
Abstract
SETD8 is the methyltransferase responsible for monomethylation of lysine at position 20 of the N-terminus of histone H4 (H4K20). This activity has been implicated in both DNA damage and cell cycle progression. Existing biochemical assays have utilized truncated enzymes containing the SET domain of SETD8 and peptide substrates. In this report, we present the development of a mechanistically balanced biochemical assay using full-length SETD8 and a recombinant nucleosome substrate. This improves the binding of SAM, SAH, and sinefungin by up to 10,000-fold. A small collection of inhibitors structurally related to SAM were screened and 40 compounds were identified that only inhibit SETD8 when a nucleosome substrate is used.
Introduction
Genomic instability is a characteristic marker of most human cancers. 1 DNA damage and the associated repair mechanisms play an important role in carcinogenesis. Inefficient repair of damaged DNA can lead to mutations, translocations, amplifications, deletions, and epigenetic modifications that can be oncogenic. 2 Genomic instability can lead to the activation of the DNA damage response (DDR) pathway, and constitutive activation of DDR proteins has been reported in human tumors but not in normal tissues. 2 In addition, double-strand breaks (DSBs) of DNA lead to oncogenic chromosomal rearrangements when repaired improperly. 3 The regulation of the cell cycle, like the control of genome integrity, is a well-coordinated process in cells that includes posttranslational modification of chromatin, which is important for DNA replication, mitotic condensation, and cell division. 4 SETD8 (also known as SET8, PR-SET7, or KMT5A) plays an important role in both DDR and chromatin-associated regulation of the cell cycle.5–7 In embryonic stem cells and differentiated cells, loss of SETD8 results in massive DNA damage and accumulation of cells in the G2/M phase.7–9 Furthermore, SETD8 and its histone methyltransferase activity are required for the DSB response and p53 binding protein 1 (p53BP1) recruitment in the DSB area.10,11 p53BP1 is known to be a critical factor in mammalian DDR for nonhomologous end joining (NHEJ)–directed repair. 3
The role of SETD8 in these oncolytic events has initiated interest in the identification of tool compounds to probe this and other biological processes that it may regulate. Nahuoic acid A, a natural product produced by Streptomyces sp., was the first published inhibitor of SETD8 with a Ki of 2 µM.
12
It is competitive with S-(5′-adenosyl)-
Although SETD8 can methylate nonhistone proteins,17–19 the most commonly studied site of methylation is lysine at position 20 on the N-terminus of human histone H4 (H4K20).5,6,20,21 The methyltransferase activity resides in a small 157–amino acid domain at the C-terminus of SETD8, called the Su(var)3-9 and “Enhancer of zeste” and Trithorax (SET) domain. SETD8 is believed to recognize seven amino acids from positions 17–23 on histone H4. SETD8 is the only enzyme known to monomethylate H4K20.5–7 Two other methyltransferases, SUV4-20H1 and SUV4-20H2, recognize and methylate H4K20me1 and H4K20me2, respectively. 22 A majority of biochemical studies using SETD8 have utilized the truncated SET domain and peptide substrates corresponding to H4K20.12–16 These assays have successfully identified good chemical tools for SETD8, including UNC0379. While little is known about the larger 194–amino acid N-terminal region, it is reported to bind phosphorylated tyrosine at position 72 on histone H4. 23 The N-terminal domain also contains a basic patch of residues that bind nucleosomes. 24 These and additional studies have suggested that nucleosomes are an optimal substrate for full-length SETD8. 25 However, there are no reports of the development and characterization of an assay using nucleosomes suitable for inhibitor identification and characterization.
The identification of high-quality, physiologically relevant starting points is critical to accurately probe the role of SETD8 in the cellular pharmacology of H4K20 methylation. In this report, a nucleosome substrate dramatically improved the binding of SAM to full-length SETD8. Inhibitors structurally related to SAM showed a similar improvement in potency. This suggests that nucleosome binding to SETD8 may dramatically alter the architecture of the SAM pocket, which could lead to the identification of novel inhibitors.
Materials and Methods
Materials
Full-length SETD8 (amino acids 1–352) and the SET domain of SETD8 (amino acids 195–352) were expressed in Escherichia coli with an N-terminal His tag. Both proteins were purified using nickel chelating and size exclusion chromatography to >95% purity. The His tag was cleaved from the SET domain protein. SAM was obtained from Sigma (St. Louis, MO; cat. A7007). 3H-SAM was obtained from PerkinElmer (Waltham, MA; cat. NET155001MC). S-(5′-adenosyl)-
Peptide Mass Spectroscopy Assay
The KM of SAM and peptide substrate for truncated and full-length SETD8 was determined using mass spectroscopy analysis of peptide substrate and monomethylated product concentrations. The final buffer contained 50 mM Tris-HCl, 1 mM DTT, and 0.005% Triton X-100 at pH 8.5 in nuclease-free water. SAM or peptide substrate concentrations were varied and tested with a fixed concentration of 10 µM peptide substrate or 50 µM SAM, respectively. Reactions were initiated by the addition of 50 nM truncated SETD8 or 125 nM full-length SETD8. The reactions were terminated by the addition of 1% formic acid at different time points. The initial linear rate of enzyme activity was determined and fit to the Henri–Michaelis–Menten equation: rate = Vmax[substrate]/(KM + [substrate]). The results of the enzyme kinetic studies were used to select a reaction time and SAM, peptide, and SETD8 concentrations that produced initial linear velocities at or below the KM of SAM and peptide substrate. This resulted in two assays containing 300 µM SAM, 1 µM peptide substrate, and either 3.5 nM truncated SETD8 or 20 nM full-length SETD8 incubated for 2 h at room temperature.
