
Abstract
The concept of ligand-receptor binding kinetics is emerging as an important parameter in the early phase of drug discovery. Since the currently used kinetic assays are laborious and low throughput, we developed a method that enables fast and large format screening. It is a so-called dual-point competition association assay, which measures radioligand binding at two different time points in the absence or presence of unlabeled competitors. Specifically, this assay yields the kinetic rate index (KRI), which is a measure for the binding kinetics of the unlabeled ligands screened. As a prototypical drug target, the adenosine A1 receptor (A1R) was chosen for assay validation and optimization. A screen with 35 high-affinity A1R antagonists yielded seven compounds with a KRI value above 1.0, which indicated a relatively slow dissociation from the target. All other compounds had a KRI value below or equal to 1.0, predicting a relatively fast dissociation rate. Several compounds were selected for follow-up kinetic quantifications in classical kinetic assays and were shown to have kinetic rates that corresponded to their KRI values. The dual-point assay and KRI value may have general applicability at other G-protein-coupled receptors, as well as at drug targets from other protein families.
Introduction
The traditional paradigm of drug discovery places an emphasis on dose-dependent assessments (i.e., affinity or potency) to identify lead compounds, which are usually performed under equilibrium conditions.1,2 With this approach, ligand-receptor binding kinetics is usually overlooked. However, the importance of this latter aspect is increasingly recognized since several lines of research have retrospectively suggested that the binding kinetics, especially the lifetime of the ligand-receptor interaction, is a critical differentiator and predictor for drug efficacy and safety.1–5 This emerging paradigm shift from a classical affinity-based approach emphasizes ligand-receptor residence time (RT, the reciprocal of the dissociation rate constant koff), 6 which is a measure for the duration that a ligand stays in complex with its target.
To date, several methods have been developed to investigate ligand-receptor residence times. The most common and accurate way is to radiolabel a compound of interest with low nanomolar affinity and directly measure its association and dissociation rates in kinetic radioligand binding experiments. In the context of drug-target residence time, two examples are radiolabeled candesartan at the angiotensin AT1 receptor and, more recently, olodaterol at the β2 adrenergic receptor.7,8 Both were found to dissociate slowly from their respective targets. However, this process of radiolabeling a ligand is both time-consuming and labor intensive and thus offers limited applications for these types of kinetic experiments. Therefore, alternative approaches have been developed to determine the binding kinetics of unlabeled ligands. In these approaches, the effect of unlabeled competitors on the association processes of a coincubated or subsequently added radioligand is measured; although the first method allows for quantification of binding kinetics, the latter only provides a qualitative result.9,10 Nevertheless, these current methods are laborious and low throughput as well. Thus, the availability of a high-throughput kinetic binding assay in the early phases of drug discovery is vital, when optimization of ligand-receptor binding kinetics is desired.
In the present study, we introduce an assay to perform kinetic binding screens—namely, the dual-point competition association assay. This method is based on the theory developed by Motulsky and Mahan, 11 in which an unlabeled competitor is coincubated with a radioligand during a kinetic association experiment. If the competitor dissociates faster from its target than the radioligand, the specific binding of the radioligand will slowly and monotonically approach its equilibrium in time ( Fig. 1 , curve C). However, when the competitor dissociates slower, the association curve of the radioligand will consist of two phases, starting with a typical “overshoot” and then a decline until a new equilibrium is reached ( Fig. 1 , curve B). In the dual-point competition association assay, we select two time points to measure radioligand binding: (1) at which the radiolabeled ligand just reaches equilibrium under control conditions in the absence of an unlabeled ligand ( Fig. 1 , curve A and t1) and (2) at which the incubation time is long enough for the labeled and unlabeled compound to equilibrate with the target ( Fig. 1 , curves B and C, t2). Next, we calculate the ratio of the binding at the first time point (Bt1) and that at the second time point (Bt2), which we define as the kinetic rate index (KRI) value for a certain unlabeled compound. In this manner, the compounds that quickly dissociate from their target will have a ratio below or equal to 1 ( Fig. 1 , curve C). Conversely, compounds that dissociate slowly from their target, compared with the radioligand used, will have a KRI value larger than 1, resulting from the typical “overshoot” in the association curve ( Fig. 1 , curve B).
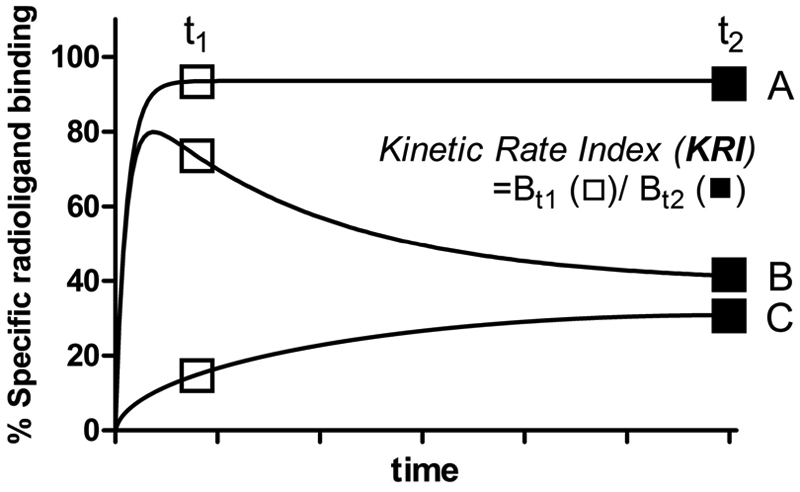
Schematic presentation of the background for the dual-point competition association assay. Curve A: a radioligand association curve without a coincubation of unlabeled competitor. Curve B: a coincubated unlabeled competitor dissociates slower than the radioligand used (k2 > k4). Curve C: a coincubated unlabeled competitor dissociates faster than the radioligand used (k2 < k4). Bt1: specific radioligand binding at the first time point (t1); Bt2: specific radioligand binding at the second time point (t2). Kinetic rate index (KRI) is defined as Bt1/Bt2.
