
Abstract
The dissociation rates of unlabeled drugs have been well studied by kinetic binding analyses. Since kinetic assays are laborious, we developed a simple method to determine the kinetic binding parameters of unlabeled competitors by a preincubation endpoint assay. The probe binding after preincubation of a competitor can be described by a single equation as a function of time. Simulations using the equation revealed the degree of IC50 change induced by preincubation of a competitor depended on the dissociation rate koff of the competitor but not on the association rate kon. To validate the model, an in vitro binding assay was performed using a smoothened receptor (SMO) and [3H]TAK-441, a SMO antagonist. The equilibrium dissociation constants (KI) and koff of SMO antagonists determined by globally fitting the model to the concentration–response curves obtained with and without 24 h preincubation correlated well with those determined by other methods. This approach could be useful for early-stage optimization of drug candidates by enabling determination of binding kinetics in a high-throughput manner because it does not require kinetic measurements, an intermediate washout step during the reaction, or prior determination of competitors’ KI values.
Introduction
Drug actions have been traditionally defined by drug affinity, which is examined under equilibrium conditions. Under these circumstances, kinetic ligand–protein binding properties may be overlooked. However, the kinetic binding characteristics, which reflect drug–target residence times of drugs, are currently attracting considerable interest because of the potential relevance to drug efficacy, duration of action, and safety.1–4 In these respects, the importance of the structure–kinetic relationship (SKR) has become increasingly recognized in addition to the importance of the traditional affinity- or potency-driven structure–activity relationship, and challenges in the reevaluation of drugs using SKR studies have recently been reported for CRF1, 5 CCR2, 6 and adenosine A2A 7 receptors.
The most direct method for determination of kinetic properties is to measure the association and dissociation rates of a radiolabeled test compound. Although this approach provides the most precise kinetic parameters, it is not practical for screening studies because of the cost and time-consuming radiolabeling process. Therefore, several studies have used the competition association assay described by Motulsky and Mahan 8 to estimate the kinetic rate constants of unlabeled compounds.9–14 In this approach, an unlabeled compound is added simultaneously with a kinetically characterized radioligand to the target protein, and binding of the radioligand is measured over time. If the competitor dissociates from the target slower than the radioligand, the radioligand association binding decreases time dependently until equilibrium is reached after an initial “overshoot.” In contrast, when the competitor dissociates faster, radioligand association binding is delayed in a monophasic manner. This method enables estimation of unlabeled competitors’ kinetic constants for several receptors, whereas the throughput of this approach is not very high because kinetic measurements of a number of concentrations are required; therefore, a modified approach has been reported. Guo et al. reported a dual-point competition association in which radioligand binding is measured at two time points in the absence and presence of unlabeled competitors, and the dissociation rates of competitors are ranked according to the ratio of binding at the first time point to that at the second time point. 15 This approach allows semiquantitative estimation of a competitor’s dissociation rates in a high-throughput manner.
Alternatively, dissociation rates of unlabeled competitors can also be assessed using an indirect kinetic radioligand binding assay, “delayed association.” 16 In this approach, a saturating concentration of an unlabeled competitor is preincubated with the material containing the receptor to occupy most of the binding sites, followed by washing of the material to remove the free competitors. Then, a fixed concentration of the radioligand is added and its binding is measured over time to estimate the dissociation rate of the competitor. Packeu et al. introduced a convenient “two-step” competition binding approach, a simplified version of the delayed association equation. 17 The method comprises a preincubation of various concentrations of unlabeled competitor with the receptor-containing preparation, a brief wash, and a final fixed-time incubation with a single concentration of radioligand. The dissociation rate of the competitor can be evaluated from the upward shift of the curve. This method with an intermediate wash step is especially suited for binding studies with intact plated cells.
To date, several approaches mentioned above have been developed to effectively assess the binding kinetics of unlabeled competitors by preincubation-based methods, whereas there is not enough quantitative information about how the individual rate constants kon and koff affect the inhibition activity by preincubation at a preequilibrium condition. In this study, we examined simulations to determine the effects of preincubation of unlabeled competitors without an intermediate washout step on the concentration–response curve (CRC) using a preequilibrium competition assay based on the law of mass action. The validity of the model was assessed by performing radioligand binding experiments and a label-free binding assay using a smoothened receptor (SMO), a seven-transmembrane receptor that is involved in Hedgehog signaling, and TAK-441, a potent SMO antagonist, as a model. The results of these experiments suggested that this new analytical approach could be used to determine the kinetic parameters of unlabeled compounds by performing an endpoint assay with and without preincubation of the compounds, and that it does not require any kinetic measurements, an intermediate wash step during the reaction, and an equilibrium condition.
Materials and Methods
Materials
All SMO antagonists other than cyclopamine used in this study were synthesized by Takeda Pharmaceutical Company. Cyclopamine was purchased from BIOMOL (Plymouth Meeting, PA). [3H]TAK-441 was obtained from BioBridge (Chiba, Japan). Other materials were obtained from Wako Pure Chemical Industries (Osaka, Japan) unless noted otherwise.
Preparation of SMO-Expressing Cell Membrane
Human wild-type SMO gene was transfected into FreeStyle 293 cells using NeoFection (ASTEC, Fukuoka, Japan) and cultured for 3 days at 37 °C in the presence of 5% CO2 in FreeStyle 293 Expression Medium (Life Technologies, Carlsbad, CA). The cells were washed with phosphate-buffered saline (PBS) and suspended in a suspension buffer containing 50 mM HEPES (pH 7.4), 1 mM EDTA, and a protease inhibitor mixture (Roche Diagnostics, Basel, Switzerland). The cells were homogenized and centrifuged at 950g for 10 min at 4 °C, and then the resulting supernatant was centrifuged at 140,000g for 60 min at 4 °C. The pellet was washed with PBS and resuspended in suspension buffer. The membrane fractions were frozen in aliquots at −80 °C until use.