DNA Binding Assays
The KD of DNA for SETD8 was determined by FP and confirmed in an activity assay. An enzyme titration of full-length SETD8 was tested against 1 nM of fluorescein-labeled 75 bp DNA in a buffer containing 50 mM Tris-HCl, 10 mM DTT, and 0.01% Triton X-100, pH 8.5, in nuclease-free water. The fluorescence emissions in the parallel and perpendicular channels were used to determine the millipolarization (mP) at each enzyme concentration. This was fit to a four-parameter logistic model, mP = Min + (Max – Min)/(1 + (EC50/[Enzyme])slope), to determine the KD. In the activity assay, unlabeled DNA was titrated and added to 1 nM full-length SETD8, 10 µM SAM, and 10 µM peptide substrate in the above buffer for 2 h at room temperature. The concentration of peptide substrate and methylated product was determined by mass spectroscopy. The enzyme activity versus concentration of DNA was fit to a four-parameter logistic model, Activity = Min + (Max – Min)/(1 + (EC50/[DNA])slope), to determine the EC50.
Peptide/DNA SPA Assay
The KM of SAM and peptide substrate for full-length SETD8 in the presence of DNA was determined by mass spectroscopy. All reactions were carried out in the following nuclease-free buffer: 50 mM Tris-HCl, 1 mM DTT, and 0.005% Triton X-100 at pH 8.5. The final reactions contained 0.15 nM full-length SETD8 enzyme and 2 nM DNA. SAM or peptide substrate concentrations were varied and tested with a fixed concentration of either 0.4 µM peptide substrate or 3 µM SAM, respectively. Enzyme reactions were stopped at different time points using 1% formic acid. The initial linear rates of enzyme activities were replotted versus substrate concentration. KM values were determined by performing nonlinear regression of the data to the Henri–Michaelis–Menten equation.
Compound inhibition of SETD8 was measured in a scintillation proximity assay (SPA) using streptavidin-coated yttrium silicate (YSi) beads (PerkinElmer cat. RPNQ0012) and 3H-SAM. The assay was conducted in a buffer system of 50 mM Tris-HCl, 1 mM DTT, and 0.005% Triton X-100 at pH 8.5 in nuclease-free water. The final assay contained compound with 1 µM 3H-SAM, 2 nM DNA, and 0.4 µM biotinylated peptide substrate. Enzyme was added last to a final concentration of 0.8 nM SETD8 and incubated at room temperature for 2 h. The reaction was stopped with a 0.5 mg/mL solution of streptavidin SPA beads in 3 M guanidine HCl. After an equilibration time of 30 min, the plates were read on the Trilux (PerkinElmer Wallac, Model 1450). The percent inhibition at each inhibitor concentration was calculated based on a maximum inhibition control lacking SETD8 and a minimum inhibition control where there was no inhibitor present.
Nucleosome Assays
The KM of SAM and nucleosome substrate was determined in a filter binding assay format. The final assay buffer contained 50 mM Tris-HCl, 50 mM NaCl, 10 mM DTT, and 0.005% Triton X-100 at pH 8.5 in nuclease-free water. SAM concentrations were varied and tested with a fixed concentration of either 1 µg/mL recombinant nucleosomes or 8 µg/mL HeLa nucleosomes. Nucleosome concentrations were varied and tested with a fixed concentration of 50 nM 3H-SAM. Reactions were initiated with 0.2 nM full-length SETD8 and terminated at different time points by adding 20% trichloroacetic acid (TCA). The reactions were performed in a Costar (no. 3632) 96-well assay plate and the contents transferred to a Millipore (no. MSFCN6B50) 96-well filtration plate. The plate was washed with 10% TCA four times and 100% ethanol once. The plate was dried at 42 °C for 1 h, adding 70 µL of MicroScint-O, and read on a Trilux. Compounds were tested as described above using either 1 µg/ mL recombinant or 8 µg/mL HeLa nucleosomes with 50 nM 3H-SAM and 0.2 nM full-length SETD8 incubated for 3 h at room temperature.