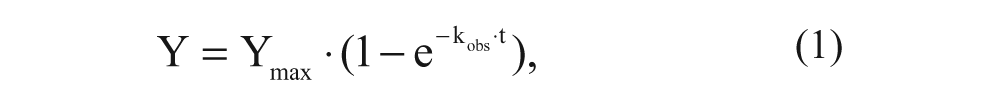
In the present study, we have used the adenosine A1 receptor (A1R) as a prototypic target to illustrate the utility of the dual-point competition association screening method and the resulting KRI values. The adenosine A1R is one of four adenosine receptors subtypes (i.e., A1, A2A, A2B, and A3 receptors), which belong to the superfamily of G-protein-coupled receptors (GPCRs).12,13 The A1R is a promising therapeutic target since it has clinical relevance in neurological disorders, such as cognition deficits, and is involved in cardiovascular preconditioning.14,15 A tool compound—namely, FSCPX—was used to set up the dual-point screening assay since it is a known irreversibly binding antagonist for the A1R.
16
In total, 35 in-house synthesized A1R antagonists were screened using the dual-point competition association assay.17,18 Eight compounds with divergent KRI values were selected for extensive kinetic assessments. Moreover, we decided to radiolabel one of the slowly dissociating antagonists (namely, LUF5962 [compound
Materials and Methods
Chemicals and Reagents
[3H]1,3-dipropyl-8-cyclopentyl-xanthine ([3H]DPCPX, specific activity 116.7 Ci/mmol) was purchased from ARC, Inc. (St. Louis, MO). Adenosine deaminase (ADA) was purchased from Boehringer Mannheim (Mannheim, Germany). CHAPS (3-[(3-cholamidopropyl)-dimethylammonio]-1-propanesulfonate) was obtained from Carl Roth GmbH (Karlsruhe, Germany). DPCPX (selective A1R antagonist 19 ), PEI (polyethyleneimine), and bovine serum albumin (BSA) were from Sigma (St. Louis, MO). Bicinchoninic acid (BCA) and BCA protein assay reagent were obtained from Pierce Chemical Company (Rockford, IL). All 35 A1R antagonists were synthesized in our laboratory as described previously.17,18 Chinese hamster ovary (CHO) cells stably expressing the hA1R were obtained from Prof. Steve Hill (University of Nottingham, UK). All other chemicals were of analytical grade and obtained from standard commercial sources.
Preparation of [3H]LUF5962
[3H]8-cyclopentyl-2,6-diphenyl-9H-purine ([3H]LUF5962, specific activity 51.7 Ci/mmol) was custom-labeled by RC Tritec (Teufen, Switzerland). Unlabeled LUF5962 was provided as a dehydrogenated precursor compound—namely, 8-cyclopent-3-en-1-yl-2,6-diphenyl-9H-purine. This compound was dissolved in ethanol, 10% palladium on carbon was added to the solution, and the reaction mixture was tritiated with tritium gas. After removal of labile tritium and purification by high-performance liquid chromatography (HPLC), the one-stage synthesis yielded [3H]LUF5962 with a specific activity of 54.0 Ci/mmol (2.0 TBq/mmol). The radiochemical purity was >97%, as determined by HPLC.
[3H]LUF5962 Binding Assay Optimization
The assay conditions for [3H]LUF5962 binding to CHOhA1R membranes were optimized according to a general radioligand binding protocol in our laboratory. 20 Initial experiments were performed with 1.0 nM (approximately 12 000 disintegrations per minute [DPM]) [3H]LUF5962 and 5 µg CHOhA1R membranes in a simple buffer of low ionic strength (25 mM Tris-HCl, pH 7.4), to which 5 mM MgCl2 was added. Next, we studied further buffer components, filters, filter pretreatment, and different radioligand and membrane concentrations to improve the window of specific binding. First, a relatively low concentration (1.0 nM) of [3H]LUF5962 was enough for an appreciable window of specific binding, which was improved by the addition of 0.1% CHAPS due to a decrease in nonspecific binding, whereas the addition of 0.1% BSA had no effect. Second, coating GF/B or GF/C glass fiber filters with PEI to separate free from membrane-bound radioligand did not significantly reduce the nonspecific binding (data not shown). Taken together, it was decided to add 0.1% CHAPS in the assay buffer and use uncoated GF/B filters and 5 µg of membranes to yield a desired window of approximately 2000 DPM. Total binding of the radioligand was around 35% of the amount added and thus resulted in radioligand depletion that was unavoidable due to relatively high nonspecific binding of [3H]LUF5962 even under optimized conditions. Finally, initial kinetic association experiments taught us that the optimal incubation time to reach equilibrium was 30 min at 25 °C.