[3H]TAK-441 Radioligand Binding Assay
Binding assays were performed in 96-well formats with a final assay volume of 0.1 mL. For competition experiments, cell membranes (2 μg protein/well) were incubated with a specific concentration of [3H] TAK-441 and test compounds for 24 h at room temperature in assay buffer (50 mM HEPES [pH 7.4], 200 mM NaCl, 10 mM MgCl2, and 0.2% bovine serum albumin). The reaction was terminated by rapid filtration through polyethyleneimine-coated GF/C filter plates (PerkinElmer, Waltham, MA) using a cell harvester (PerkinElmer). The filter plates were washed five times with 50 mM Tris-HCl and dried at 42 °C. Microscint 0 (PerkinElmer) was added to each well, and radioactivity was measured using a TopCount microplate scintillation counter (PerkinElmer).
For the preincubation endpoint (PIE) assay, cell membranes (2 μg protein/well) were incubated with test compounds for 0–24 h at room temperature in 95 μL of assay buffer. Then, 5 μL of a specific concentration of [3H]TAK-441 (20-fold of final concentration, to minimize the change in the volume of the assay) was added and incubated for a specific time (15–30 min) at room temperature. Under the assay condition of no preincubation (preincubation time = 0 h), [3H]TAK-441 and test compounds were simultaneously added to the cell membrane and incubated for a specific time at room temperature. Cell membranes were then transferred to polyethyleneimine-coated GF/C filter plates using a cell harvester to terminate the reaction. The filter plates were washed five times with 50 mM Tris-HCl and dried at 42 °C, and radioactivity was measured as described above.
Affinity Selection–Mass Spectrometry Binding Assay
The binding assay using affinity selection–mass spectrometry (AS–MS) was performed at room temperature in a final volume of 0.45 mL using the same membranes as used in the [3H]-TAK-441 radioligand binding assay. The membranes (50–100 µg mL−1 of protein) were incubated with test compounds for 1.5 h in assay buffer (50 mM HEPES [pH 7.4], 200 mM NaCl, 10 mM MgCl2, and 0.005% Tween-20). Then, an excess amount of the competitor compound (50 μM of TAK-441) was added to initiate dissociation of the test compounds. The reaction was terminated by separation of bound from free compound at each time point using a 96-well ultrafiltration membrane (MSHVN45; Millipore Corp., Billerica, MA) packed with Sephadex G50-Superfine. Then, 18 µL of flow-through was mixed with 50 µL of water and acetonitrile (30:70) to denature the receptor–compound complexes. Liberated compounds were quantified by liquid chromatography–mass spectrometry using an API5000 LC/MS/MS system equipped with an electrospray ionization interface (AB SCIEX, Tokyo, Japan). Compounds were separated on a reverse-phase column (Unison UK-C18, 30 × 2.0 mm; Imtakt, Kyoto, Japan) at a flow rate of 1.0 mL min−1 using a linear gradient. The mobile phase consisted of solvent A (10 mM ammonium formate containing 0.2% formic acid) and solvent B (acetonitrile containing 0.2% formic acid). The following binary gradient was used: 0 min, 5% B; 0–0.02 min, linear gradient to 95% B; and 0.4 min isocratic elution at 95% B. At the end of the gradient, the column was washed with 5% B for 0.55 min. The column oven temperature was set at 50 °C. The mass transitions (Q1/Q3) used for compounds
Data Analysis
All data were analyzed using Prism 5 (GraphPad Software, Inc., San Diego, CA). To simplify the analysis, we assumed that the amounts of the probe and competitor bound to the receptor were much less than their free concentrations. This situation is referred to as “zone A”; that is, the concentrations of the free probe and competitor did not change significantly throughout the experiments.
Dissociation binding data were analyzed by fitting with the one-phase decay model:
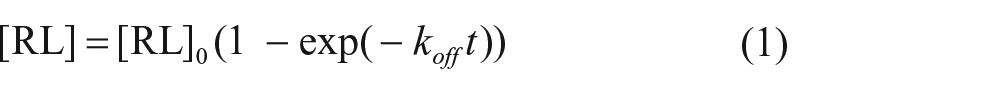
where [RL]0 is the specific binding of the ligand at time 0. The dissociation half-life (t1/2) was determined using the following equation:
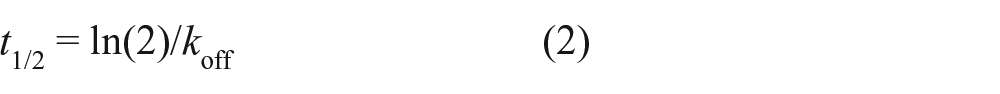
In this study, we used a novel model to estimate kinetic parameters of unlabeled competitors according to the law of mass action. During the first phase (preincubation phase, Fig. 1 ), competitors (I) bind to the receptors (R):

where k3 and k4 are the association and dissociation rate constants of the competitor, respectively.