Results
Kinetic Parameters Using Peptide Substrate
The ability of SETD8 to methylate peptide substrate corresponding to positions 15–25 of the N-terminus of histone H4 was evaluated. This peptide sequence covers the primary recognition sites for the SET domain of SETD8 (truncated SETD8), reported to be positions 17–23.27,28 This peptide substrate was tested against full-length SETD8 and the SET domain of SETD8. The formation of monomethylated peptide was monitored by mass spectroscopy over time to determine the initial linear rate of enzyme activity at each concentration of peptide substrate or SAM. These rates were fit to the Henri–Michaelis–Menten equation to determine the KM (
Fig. 1A
and
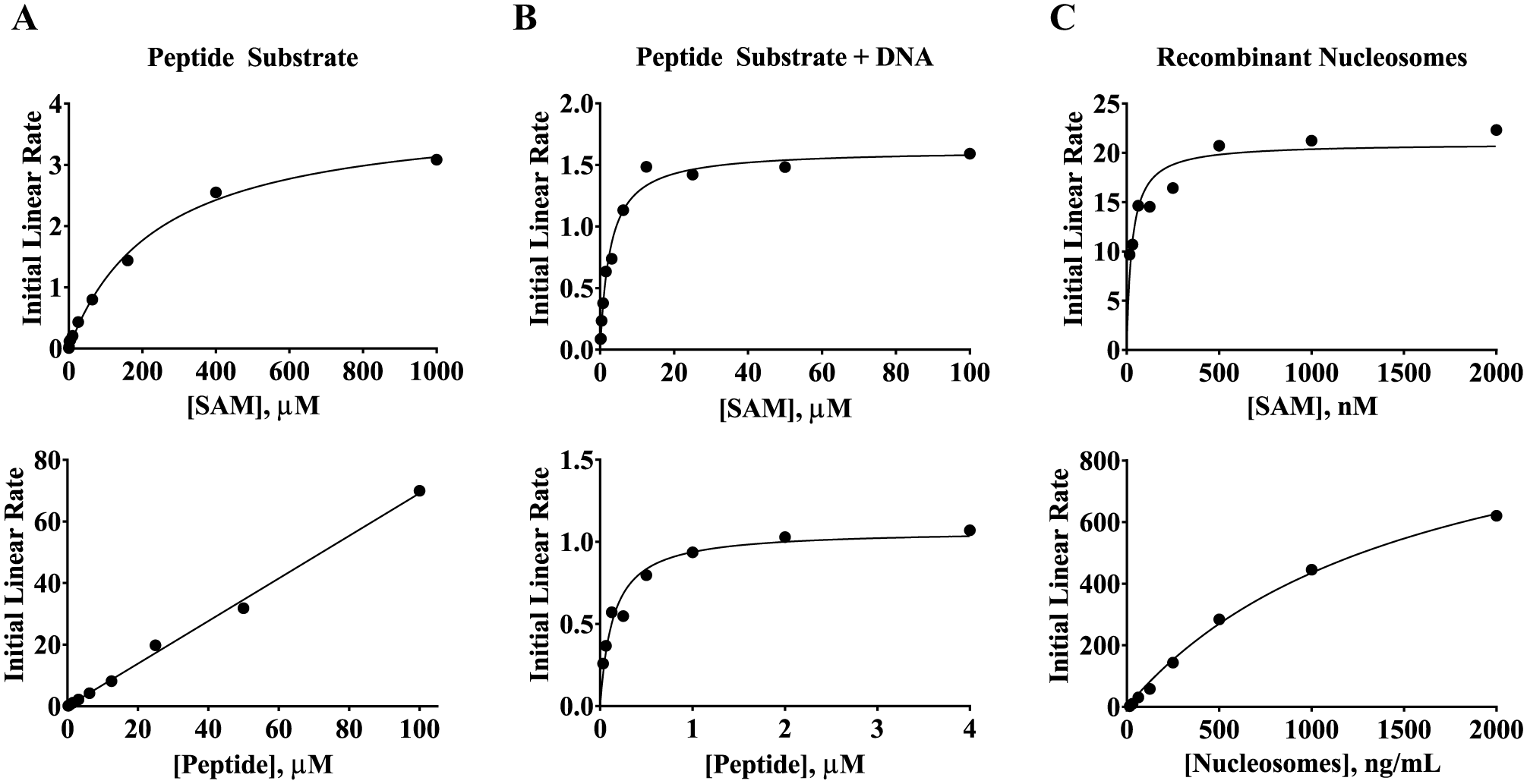
The KM values of SAM and substrate were lower when full-length SETD8 and nucleosome substrates were used. (
Characterization of DNA Binding
Visual inspection of the crystal structure of the nucleosome complex revealed that lysine 20 in histone H4 is directly adjacent to DNA that is wrapped around the histone core. 30 Based on this close proximity, SETD8 could contain some DNA recognition elements, and a recent publication identified a small basic patch adjacent and N-terminal to the SET domain that binds nucleosomes. 24 To evaluate the DNA binding of SETD8, a collection of fluorescein-labeled DNA sequences were tested for binding to full-length SETD8 by FP. The most optimal DNA sequence was a 75 bp segment from small nuclear riboprotein-associated peptide N (SNRPN) exon 1. 26 DNA sequences of 20 bp or fewer produced poorer binding and less activation of enzyme activity. The 75 bp DNA produced an approximately 250 mP increase consistent with binding to full-length SETD8 ( Fig. 2A ). The KD of the interaction was 10.7 ± 0.7 nM, approaching the concentration of DNA used in the assay. To understand the consequences of this binding event, the 75 bp DNA sequence was titrated into an activity assay using full-length SETD8 at subsaturating levels of histone H4(15–25) and SAM. Under these conditions, only monomethylated product was observed. The presence of DNA increased enzyme activity by more than 30-fold, yielding an EC50 of 40.9 ± 9.2 nM that correlated well to the binding observed in the FP assay ( Fig. 2B ). The KM of peptide substrate and SAM in the presence of 2 nM DNA was then determined to understand how DNA was improving enzyme activity. The Henri–Michaelis–Menten plot of initial linear velocities produced a quality fit with a KM(peptide) of 0.142 ± 0.029 µM and KM(SAM) of 2.72 ± 0.35 µM ( Fig. 1B ). This represents an approximately 700-fold decrease in the KM of peptide substrate and an 88- to 133-fold decrease in the KM of SAM compared to the assays lacking DNA.