Cell Culture and Membrane Preparation
Chinese hamster ovary cells stably expressing the human adenosine A1 receptor (CHOhA1R) were grown in Ham’s F12 medium containing 10% (v/v) normal adult bovine serum, streptomycin (100 µg/mL), penicillin (100 IU/mL), and G418 (0.4 mg/mL) at 37 °C in 5% CO2. Cells were subcultured twice weekly at a ratio of 1:20 on 10-cm ø culture plates. For membrane preparation, cells were subcultured 1:10 and then transferred to 15-cm ø plates. Cells grown to 80% to 90% confluency were detached from plates by scraping them into 5 mL phosphate-buffered saline (PBS), collected, and centrifuged at 700 g (3000 rpm) for 5 min. Cell pellets derived from 30 plates were pooled and resuspended in 20 mL of ice-cold 25 mM Tris-HCl buffer (pH 7.4). An UltraThurrax (Heidolph Instruments, Schwabach, Germany) was used to homogenize the cell suspension. Membranes and the cytosolic fraction were separated by centrifugation at 100 000 g (31 000 rpm) in a Beckman Optima LE-80K ultracentrifuge (Beckman Coulter, Fullerton, CA) at 4 °C for 20 min. The pellet was resuspended in 15 mL of the Tris-HCl buffer, and the homogenization and centrifugation step was repeated. Tris-HCl buffer (10 mL) was used to resuspend the pellet, and ADA was added (0.8 IU/mL) to break down endogenous adenosine. Membranes were stored in 250-µL aliquots at −80 °C. Concentrations of the membrane protein were measured using the BCA method. 21
Radioligand Saturation and Displacement Assays
Membrane aliquots containing 5 µg of protein were incubated in a total volume of 100 µL of assay buffer (25 mM Tris-HCl [pH 7.4], supplemented with 5 mM MgCl2 and 0.1% [w/v] CHAPS) at 25 °C for 1 h ([3H]DPCPX) or 3 h ([3H]LUF5962). Notably, optimal incubation times were determined with equation (5) under “Data Analysis,” which ensured that binding of the radioligand at the lowest concentration also reached equilibrium. For saturation experiments, a range of concentrations of [3H]DPCPX (~0.2–20 nM) or [3H]LUF5962 (~0.1–13 nM) were used, respectively. In an initial experiment, nonspecific binding was determined at 12 concentrations of radioligand (i.e., [3H]DPCPX ~0.2–20 nM and [3H]LUF5962 ~0.1–13 nM) in the presence of 100 µM N 6 -cyclopentyladenosine (CPA, selective A1R agonist 22 ). Since the nonspecific binding of both radioligands proved to be linear for these concentrations (R2 > 0.95, p < 0.0001; data not shown), it was decided to use only three, evenly spaced, concentrations of radioligand to determine nonspecific binding in following saturation experiments. Displacement experiments were performed using 11 concentrations of competing ligands in the presence of 2.5 nM [3H]DPCPX. At this concentration, total radioligand binding did not exceed 10% of that added to prevent ligand depletion. Nonspecific binding was determined in the presence of 100 µM CPA. Incubations were terminated by rapid vacuum filtration to separate the bound and free radioligand through 96-well GF/B filter plates using a PerkinElmer Filtermate-harvester (Perkin Elmer, Groningen, the Netherlands) or through Whatman GF/B filters (Whatman International, Maidstone, UK) using a Millipore manifold (Millipore, Billerica, MA). Filters were subsequently washed three times with ice-cold wash buffer (25 mM Tris-HCl [pH 7.4], supplemented with 5 mM MgCl2). The filter-bound radioactivity was determined by scintillation spectrometry using the P-E 1450 Microbeta Wallac Trilux scintillation counter (PerkinElmer) for 96-well GF/B filter plates or by a liquid scintillation counter (Tri-Carb 2900 TR; PerkinElmer) for Whatman GF/B filters.
Radioligand Association and Dissociation Assays
Association experiments were performed by incubating membrane aliquots containing 5 µg of protein in a total volume of 100 µL of assay buffer at 25 °C with 2.5 nM [3H]DPCPX or 1.0 nM [3H]LUF5962. The amount of radioligand bound to the receptor was measured at different time intervals during a total incubation of 20 min for [3H]DPCPX or 1 h for [3H]LUF5962. Dissociation experiments were performed by preincubating membrane aliquots containing 5 µg of protein in a total volume of 100 µL of assay buffer for either 20 min for [3H]DPCPX or 1 h for [3H]LUF5962. After the preincubation, radioligand dissociation was initiated by the addition of 10 µM unlabeled CPA. The amount of radioligand still bound to the receptor was measured at various time intervals for a total of 1 h for [3H]DPCPX or 2 h for [3H]LUF5962 to ensure that they were fully dissociated from hA1R. Incubations were terminated and samples were obtained as described under “Radioligand Saturation and Displacement Assays.”