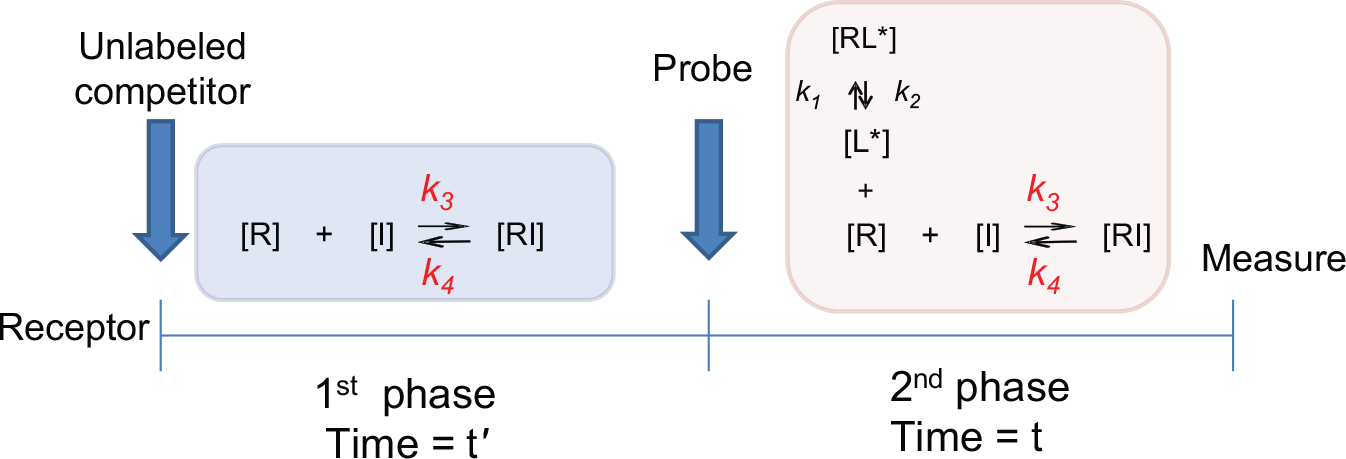
Assay scheme of the PIE approach.
At time t′ (t = 0), the amount of the receptor–competitor complex is expressed as follows:
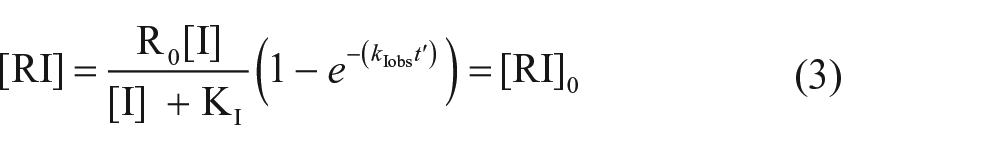
where KI is the equilibrium dissociation constant of the competitor and . The second phase is started by addition of the probe (L), and the interactions between the probe, competitor, and receptor are described as follows:


where k1 and k2 are the association and dissociation rate constants of the probe, respectively. When the time after addition of the probe equals zero, there is no probe binding. The kinetics of binding reactions of the probe and competitor are described by the following equations:



where [R] is the concentration of the free receptor and R0 is the total concentration of the receptor. Solving these differential equations (see supplementary information) yields an equation to describe the amount of probe binding ([RL]) as a function of time (t):
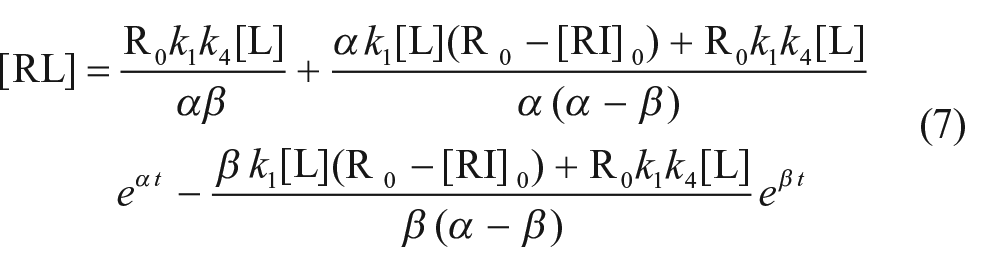
where
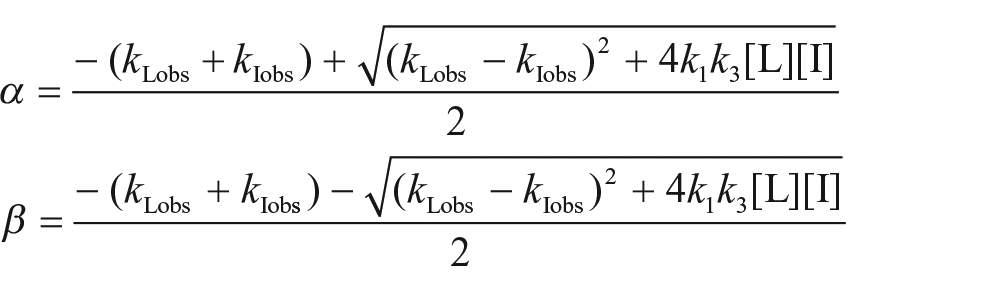

For the PIE assay
(with and without 24 h preincubation), eq 7 was globally fitted to the data. The kinetic equilibrium dissociation constant of the unlabeled competitor was determined as k4/k3.
