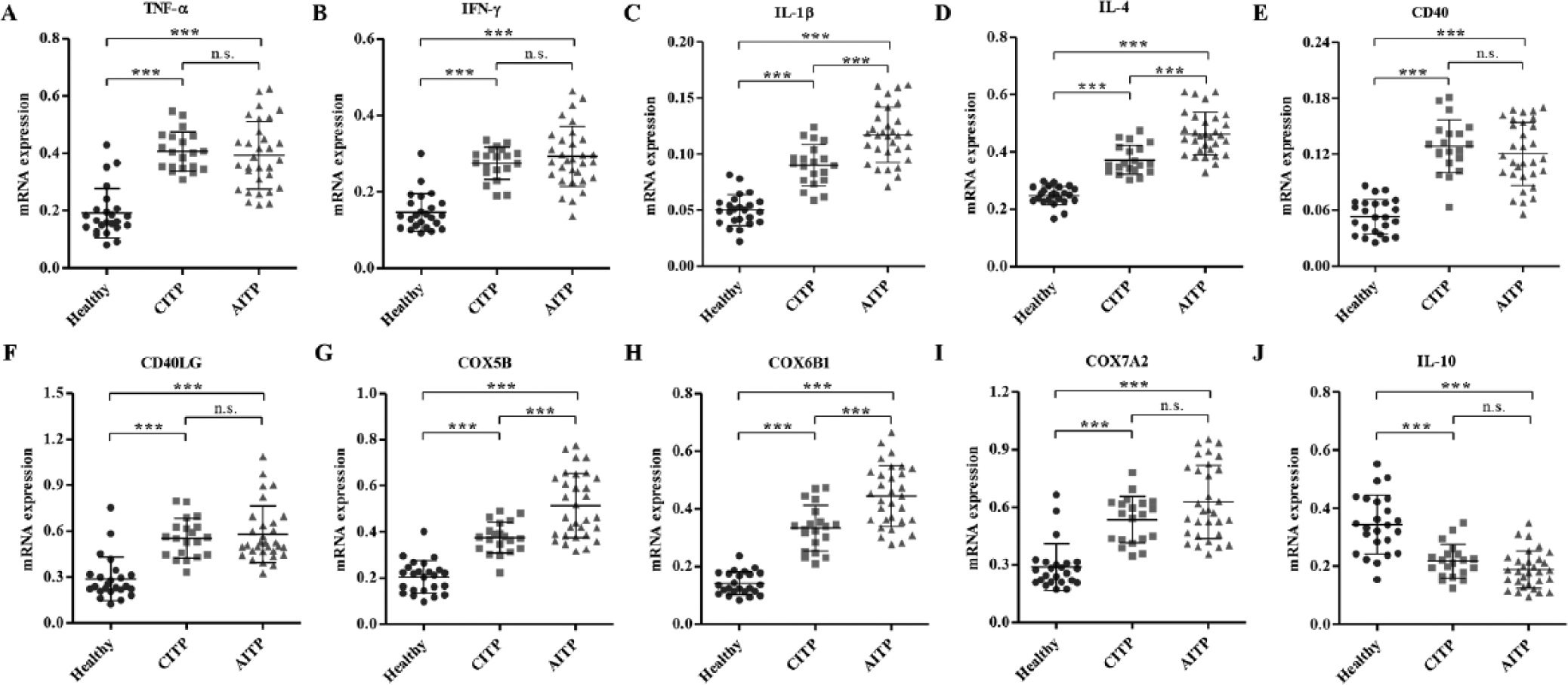
Expression of genes involved in the signaling pathways enriched in immune thrombocytopenia (ITP) patients. To verify the results from microarray analysis, we analyzed the messenger RNA expression of 10 selected genes in peripheral leukocytes among 20 patients with chronic ITP (CITP), 31 patients with acute ITP (AITP), and 24 healthy controls with quantitative real-time PCR and confirmed the upregulation of tumor necrosis factor (TNF)–α (
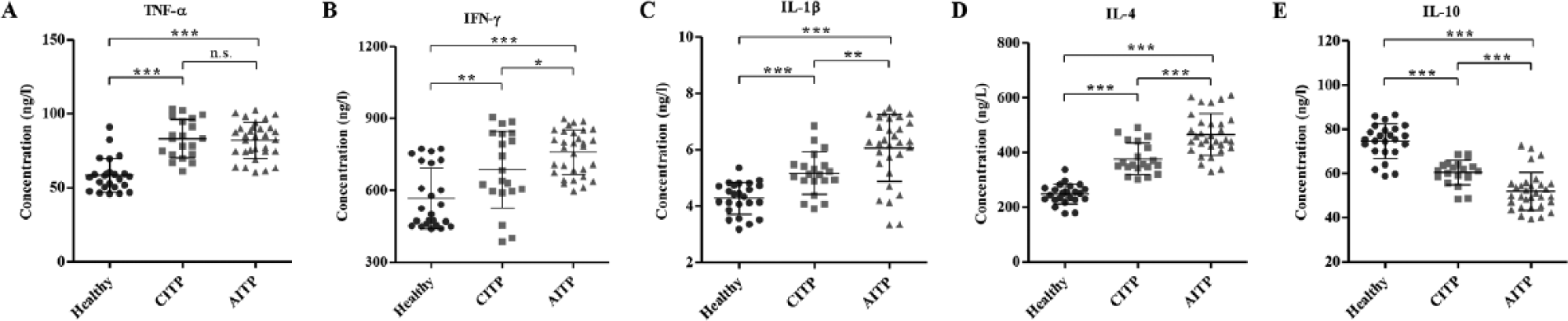
Concentrations of tumor necrosis factor (TNF)–α, interferon (IFN)–γ, interleukin (IL)–1β, IL-4, and IL-10 in immune thrombocytopenia (ITP) patients and healthy controls. To verify the results from microarray analysis, we analyzed the concentrations of TNF-α, IFN-γ, IL-1β, IL-4, and IL-10 in peripheral leukocytes among 20 patients with chronic ITP (CITP), 31 patients with acute ITP (AITP), and 24 healthy controls with enzyme-linked immunosorbent assay and confirmed the upregulation of TNF-α (
Discussion
Autoimmune diseases consist of variable and serious illnesses with debilitating effects on more than 5% of the population in the United States. 19 Loss of tolerance is central in these diseases, where the immune system is dysregulated and attacks organs and cells instead of protecting them. ITP is an autoimmune disease characterized by prematurely destroyed platelets; it is common in children, with increased morbidity. 20 To better understand which genes are important in ITP, we performed mRNA microarray analysis of peripheral leukocytes from ITP and healthy controls. Several studies have shown differentially expressed genes in serum and plasma from peripheral blood in ITP patients.16,21
We identified differently expressed genes in peripheral leukocytes between ITP patients and healthy controls. Genes involved in type I diabetes mellitus, intestinal immune network for IgA production, and oxidative phosphorylation were overexpressed in ITP patients compared with healthy controls. These findings suggest that ITP is associated with an unspecific and wider inflammatory gene activation profile. Diabetes mellitus and graft versus host disease are associated with thrombocytopenic purpura and increased the incidence of it.22,23 Although thrombocytopenia is a common feature of systemic lupus erythematosus (20%–40%), severe thrombocytopenia is comparatively rare (~5%) and can predate other features of systemic lupus erythematosus. 24