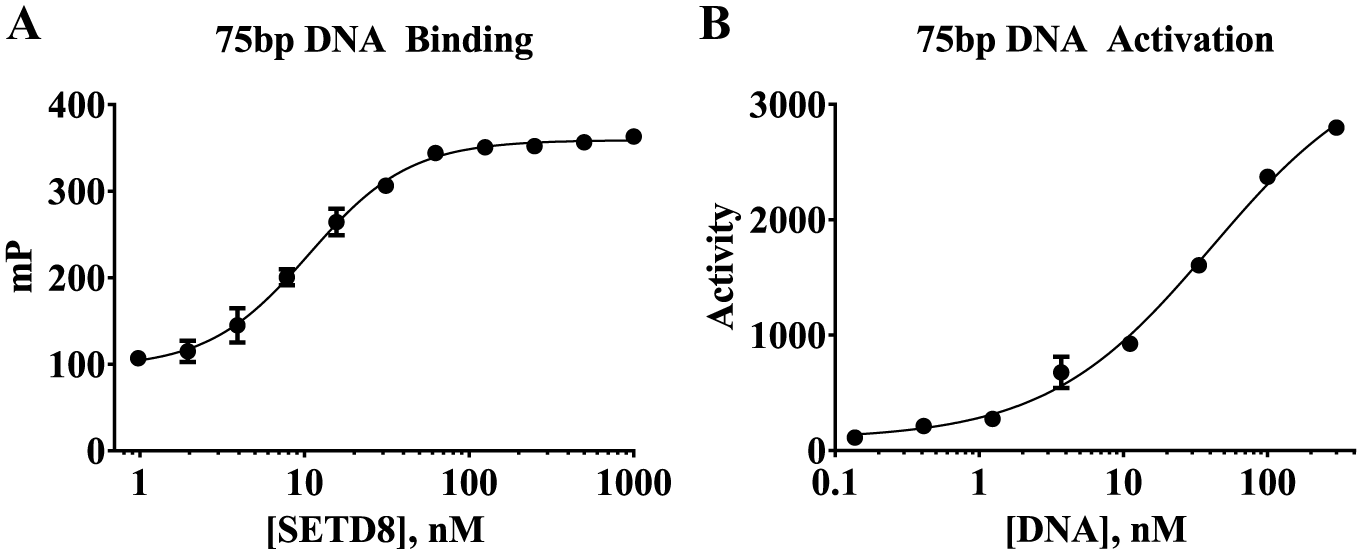
DNA can bind to full-length SETD8 and increase its enzyme activity toward peptide substrate. (
Kinetic Parameters Using Nucleosome Substrates
After observing that full-length SETD8 is able to bind DNA with high affinity, nucleosome substrates were hypothesized to bind better than peptide substrate. This is consistent with a report showing full-length SETD8 is more active against nucleosomes than either the histone octamer or peptide substrate corresponding to positions 1–29 of histone H4.
25
Recombinant human mononucleosomes, free of any posttranslational modifications, and mono/dinucleosomes purified from HeLa cells, which may contain some posttranslational modifications, were tested. The initial linear velocities and KM of SAM and each nucleosome were measured for full-length SETD8. The KM values of recombinant and HeLa nucleosomes were 1580 ± 214 ng/mL and 13,133 ± 2869 ng/mL, respectively (
Fig. 1C
and
Characterization of SETD8 Inhibitors
After establishing the kinetic parameters for mechanistically balanced assay conditions, 31 a collection of inhibitors were tested to further probe the impact of the methyl acceptor substrate on SETD8. Table 1 lists the final concentrations of the key reagents used in five biochemical assays evaluated: SET domain using peptide substrate, full-length SETD8 using peptide substrate, full-length SETD8 using peptide substrate and DNA, full-length SETD8 using recombinant nucleosomes, and full-length SETD8 using mono/dinucleosomes from HeLa cells. The dramatic decrease in the KM of SAM when using nucleosome substrates suggested that the architecture of the SAM pocket is heavily influenced by the choice of substrate. To determine if this observation would translate to improved binding for inhibitors structurally related to SAM, the IC50 of SAH and sinefungin was measured in these five assays. Figure 3A shows that up to 100 and 1000 µM SAH are unable to inhibit full-length SETD8 and the SET domain, respectively, when using peptide substrate. However, when DNA was added to the peptide assay, the IC50 of SAH decreased to 12.0 ± 1.8 µM. When recombinant nucleosome substrates were used, the IC50 decreased to 0.254 ± 0.024 µM. The IC50 using HeLa nucleosomes was 0.079 ± 0.009 µM. A similar trend was observed for sinefungin, a structural analog of SAM. In Figure 3B , the IC50 was >100 µM when using peptide substrate, 12.6 ± 7.3 µM when DNA was included, 0.273 ± 0.052 µM when using recombinant nucleosomes, and 0.356 ± 0.106 µM when using HeLa nucleosomes. For these and all subsequent IC50 determinations, the Z′ was >0.40 on each assay plate.
Conditions for SETD8 Assays.