In the competitive binding experiments, the half-maximal inhibition concentration (IC50) values were determined by nonlinear regression analysis and converted to KI values using the Cheng–Prusoff equation, KI = IC50/(1 + [L]/KD). 18
Simulations
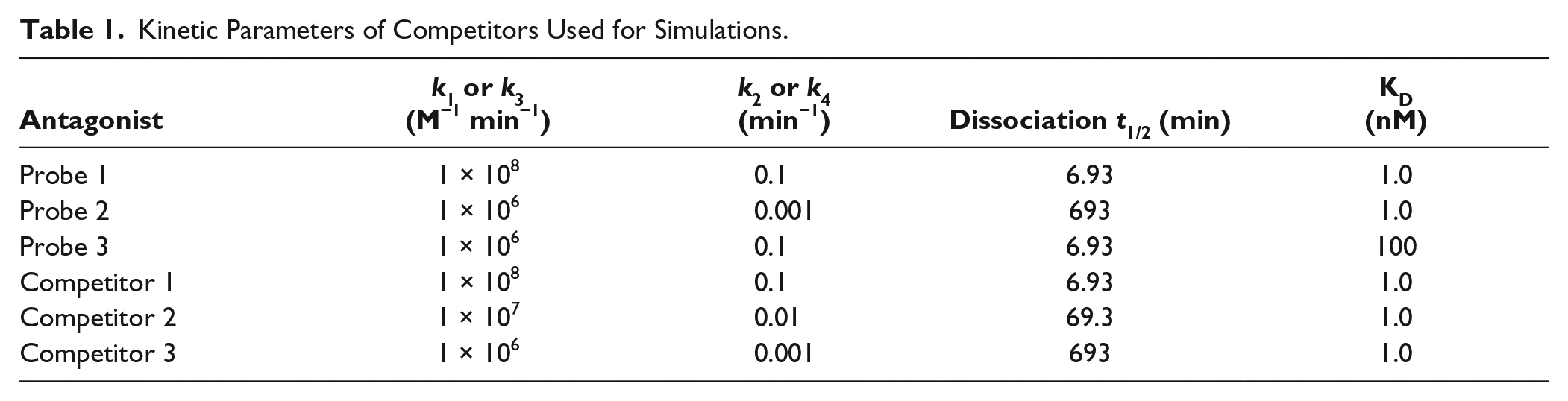
The effects of preincubation on competition IC50 curves were simulated using Prism 5 software and eq 7. The kinetics parameters of the virtual probe ligands are shown in Table 1 . IC50 values were determined using a variable-slope model because the slope of the line was not always unity at a preequilibrium condition even though the one-site binding model was assumed. 8 For simulation of actual data of [3H]TAK-441 binding, k1 and k2 values were fixed to the reported values (1.5 × 106 M−1 min−1 and 9.7 × 10−4 min−1, respectively). 19
Kinetic Parameters of Competitors Used for Simulations.
Results
Simulations
We first conducted a number of simulations to understand the effect of competitor preincubation on the CRCs in a nonequilibrium competition assay using probes with different kinetic parameters (fast- and slowly dissociating probes). The effect of preincubation of unlabeled competitors on the binding of a fast-dissociating RI probe (probe 1, k1 = 1 × 108 M−1 min−1, k2 = 0.1 min−1, KD = 1 nM) was simulated using eq 7 under nonequilibrium conditions (reaction time t was set to 15 min). In the simulations, various competitors (competitors 1–3) with different off rates but the same KD value (1 nM) were examined (
Table 1
). As shown in
Figure 2A
–
C
, preincubation of a fast off-rate compound (competitor 1, koff = 0.1 min−1) slightly affected the competition CRC, whereas preincubation of a moderate off-rate compound (competitor 2, koff = 0.01 min−1) and slow off-rate compound (competitor 3, koff = 0.001 min−1) caused a leftward shift of CRC in a