To verify some of the differences observed in the mRNA microarray analysis, we performed qRT-PCR and ELISA in another independent sample of peripheral leukocytes of healthy controls and ITP patients, including CITP and AITP patients. We analyzed the expression of HLA-DRB5, IGHV3-66, IFI27, FAM212A, PLD5, MMP8, SLC1A3, CRISP3, THBS1, and FMN1 in ITP patients, which showed the similar results of our microarray analysis. Importantly, the mRNA expression levels of genes were notably different between AITP and CITP groups, with the exception of HLA-DRB5, IGHV3-66, and THBS1, suggesting that these factors may be associated with the pathological progression of ITP. HLA class II heterodimer β5 (HLA-DRB5) plays a central role in the immune system by presenting peptides derived from extracellular proteins 25 and is associated with risk of systemic lupus erythematosus in the Chinese Han population, 26 which is in agreement with our findings that HLA-DRB5, upregulated in systemic lupus erythematosus, is enriched in ITP patients. A recent study demonstrated that IFI27 is more likely to be upregulated in lupus than in another autoimmune condition, idiopathic thrombocytopenic purpura. 27 Two B-cell costimulatory genes, CD40 and CD40LG, contribute to the production of IgA, which is also similar to our data in the GSEA. 28 The increased expression of CD40LG was not consistent with our previous microarray analysis. However, the most likely explanations are that the qRT-PCR assay we used is more sensitive than the microarray method and the number of patients we collected in the qRT-PCR assay (n ≥ 20) was larger than that in microarray assay (n = 3). Nevertheless, the correlation between these genes and pathogenesis of ITP have not been well identified, and this is the first time that we have identified the increased expression of these genes in ITP patients.
We also analyzed other cytokine levels in the two phases of the disease. We found that the mRNA and protein expressions of TNF-α, IFN-γ, IL-1β, and IL-4 were elevated in peripheral leukocytes of ITP patients compared with healthy controls, with the exception of a decreased expression of IL-10. Moreover, there were significant differences in protein expression of these cytokines between CITP and AITP but no difference in protein expression of TNF-α measured by ELISA, suggesting an important role of these cytokines in the pathophysiologic mechanism in ITP. In contrast, Ma et al. 18 reported that IFN-γ levels in plasma had no statistical