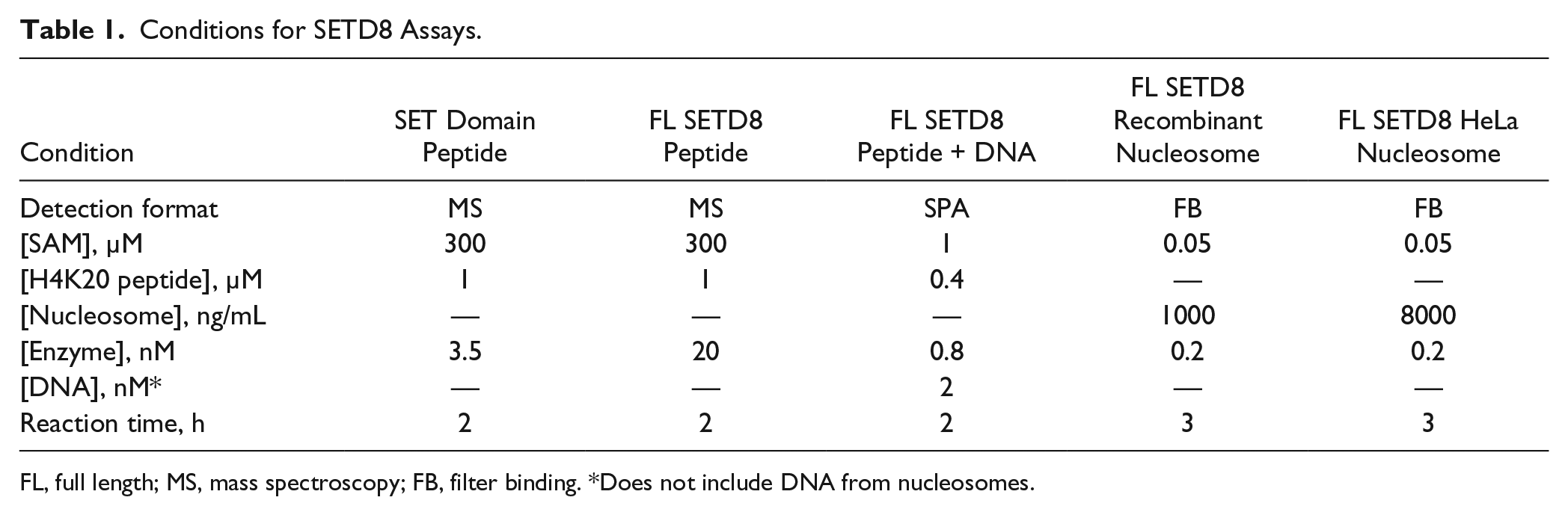
FL, full length; MS, mass spectroscopy; FB, filter binding. *Does not include DNA from nucleosomes.
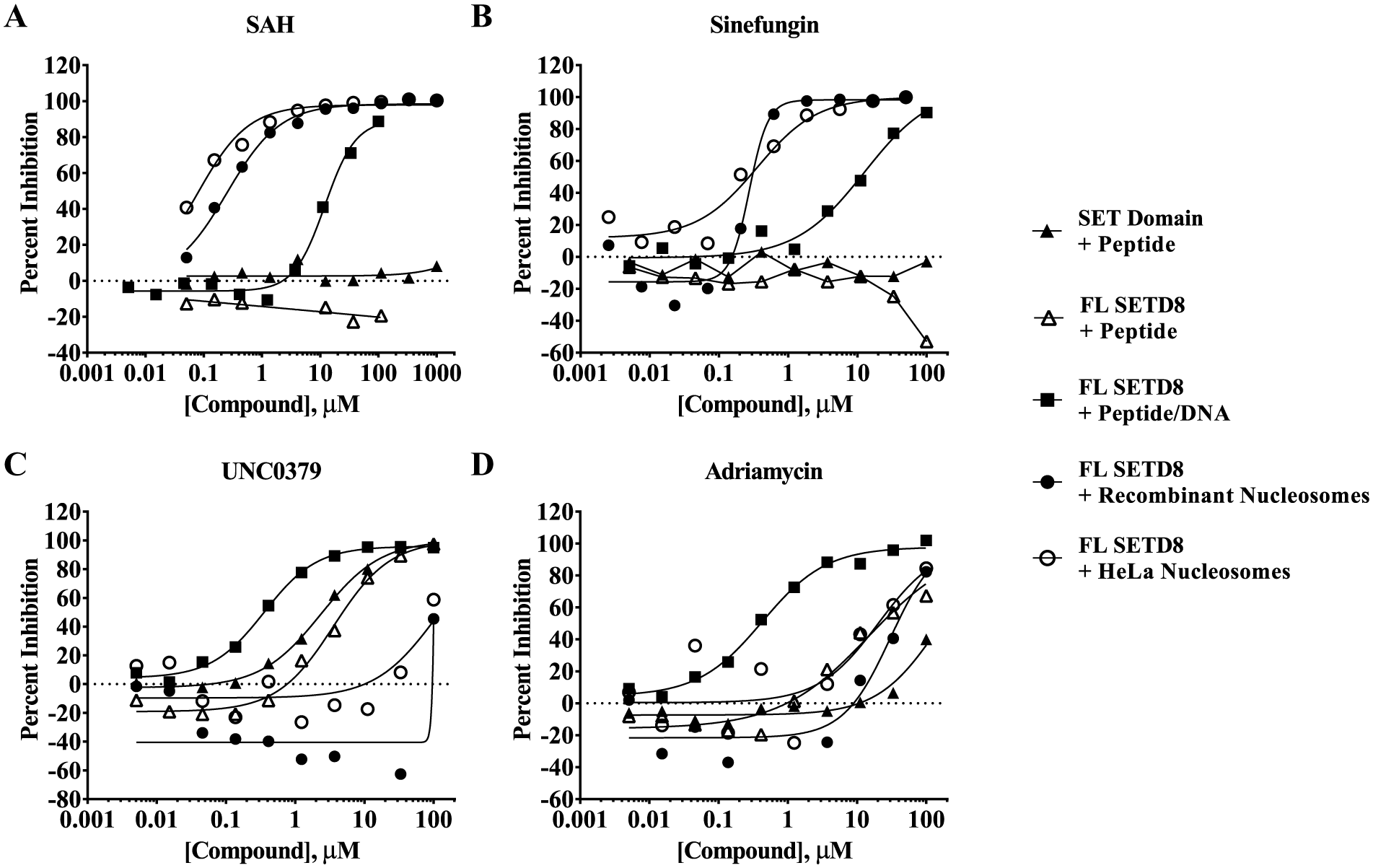
Inhibition profile of four inhibitors against truncated (SET domain) and full-length (FL) SETD8 using peptide substrate, full-length SETD8 using peptide substrate with DNA, full-length SETD8 using recombinant nucleosomes, or full-length SETD8 using HeLa nucleosomes. The IC50 values of (
The reported IC50 of 7.3 µM for UNC0379 correlates reasonably well with the KD values reported using ITC (KD = 18.3 µM) and SPR (KD = 36.0 µM).15,16 When tested in these assays using peptide substrate, the IC50 values in Figure 3C were 2.36 ± 0.14 µM using truncated SETD8 and 3.59 ± 0.43 µM when using full-length SETD8. When DNA was added to the peptide substrate assay, the IC50 lowered to 0.356 ± 0.038 µM. However, when nucleosome substrates were used, the IC50 values increased to >100 µM (with approximately 50% inhibition observed at 100 µM). Some modest enzyme activation was observed at lower concentrations of UNC0379 when using recombinant nucleosomes.
Given that three of the five assays utilize DNA, the impact of adriamycin (doxorubicin), a known DNA intercalator, was tested. 32 Figure 3D shows that it was the most potent against the peptide substrate assay containing DNA with an IC50 of 0.428 ± 0.079 µM. The IC50 was 30.3 ± 34.4 µM using recombinant nucleosomes and 20.4 ± 12.0 µM using HeLa nucleosomes. It was inactive (IC50 > 100 µM) when using truncated SET8. If this compound is reflective of a broader class of DNA intercalators, then the peptide/DNA assay is more sensitive than the nucleosome assays to inhibition resulting from DNA intercalation. The modest potency (IC50 of 14.8 ± 4.1 µM) in the peptide substrate assay using full-length SETD8 suggests that adriamycin may have some specific or nonspecific activity other than DNA intercalation.
Design of the SAM/SAH Focused Library
A collection of 1569 compounds, structurally related to SAM, was assembled. The primary rationale for building and testing this library was to determine if the improved binding of SAM, SAH, and sinefungin in the presence of nucleosomes would translate to a larger collection of related inhibitors. It also provided an opportunity to identify new SETD8 actives. The compound selection strategy for this set was heavily based on substructure searching within the Lilly Compound Collection. A majority of the compounds included in this cassette contained substituted cyclic ether and a