preincubation time-dependent manner. The IC50 values at t′ = 0 (without preincubation) were 2.21, 11.2, and 107 nM for competitors 1, 2, and 3, respectively. Under a long preincubation condition (t′ = 1440 min), the observed IC50 values were 1.43, 1.06, and 1.11 nM for competitors 1, 2, and 3, respectively; these values were almost the same among the three compounds and were very close to their KD values (1 nM). The IC50 shift ratios with and without 1440 min preincubation were 1.5, 11, and 96 for competitors 1, 2, and 3, respectively. The effects of preincubation of unlabeled competitors on the IC50 values were nearly the same regardless of the probe on rate or off rate (
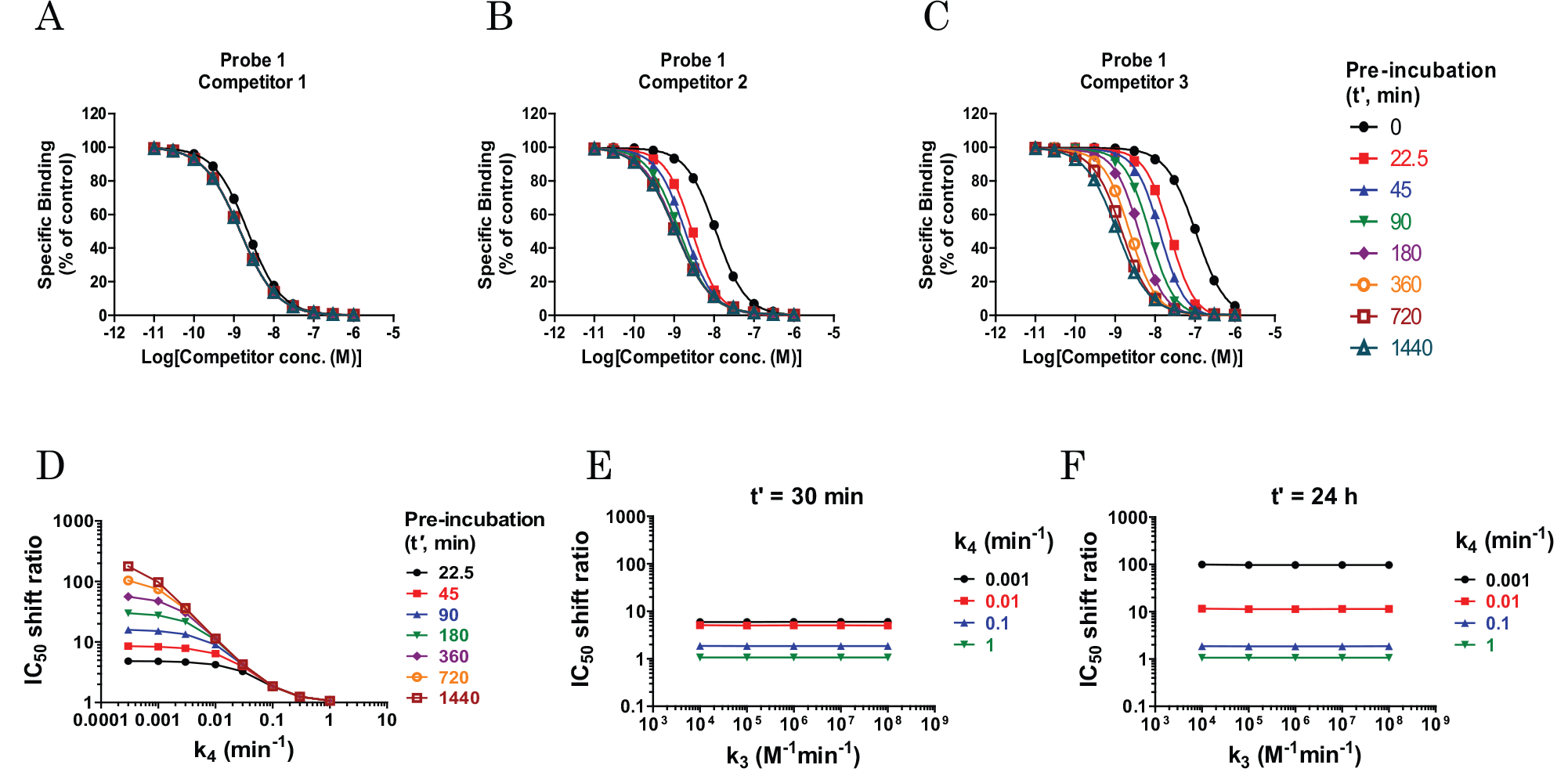
Theoretical simulations according to eq 7 depicting the effects of preincubation of competitors on the binding of the probes under nonequilibrium conditions. Receptors are preincubated with a range of concentrations, including fast (
To assess which kinetic parameters of the competitors contributed to the CRC shift in the nonequilibrium preincubation experiment, further simulation was performed. For this purpose, simulated IC50 shift ratios (IC50 at t′ = 0 divided by the IC50 values at various t′ values) were plotted as a function of k3 or k4. As shown in Figure 2D , the IC50 shift ratio was largely dependent on the dissociation rate (k4). In addition, the magnitude of the IC50 shift ratio was associated with the preincubation time (t′), particularly when the dissociation rate was small. On the other hand, the association rate (k3) did not affect the IC50 shift ratio both at a short preincubation time (t′ = 30 min) and at a long preincubation time (t′ = 24 h) ( Fig. 2E , F ). These results indicate that the slower the dissociation rate of the competitor, the greater the CRC shift caused by preincubation of the competitor.