Competition Association Assay
The binding kinetics of unlabeled ligands was quantified using the competition association assay based on the theoretical framework by Motulsky and Mahan. 11 In the standard assay, three different concentrations of unlabeled DPCPX were tested—namely, at 0.3-, 1-, and 3-fold its Ki. For unlabeled LUF5962, 1-, 3-, and 10-fold its Ki were used, whereas for FSCPX, 3-, 10-, and 30-fold its Ki were used. In the simplified one-concentration competition association assay, only a concentration of 10-fold of the Ki value was used to determine the binding kinetics of unlabeled ligands. The competition association assay was initiated by adding membrane aliquots (5 µg/well) at different time points for a total of 90 min to a total volume of 100 µL of assay buffer at 25 °C with 2.5 nM [3H]DPCPX in the absence or presence of competing ligand (10-fold Ki). Only for FSCPX was the assay time increased to 180 min. Incubations were terminated and samples were obtained as described under “Radioligand Saturation and Displacement Assays.”
Dual-Point Competition Association Assay
The dual-point competition association experiments were performed by incubating membrane aliquots containing 5 µg protein for 15 min or 120 min in a total volume of 100 µL of assay buffer with 2.5 nM [3H]DPCPX and for 30 min or 120 min with 1.0 nM [3H]LUF5962 in the absence or presence of unlabeled ligands. The amount of radioligand bound to the receptor was measured after coincubation of the unlabeled A1R antagonists at 10-fold their respective Ki values. Incubations were terminated after either 15 min or 120 min for [3H]DPCPX and 30 min or 120 min incubation for [3H]LUF5962, and samples were obtained as described under “Radioligand Saturation and Displacement Assays.” KRI values of unlabeled ligands were calculated by using equation (6), as mentioned below in “Data Analysis.”
Data Analysis
All experimental data were analyzed by using GraphPad Prism 5.0 (GraphPad Software, Inc., San Diego, CA). All values obtained are means of at least three independent experiments performed in duplicate. KD and Bmax values of [3H]DPCPX or [3H]LUF5962 at hA1R membranes were obtained by computational analysis of saturation curves. IC50 values obtained from competition displacement binding data were converted into Ki values using the Cheng-Prusoff equation. 23 Association data were fitted using one-phase exponential association as follows:
where t is a given time, Y is the amount of specific radioligand binding, Ymax is the specific radioligand binding at equilibrium, and kobs is the observed rate constant to approach equilibrium. The time for a radioligand to reach half of the Ymax (t1/2, association, the association half-life) is
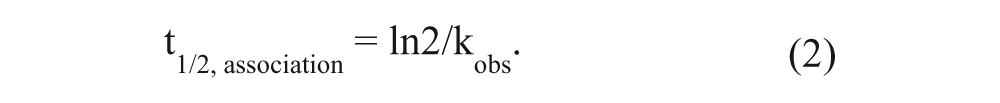
Values for kon were obtained by converting kobs values as follows:
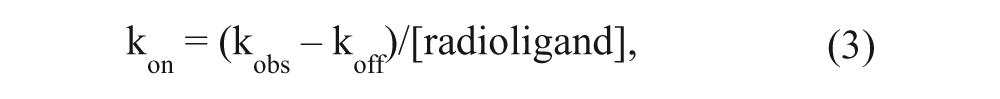
where koff was determined in independent dissociation experiments. Dissociation data were fitted using one-phase exponential decay.
In the dual-point competition association assay, t1 was set at the time that radioligand binding just reached equilibrium. Specifically, we have defined the time to reach equilibrium, when 99.5% of total binding was reached (i.e., at 8-fold the association half-life). Thus,
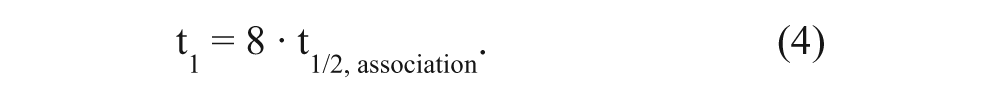
Taken together, t1 was determined by combining equations (2), (3), and (4) as follows:
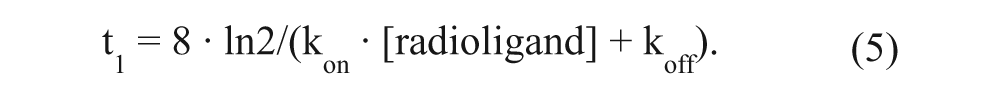
The second time point (t2) was arbitrarily set at a time long enough for the binding of the unlabeled ligand to reach a plateau. KRI values were calculated by dividing the specific radioligand binding measured at t1 (Bt1) by its binding at t2 (Bt2) in the presence of unlabeled competing ligands as follows:

Association and dissociation rates for unlabeled ligands were calculated by fitting the data in the competition association model using “kinetics of competitive binding” in Prism 5 as follows 11 :
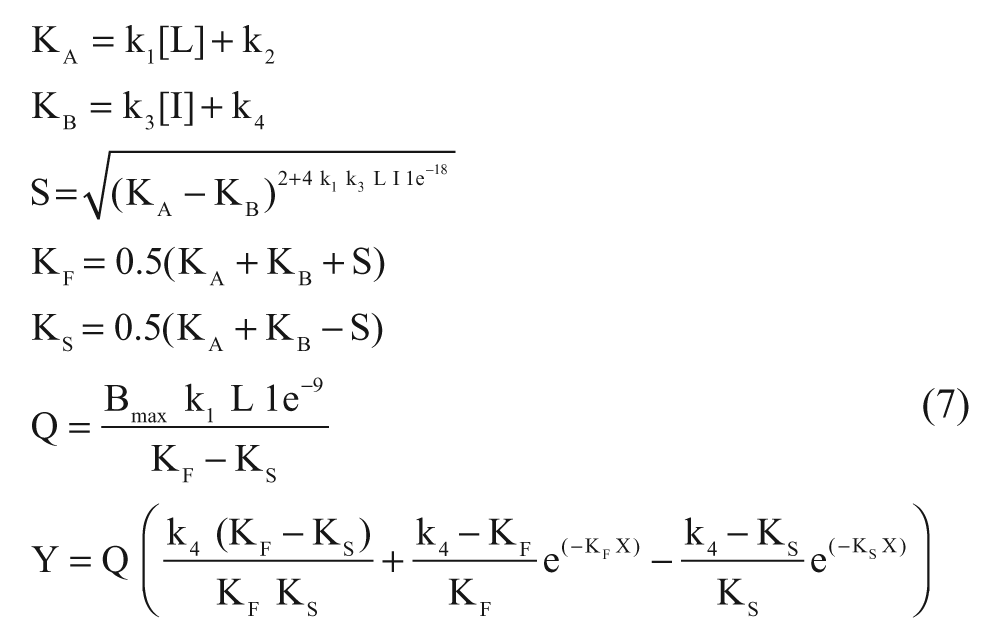
where X is the time (min), Y is the specific binding (DPM), k1 is the kon (M−1·min−1) of [3H]DPCPX predetermined in association experiments, k2 is the koff (min−1) of [3H]DPCPX predetermined in dissociation experiments, L is the concentration of [3H]DPCPX used (nM), Bmax is the total binding (DPM), and I is the concentration of unlabeled ligand (nM). Fixing these parameters into equation (7) allows the following parameters to be calculated: k3 is the kon (M−1·min−1) of the unlabeled ligand, and k4 is the koff (min−1) of the unlabeled ligand. The association and dissociation rates were used to calculate the “kinetic KD” as follows:

Results and Discussion
Quantification of the KD and Bmax of [3H]DPCPX and [3H]LUF5962 in Saturation Binding Experiments
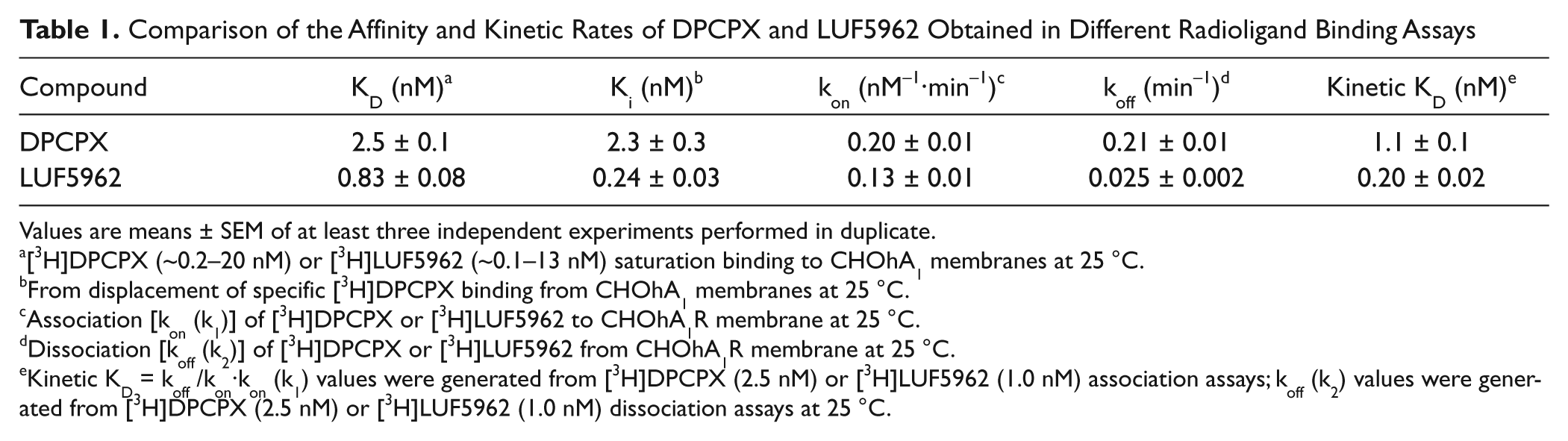
Saturation binding experiments were performed with [3H]DPCPX and [3H]LUF5962 at 25 °C. Both [3H]DPCPX and [3H]LUF5962 bound saturably and specifically to a single class of binding sites at CHOhA1R membranes. The KD value of [3H]DPCPX obtained from the saturation experiments was 2.5 ± 0.1 nM and the Bmax value was 14.0 ± 1.0 pmol/mg protein, whereas the KD value of [3H]LUF5962 was 0.83 ± 0.08 nM and the Bmax was 17.1 ± 0.9 pmol/mg protein ( Table 1 ). It should be kept in mind that ligand depletion was an issue when using [3H]LUF5962, and thus it represents a less than ideal radioligand. However, in the paragraphs below, we discuss that it can still be effectively used in the kinetic characterization of unlabeled ligands. The KD value for [3H]DPCPX obtained in this experiment was in line with the KD value reported previously. 20 This KD was used to derive Ki values from IC50 values for unlabeled ligands (see below).