proximal heterocylic group both with and without an amino acid or an amino acid mimetic. Additionally, project teams have assembled focused libraries mimicking SAM binding motifs based on rational designs that were included to augment the substructure hits. Finally, proprietary inhibitors that have been mechanistically and biophysically confirmed to bind in the SAM pocket of other enzymes were included. Inhibitors related to scaffolds with known nuisance activities were not included in this library.
Screening Conditions and Results
The assembled SAM/SAH library was screened at 11 µM against full-length SETD8 using either peptide substrate with DNA or recombinant nucleosomes. The assays using truncated or full-length SETD8 and peptide substrate were not screened. The relatively high KM values for SAM and peptide suggested that the combination of protein and substrate produced poorly defined pockets for inhibitor discovery. The screen using peptide/DNA substrate was performed using SPA. The average Z′ was 0.71, with a minimum Z′ of 0.59 observed for one plate. The average percent inhibition for all maximum enzyme activity controls was −0.57 ± 10.06%. Using the average plus 3 standard deviations, inhibitors with >30% inhibition should be statistically significant actives. This was compared to a screen using recombinant nucleosomes in a filter binding format. The assay using nucleosomes from HeLa cells was not screened. The average Z′ was 0.70, with a minimum Z′ of 0.45 observed for one plate. For this assay, inhibitors with >57% inhibition were considered statistically significant actives (average maximum enzyme activity control = 3.32 ± 17.86%). As illustrated in Figure 4 , most compounds did not inhibit either assay with percent inhibition values that clustered near zero. A small fraction of compounds produced negative percent inhibition values, which could be due to enzyme activation or assay interference. In the screen using peptide/DNA, 26 compounds (1.6%) had >50% inhibition and 6 compounds (0.4%) had >80% inhibition. Far more actives were identified in the screen using the nucleosome substrate, where 128 compounds (8.2%) had >50% inhibition and 46 compounds (2.9%) had >80% inhibition. Taken together, only one compound produced >80% inhibition in both assays. However, 40 compounds showed some evidence that they preferred SETD8 when the nucleosome substrate was used (>80% inhibition using nucleosome and <50% inhibition using peptide/DNA). Only two compounds showed any evidence that they preferred SETD8 when using peptide/DNA.