In the simulations, the brief reaction time (t = 15 min in
Fig. 2
) was shown to be an important factor for determination of kinetic parameters because once equilibrium was reached, no IC50 shift induced by preincubation was observed. Indeed, the longer the reaction time (t) after adding the probe, the smaller the IC50 shift ratio caused by preincubation (
Effect of Preincubation of a Competitor on [3H]TAK-441 Binding
For validation of the preincubation methodology, we use SMO as a model. The data were generated after the simulations had been completed. The effect of the preincubation time of unlabeled TAK-441, a SMO antagonist, on the competition of [3H]TAK-441 binding was examined and compared with simulated data. As expected by the simulations above, preincubation caused the CRCs to be shifted to the left in a time-dependent manner. The CRCs showed good agreement with the simulated data in which k3 and k4 were set to the same values as k1 and k2, respectively (
Estimation of the Dissociation Rates of Unlabeled Compounds Using the PIE Approach
As shown in
Figure 2
, k4 directly affected the CRC shift ratio in the preincubation assay under preequilibrium conditions. This result indicates that it may be possible to estimate k4 values using two CRCs in the PIE assay: one determined with and one without preincubation. To confirm this hypothesis, we examined the competition assay for six SMO antagonists (chemical structures are shown in the
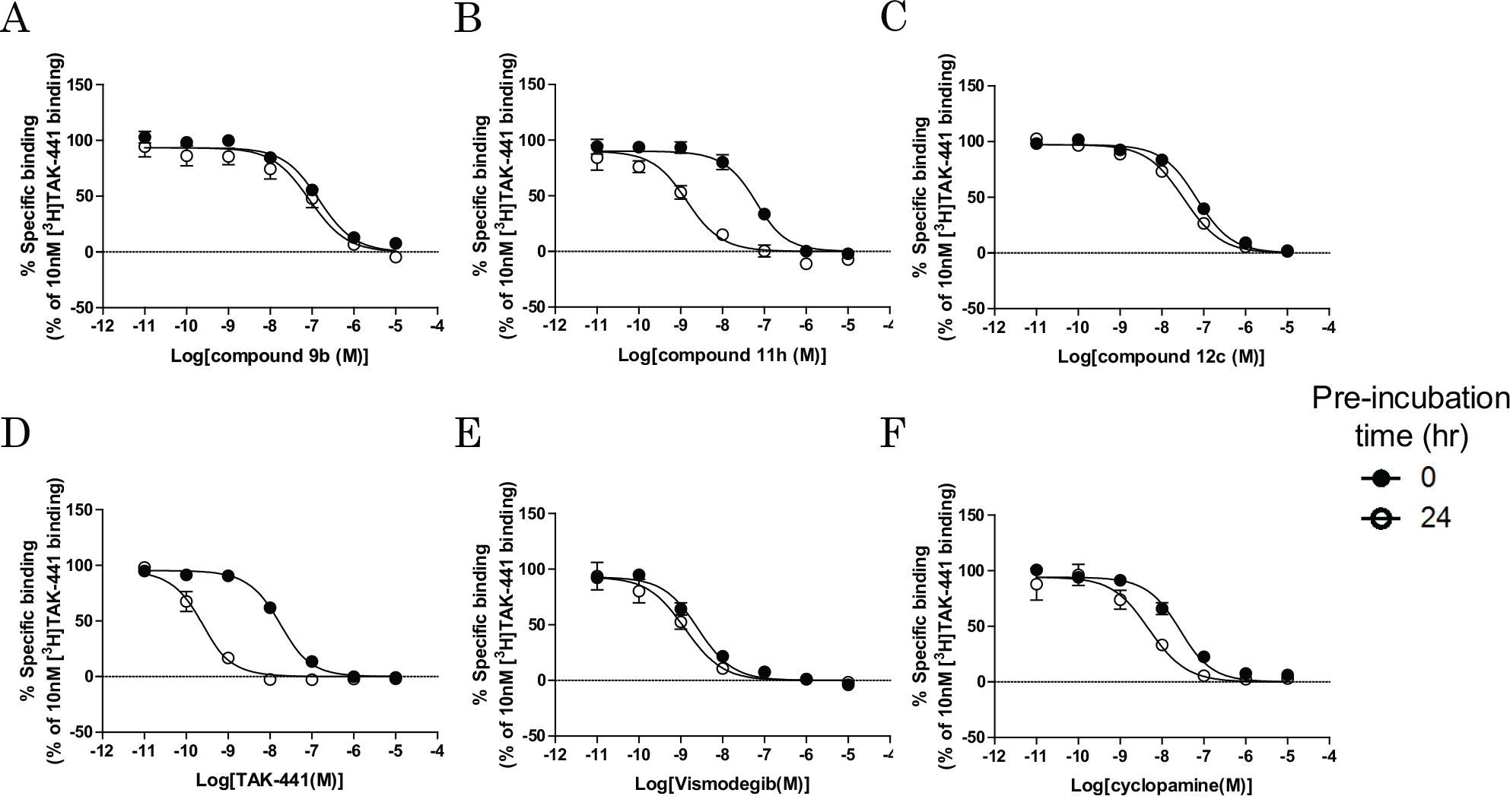
Effects of preincubation of SMO antagonists on [3H]TAK-441 binding. SMOs were preincubated with increasing concentrations of SMO antagonists for 0 (●, no preincubation) or 24 h (○) at room temperature, and then 30 nM (
Kinetic Binding Parameters of Various SMO Antagonists.
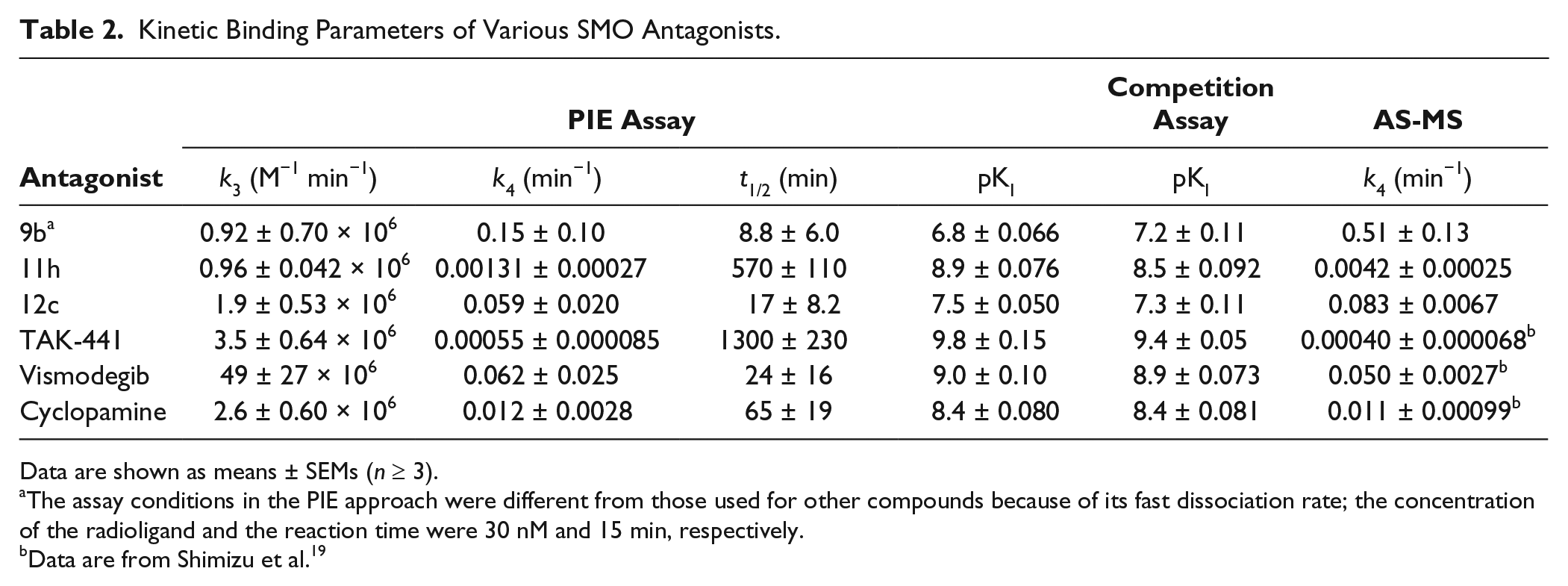
Data are shown as means ± SEMs (n ≥ 3).