Comparison of the Affinity and Kinetic Rates of DPCPX and LUF5962 Obtained in Different Radioligand Binding Assays
Values are means ± SEM of at least three independent experiments performed in duplicate.
[3H]DPCPX (~0.2–20 nM) or [3H]LUF5962 (~0.1–13 nM) saturation binding to CHOhA1 membranes at 25 °C.
From displacement of specific [3H]DPCPX binding from CHOhA1 membranes at 25 °C.
Association [kon (k1)] of [3H]DPCPX or [3H]LUF5962 to CHOhA1R membrane at 25 °C.
Dissociation [koff (k2)] of [3H]DPCPX or [3H]LUF5962 from CHOhA1R membrane at 25 °C.
Kinetic KD = koff /kon·kon (k1) values were generated from [3H]DPCPX (2.5 nM) or [3H]LUF5962 (1.0 nM) association assays; koff (k2) values were generated from [3H]DPCPX (2.5 nM) or [3H]LUF5962 (1.0 nM) dissociation assays at 25 °C.
Quantification of the Association [kon (k1)] and Dissociation Rates [koff (k2)] of [3H]DPCPX and [3H]LUF5962 at CHOhA1R Membranes
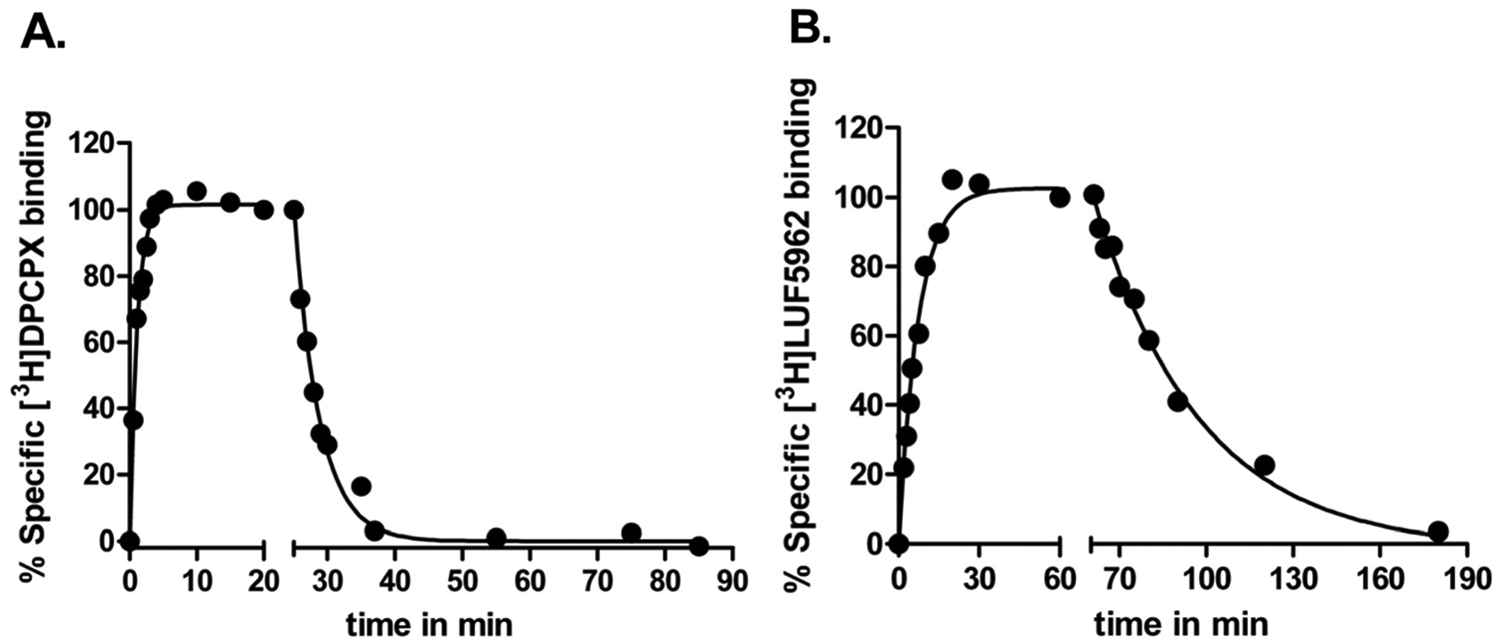
Receptor association (kon [k1]) and dissociation rates (koff [k2]) of [3H]DPCPX and [3H]LUF5962 were determined in standard radioligand association and dissociation experiments. Association and dissociation assays were conducted at 25 °C for both radioligands. The observed association rates (kobs) were converted to kon values by using equation (3), described in “Materials and Methods.” The binding of [3H]DPCPX and [3H]LUF5962 approached equilibrium after approximately 15 and 30 min, respectively ( Fig. 2 ). Both [3H]DPCPX and [3H]LUF5962 had relatively fast association rates of 0.20 ± 0.01 nM−1·min−1 and 0.13 ± 0.01 nM−1·min−1, respectively. Binding of both radioligands was reversible after the addition of 10 µM CPA and complete dissociation was reached after approximately 25 min for [3H]DPCPX and 120 min for [3H]LUF5962 ( Fig. 2 ). The dissociation rate of [3H]LUF5962 was 8-fold lower than that of [3H]DPCPX from the hA1R ( Fig. 2 and Table 1 ). The dissociation binding constants (kinetic KD) of the radioligands were derived from the dissociation and association rates. [3H]LUF5962 had an approximately 5-fold higher affinity than [3H]DPCPX, 0.20 ± 0.02 nM compared with 1.1 ± 0.1 nM, respectively.