significance between ITP patients and controls, which was similar to our microarray data analyzed in peripheral leukocytes, and Del Vecchio et al. 14 found that IL-10 levels in serum were significantly higher in AITP patients compared with either healthy controls or CITP patients. Del Vecchio et al. found that IL-10 levels in serum were significantly higher in AITP patients compared with either healthy controls or CITP patients. However, in our study we found that IL-10 concentration in peripheral leukocytes were significantly lower in AITP patients compared with either healthy controls or CITP patients. Although there is no direct evidence supporting the hypothesis that ITP is associated with TNF-α, IL-1β, and IL-4 concentration in blood, it is the first time that we have observed the differential expression of these cytokines in ITP patients.
Finally, the mRNA expression of COX5B and COX6B1 was significantly higher in AITP patients than in CITP patients, suggesting that COX5B and COX6B1 rather than COX7A2 are involved in the pathogenesis of ITP. Moreover, COX5B, COX6B1, and COX7A2 were involved in oxidative phosphorylation in our GSEA data, which is in agreement with another study. 29 These data indicate that COX5B, COX6B1, and COX7A2 are associated with the pathogenesis of ITP through the oxidative phosphorylation pathway.
Limitations of this study include the relatively small number of patients and controls, which are due to the rarity of the disease and the fund limitation of the study, and ITP patients are quite heterogeneous. However, this cohort quite accurately reflects the normal span of patients in a clinic. Moreover, the pathways should be further clarified in further investigation, especially those not involved in the immune-related signals.
In summary, our findings show that there are clear molecular differences between ITP patients and healthy controls, and gene signatures associated with ITP were also identified. The data emphasize how crucial patient selection is in clinical trials, and it can be useful to construct a new classification scheme that better assesses clinical malignancies.
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by Scientific Research Program of Science and Technology Commission of Shanghai Municipality (program number 12411952406) and Scientific Research Program of Shanghai Municipal Commission of Healthy and Family Planning (program number 201540178).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.