The assay conditions in the PIE approach were different from those used for other compounds because of its fast dissociation rate; the concentration of the radioligand and the reaction time were 30 nM and 15 min, respectively.
Data are from Shimizu et al. 19
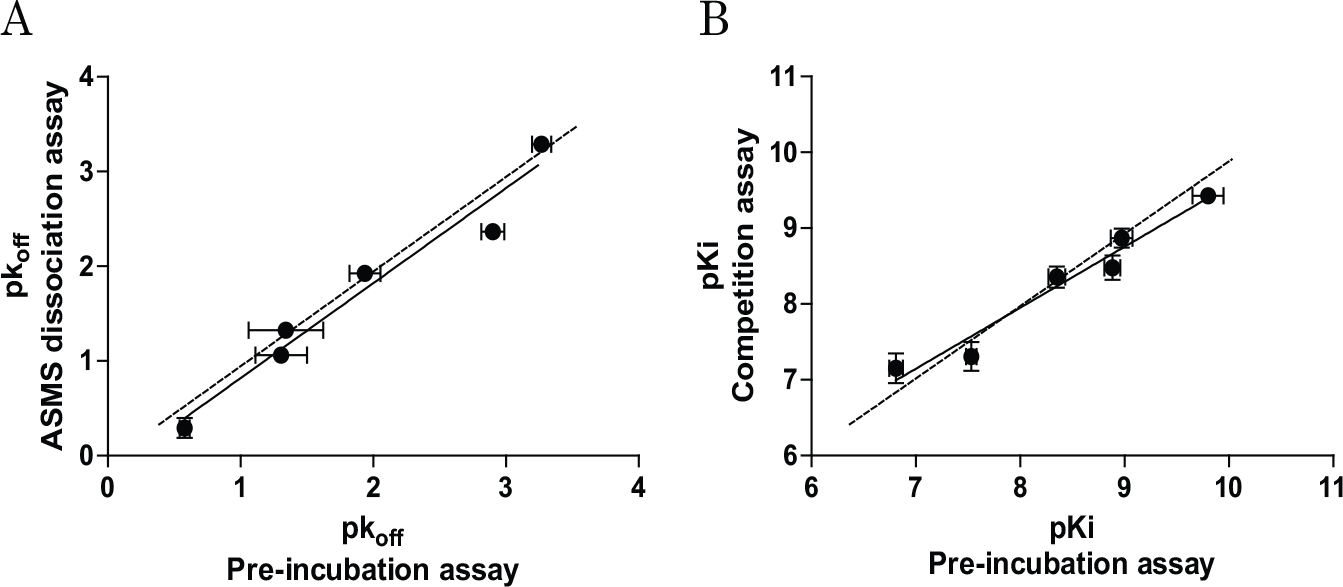
To verify the PIE approach, another method (AS-MS) for determining koff values of unlabeled compounds was also performed. In this assay, the direct dissociation rate could be determined using mass spectrometry. Each compound was first incubated with SMO-expressing membranes for 1.5 h, and then dissociation was assessed over time following addition of an excess amount of the competitor. The rank order of dissociation half-life values determined by this method was the same as that determined using the PIE approach ( Table 2 ). Importantly, the pk4 values in the PIE approach were comparable to those in the AS-MS method ( Fig. 4A , r2 = 0.96, slope = 1.0), which validated the PIE approach. For further validation, SMO antagonists were assessed using the traditional competition assay with 24 h incubation, and the determined apparent KI values ( Table 2 ) were compared with the kinetically determined KI (k4/k3) values obtained using the nonequilibrium PIE approach. There was good correlation between the pKI values determined using the nonequilibrium PIE approach and the competition assay using 24 h incubation ( Fig. 4B , r2 = 0.96, slope = 0.80).
(
Discussion
Estimation of quantitative kinetic binding profiles of drugs is of paramount importance for better understanding of pharmacology and clinical development of drugs because kinetic profiles are largely associated with in vivo efficacy, duration of action, and safety.1,4 From this point of view, optimization of drugs by affinity alone is not ideal; hence, much effort has recently been devoted to estimation of drug kinetic properties.
Shifts in IC50 curves by preincubation of a competitor have been used as an indicator of a slowly dissociating state both in a binding assay and in a functional experiment in a semiquantitative manner.20,21 However, the quantitative relationships between the IC50 shift by preincubation and the kinetic binding properties of a competitor (kon and koff) remain poorly understood. In this study, we used simulations and experiments to extensively show the effects of preincubation of competitors on probe binding at preequilibrium using an approach based on the law of mass action. This research resulted in the establishment of a new and very simple approach, PIE, to describe the kinetic parameters of unlabeled compounds in an endpoint assay. This approach does not require intermediate wash steps after preincubation of competitors, which prevents undesired dissociation of ligands from the receptor–competitor complex, particularly in fast-dissociating compounds, or avoids the presence of residual free competitors in the well after the washing step.