Association and dissociation kinetics of [3H]DPCPX (
Quantification of the Affinity (Ki) of A1R Ligands in Displacement Experiments
Displacement experiments with several A1R ligands were performed at 25 °C to determine their affinities for the hA1R. The tested hA1R antagonists showed concentration-dependent inhibition of specific [3H]DPCPX binding, and the data were best fitted to a one-site competition model. Unlabeled DPCPX and LUF5962 had affinities of 2.3 ± 0.3 nM and 0.24 ± 0.03 nM, respectively ( Table 1 ). Affinities of other antagonists shown in Table 2 were taken from previously published in-house experiments.17,18
Kinetics Rate Indexes and Binding Kinetics Obtained from the Dual-Point or Standard Competition Association Assay of DPCPX, FSCPX, and Representative hA1R Antagonists
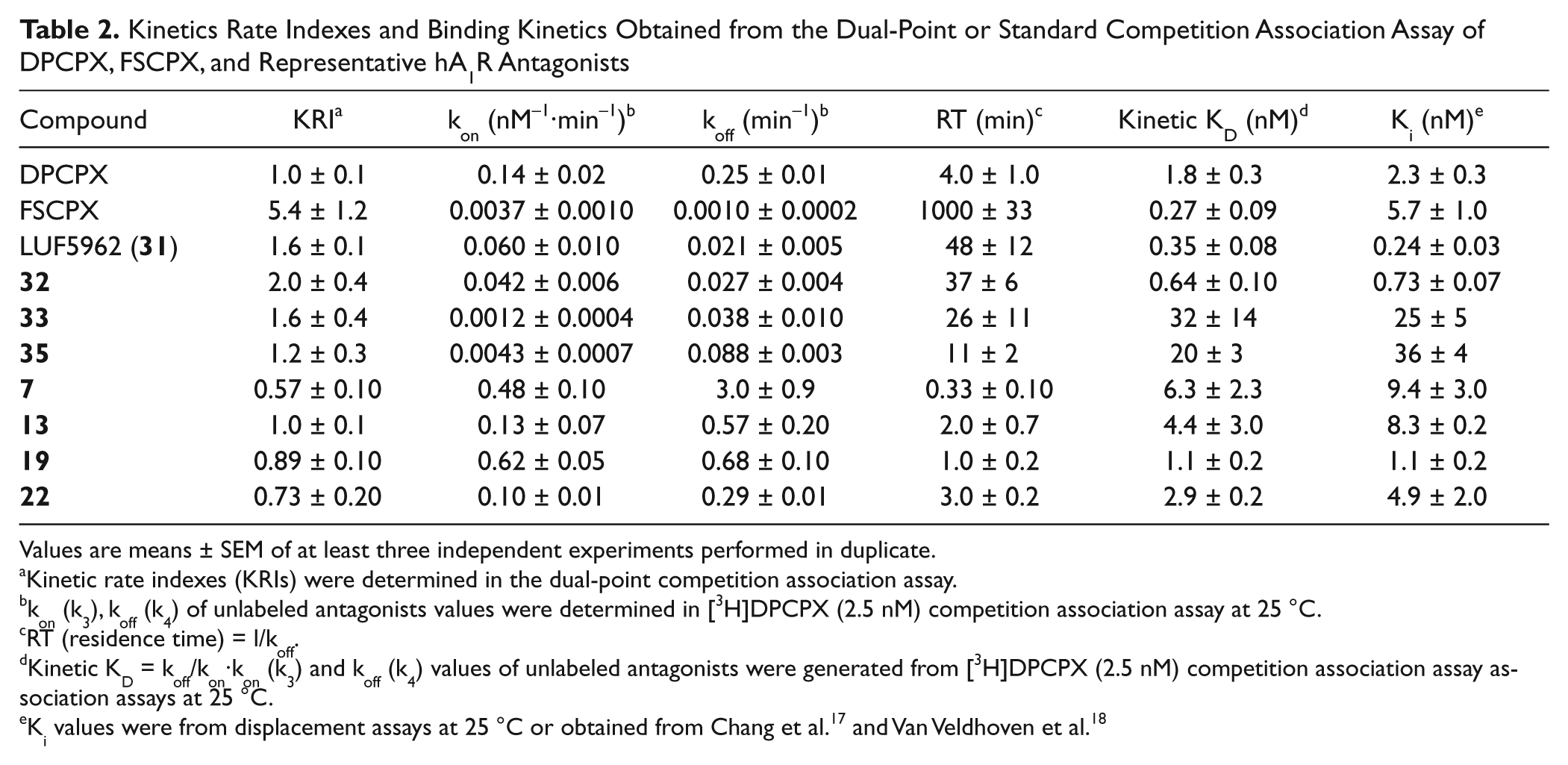
Values are means ± SEM of at least three independent experiments performed in duplicate.