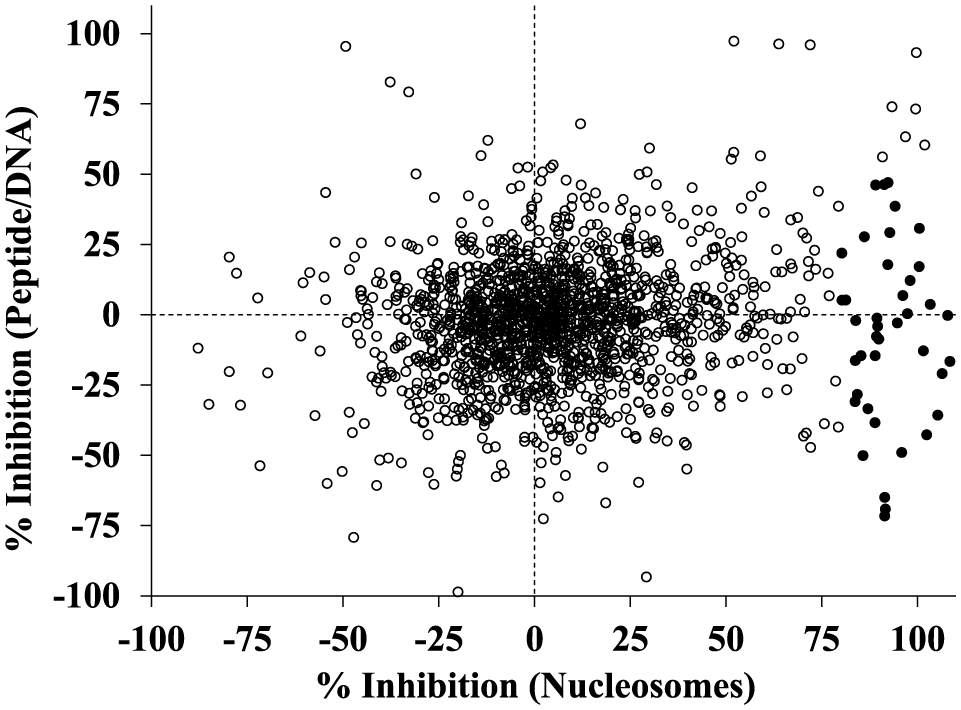
Correlation plot showing the screening results of 1569 SAM/SAH analogs tested at 11 µM against full-length SETD8 using either peptide substrate with DNA or a recombinant nucleosome substrate. A subset of these compounds (closed circles) was only active in the assay using the nucleosome substrate.
Discussion
The five biochemical assays developed reflect a continuum of conditions that may correlate with their physiological relevance. The assay using the SET domain of SETD8 and peptide substrate has the least physiological relevance because neither is found in those forms in cells. The assays using full-length SETD8 and either nucleosome substrate represent assays that may better approximate the physiological activity of SETD8 toward the H4K20 mark in cells. As summarized in Table 2 , a more than 10,000-fold difference in SAM binding and a more than 1000-fold difference in substrate binding is observed. Interestingly, the assay using peptide substrate with DNA has intermediate KM values for SAM and substrate, to suggest it is measuring a form of SETD8 between those two extremes. This continuum of binding constants correlates well with the activity of SAH and sinefungin. These two structural analogs of SAM are inactive in the assays using peptide substrate without DNA and most active when using nucleosomes. Again, significant shifts in their binding constants are observed depending on the choice of enzyme and substrate. In addition, their potency in the peptide assay containing DNA is between those two extremes.
Binding Constants for Ligands against Each SETD8 Assay.
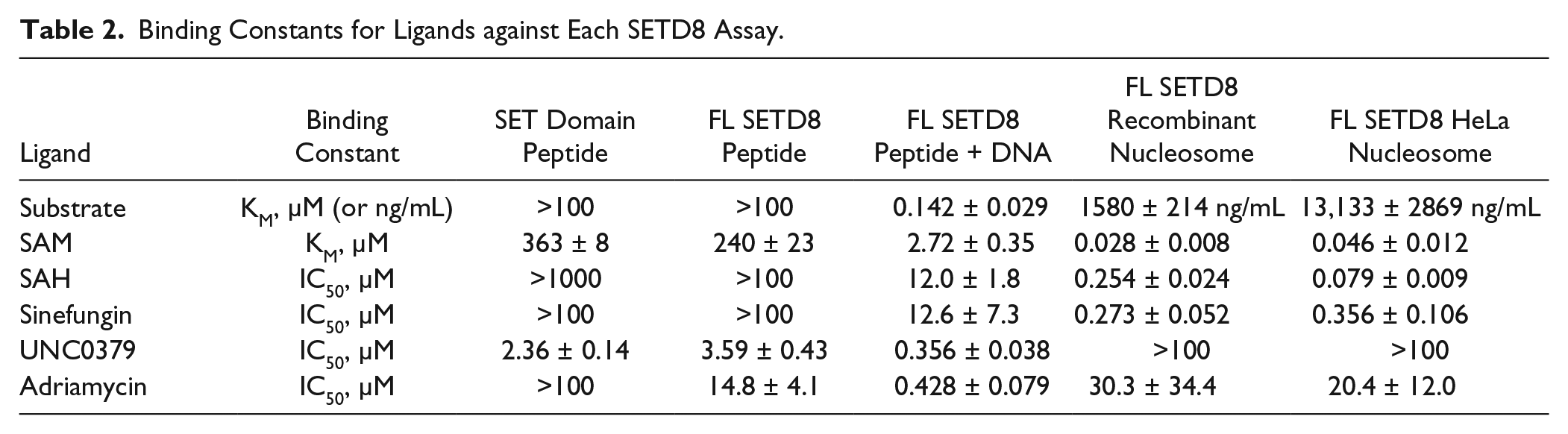
The improved binding of SAM, SAH, and sinefungin in the presence of nucleosomes suggests that structurally related inhibitors may bind best to the SETD8–nucleosome complex. This observation was reinforced in a small screen of SAM/SAH analogs where 40 compounds showed greater SETD8 inhibition using nucleosomes than using peptide with DNA. Since both assays contain DNA, it is unlikely that these 40 substrate-selective inhibitors are DNA intercalators. In addition, adriamycin was nearly 71-fold less potent in the presence of nucleosomes. These results show that a screening campaign using a nucleosome substrate could identify new actives. Although no known nuisance inhibitors were included in the library, a complete mechanistic and biophysical follow-up strategy would be necessary to confirm that these actives have a desirable mechanism of action. Of course, a biophysical assessment would need to account for the possibility that the inhibitors bind poorly to SETD8 alone and bind best to the SETD8–nucleosome complex. The identification of substrate-selective inhibitors like sinefungin, and presumably some of the 40 newly identified compounds, could have important implications for identifying, optimizing, and characterizing chemical probes to understand the cell biology of SETD8. It has also been reported to methylate nonhistone proteins, Numb and p53.17–19 The impact of these protein substrates on the binding of inhibitors, particularly those binding in the SAM pocket, may be an important consideration prior to their evaluation in cell-based assays. This type of substrate selectivity has been observed for an inhibitor of p38a with an appKi of 330 nM using MK2 as a substrate and an appKi of >20 µM using ATF-2 as a substrate. 33 These results also illustrate the importance of evaluating physiologically relevant assays using full-length enzymes and substrates in spite of examples where the impact may appear negligible. For example, SAH and sinefungin have equivalent potencies for PRC2 when tested using either peptide or nucleosome substrates. 34 In addition, the activity of NSD2 and NSD3 toward protein substrates (histone octamers, H3-H4 tetramers, and nucleosomes) depends on the construct of enzyme tested. 35 However, this has little impact on the potency of SAH and sinefungin.