This approach assumes the following: (1) both probes and competitors reversibly bind to the target 1:1, (2) probes and competitors bind to the same site and are completely competitive, (3) free concentrations of probes and competitors do not change during the assay (zone A), and (4) the protein and ligands are stable during the assay. We need to carefully select a probe with an appropriate affinity (very high-affinity ligand results in ligand depletion), with an appropriate concentration, and with an enough stability because the model loses validity if these assumptions are not met. Under these assumptions, the analyses in the present study clearly showed that the preequilibrium IC50 values decreased in a time-dependent manner. Importantly, the degree of shift in the CRC ratio due to preincubation was related to the dissociation rate of the competitor (k4) but not to the association rate (k3). In addition, the CRC shift occurred irrespective of the probe’s kinetic properties. These results suggest that this approach could be used for any target if the kinetic parameters of the probe are known.
We confirmed that only two lines of CRCs in the PIE approach, with or without preincubation, were enough to quantitatively estimate the k4 values of the competitors; the k4 values estimated by the PIE approach were highly correlated with those obtained by direct determination of the dissociation rates (
Fig. 4A
, r2 = 0.96). These values, for TAK-441, vismodegib, and cyclopamine, are also comparable with those previously determined by the competition association method (no reports for other compounds).
19
Moreover, we did not need to determine the appropriate concentrations of competitors in advance in this approach, which is necessary when using the competition kinetics approach. This k4 estimation approach using two CRCs with universal test compound concentrations could make it possible to optimize kinetic parameters of drugs in a high-throughput manner, and therefore have great value because high-throughput determination of kinetic binding parameters is critically important, particularly in early drug optimization. In addition, estimated KD values obtained using the PIE approach were also in good agreement with those determined using the general competition assay (
Fig. 4B
, r2 = 0.96). The KD values can be estimated by the IC50 values at a long preincubation condition, as shown in
Although the PIE approach is feasible for the determination of kinetic parameters of unlabeled competitors, several issues require attention. First, the quantitative performance of k4 estimation could decrease in cases of very small or large CRC shift ratios between CRCs with and without preincubation, that is, with very fast- or slowly dissociating competitors. To address this issue, a brief incubation time after adding the probe or longer preincubation time would improve the estimation of k4 of very fast-dissociating competitors or very slow ones, respectively (see below for further discussion). Second, if the receptors are degraded during incubation, accurate estimation of kinetic parameters is impossible. Therefore, the stability of the receptors needs to be known; the SMO used in this study was stable at least up to 28 h at room temperature (data not shown). Third, the degree of CRC shift was largely affected by assay conditions, including the preincubation time with a competitor and the subsequent incubation time after probe addition. Therefore, it is important to set the optimal assay conditions to maximize the k4 estimation performance. Generally, the preincubation time should be set as long as possible because the longer the preincubation time, the larger the IC50 shift ratio, especially for the slowly dissociating competitors ( Fig. 2D ). In addition, the incubation time after adding the probe should be set as short as possible to maximize the IC50 shift ratio. Simulations similar to those used in the present study would help to establish the assay conditions.
As mentioned above, a brief reaction time is important for improving the estimation of the dissociation rate of a fast-dissociating compound. In practice, however, the detectable signal may be small for a shortened reaction time because equilibrium may not be reached. A possible approach to address this issue is to increase the concentration of the probe. A high probe concentration would help to increase the signal even for shortened reaction periods, because the probe binding rate (kobs) depends on the probe concentration according to the following equation: kobs = kon[L] + koff. Although the IC50 values in traditional equilibrium competitive binding experiments increase with increasing probe concentration according to the Cheng–Prusoff equation [KI = IC50/(1 + [L]/KD)], 18 IC50 values in the PIE approach are less affected by the probe concentration. For example, the IC50 value at [L] = 100 × KD was 50.5-fold greater than that at [L] = KD at equilibrium, whereas the IC50 value at [L] = 100 × KD was only 1.4-fold greater than that at [L] = KD for a 15 min reaction without preincubation in simulations using probe 2 and competitor 3 (data not shown). These results are almost the same as those in the case of faster-dissociating compounds (competitor 1). We should also give attention to the limit of IC50 in each assay condition because the observed IC50 reaches the concentration of the receptor in the case of tight binding drugs, resulting in underestimations of the koff and KI values. In this respect, higher probe concentrations also improve the estimation of the kinetic parameters of compounds with very high affinity. Although these efforts discussed above would improve the accuracy of the PIE approach, we should always be aware of the reliability of the data; it is appropriate to assess the accuracy of the parameters by the 95% confidence interval for the mean value or standard error of the mean.
In summary, we developed the PIE approach as an alternative to the kinetic binding approach to enable high-throughput estimation of the dissociation rates of unlabeled competitors without the need for kinetic measurements. This approach also provides competitor affinity information, which makes it possible to assess both the SKR and structure–affinity relationship in early drug discovery. This rationale presented here has been applied to the case of SMO but can be extended to any binding assay if the probe and the competitor bind to the same site. Use of this approach may result in the acceleration of pharmacological studies and discovery of new drugs possessing more appropriate kinetic properties for therapeutic compounds.
Footnotes
Acknowledgements
We thank Shoichi Okubo and Takuya Sato for the preparation of plasmid and cell membranes and Yutaka Tokairin for helpful technical assistance. We also thank Satoshi Sasaki for the preparation of SMO antagonists. We also acknowledge Ikuo Miyahisa, Tomoya Sameshima, Mark S. Hixson, and Taku Sakurai for helpful discussion and Tomohiro Kawamoto, Junji Matsui, and Naoki Tarui for their support in this study.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.