The poor activity of UNC0379 against SETD8 when using nucleosome substrates was an unexpected observation. The reported SPR and ITC results using the SET domain of SETD8 suggest that it is capable of making a real and productive complex with the enzyme.15,16 Our results show that its potencies against the truncated (IC50 = 2.36 ± 0.14 µM) and full-length (IC50 = 3.59 ± 0.43 µM) enzymes are equivalent in the presence of peptide substrate. This suggests that the choice of protein construct does not impact UNC0379 activity when using peptide substrates. In addition, UNC0379 is reported to be a peptide competitive inhibitor and its binding should be mutually exclusive with substrate (peptide or nucleosome) binding. Instead of showing substrate-independent inhibition, it appears to demonstrate substrate selectivity. Given that these assays were developed to be mechanistically balanced and should have roughly an equal population of free SETD8 and nucleosome-bound SETD8, the lack of significant inhibition when using nucleosomes is surprising. Two hypotheses might explain this discrepancy. First, the true KM of nucleosomes for full-length SETD8 could be lower than estimated here, which would inflate the IC50 of UNC0379 under these conditions. The final concentration of nucleosomes in the IC50 assays is estimated to be 5 nM recombinant nucleosomes or 39 nM HeLa nucleosomes, both below our estimates of KM. However, these are just above the enzyme concentration of 0.2 nM, which could impact our ability to accurately measure the KM under steady-state conditions. Second, the nucleosome may have some exosite binding event, outside the pocket defined by the peptide substrate, that could induce a conformational change to prevent UNC0379 binding. The KM of peptide substrate decreased from >100 µM to 0.142 ± 0.029 µM when DNA was included in the assay. This suggests that DNA binding may be changing the architecture of the peptide binding pocket. However, the binding of DNA to SETD8 appears unlikely to be at this exosite interaction because UNC0379 was 10-fold more active (IC50 = 0.356 ± 0.038 µM) when DNA was included with peptide substrate in the assay. These observations suggest UNC0379 may have a more complex binding mechanism. Regardless, to the best of our knowledge, evidence that UNC0379 impacts the H4K20 methylation state in cells has not been published.15,16 The poor activity of UNC0379 in the presence of nucleosome substrates suggests that it may be difficult to achieve in cells.
In conclusion, a mechanistically balanced biochemical assay using full-length SETD8 and a nucleosome substrate was developed. This assay produced a dramatic change in the potency of SAH, sinefungin, and UNC0379. A small collection of compounds structurally related to SAM/SAH were screened, and 40 compounds showed a preference for inhibiting the SETD8–nucleosome complex. This assay can be used to identify and optimize novel inhibitors to probe the role of SETD8 in the cell biology of histone H4–lysine 20 methylation.
Footnotes
Acknowledgements
The authors thank Jingling Li, Xiliang Wang, Yue-Wei Qian, Cheyenne Logan, Mark Hilgers, and Tarun Gheyi for the expression, purification, and characterization of the proteins used in these experiments. We thank Peter Rye and William LaMarr at Agilent Technologies and Pure Honey for mass spectroscopy support. We thank Mary Mader and Robert Campbell for their comments on the manuscript.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.