Kinetic rate indexes (KRIs) were determined in the dual-point competition association assay.
kon (k3), koff (k4) of unlabeled antagonists values were determined in [3H]DPCPX (2.5 nM) competition association assay at 25 °C.
RT (residence time) = l/koff.
Kinetic KD = koff/kon·kon (k3) and koff (k4) values of unlabeled antagonists were generated from [3H]DPCPX (2.5 nM) competition association assay association assays at 25 °C.
Validation and Optimization of the Competition Association Assay at hA1R
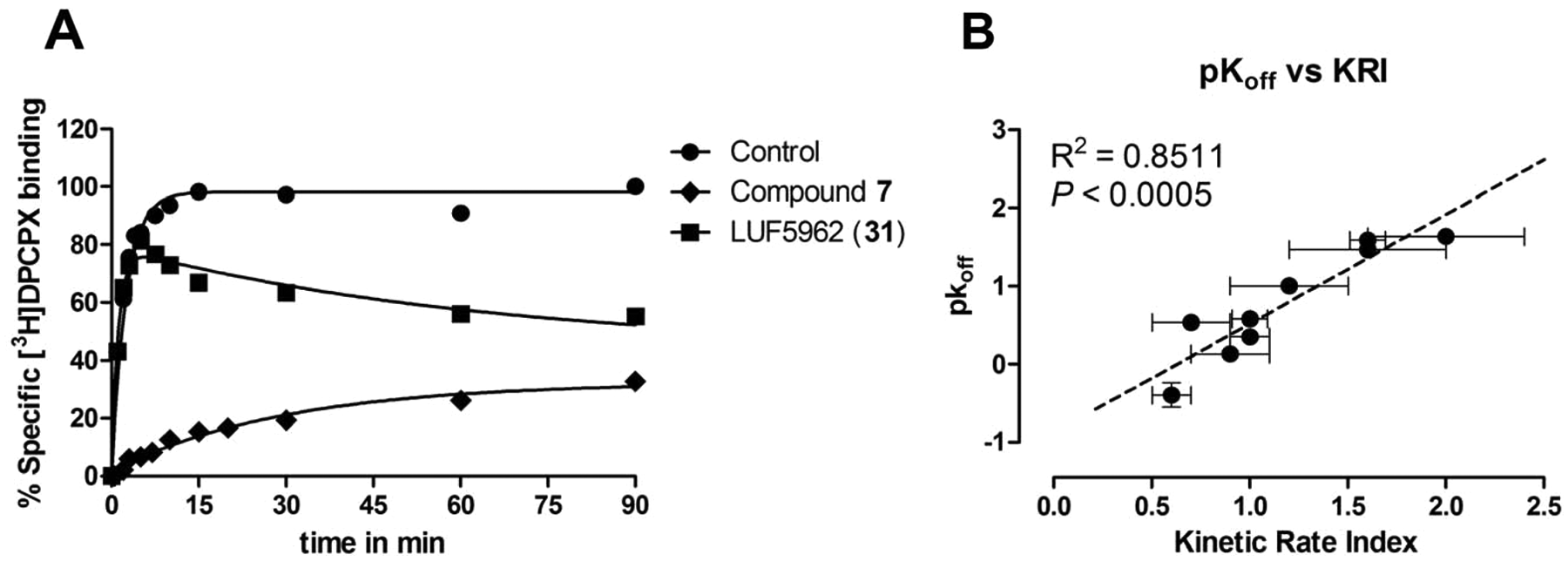
With the precharacterized kon (k1) and koff (k2) values of [3H]DPCPX binding from direct association and dissociation experiments, kon (k3) and koff (k4) values of unlabeled DPCPX and LUF5962 were determined by fitting the values into equation (7), described in “Materials and Methods.” Three different concentrations of the unlabeled reference compounds (DPCPX and LUF5962) were tested ( Fig. 3 ). Their kon (k3) and koff (k4) values determined by the competition association method were 0.14 ± 0.02 nM−1·min−1 and 0.25 ± 0.01 min−1 for unlabeled DPCPX ( Fig. 3A and Table 2 ) and 0.060 ± 0.010 nM−1·min−1 and 0.021 ± 0.005 min−1 for LUF5962 ( Fig. 3B and Table 2 ), which were in accordance with the k1 and k2 values determined in “traditional” association and dissociation experiments ( Table 1 and Fig. 2 ). Comparison of the dissociation constants and affinities obtained from the different equilibrium and kinetic experiments ( Table 1 ) further verified that the competition association assay could be accurately used to determine the binding kinetics of unlabeled A1R ligands.
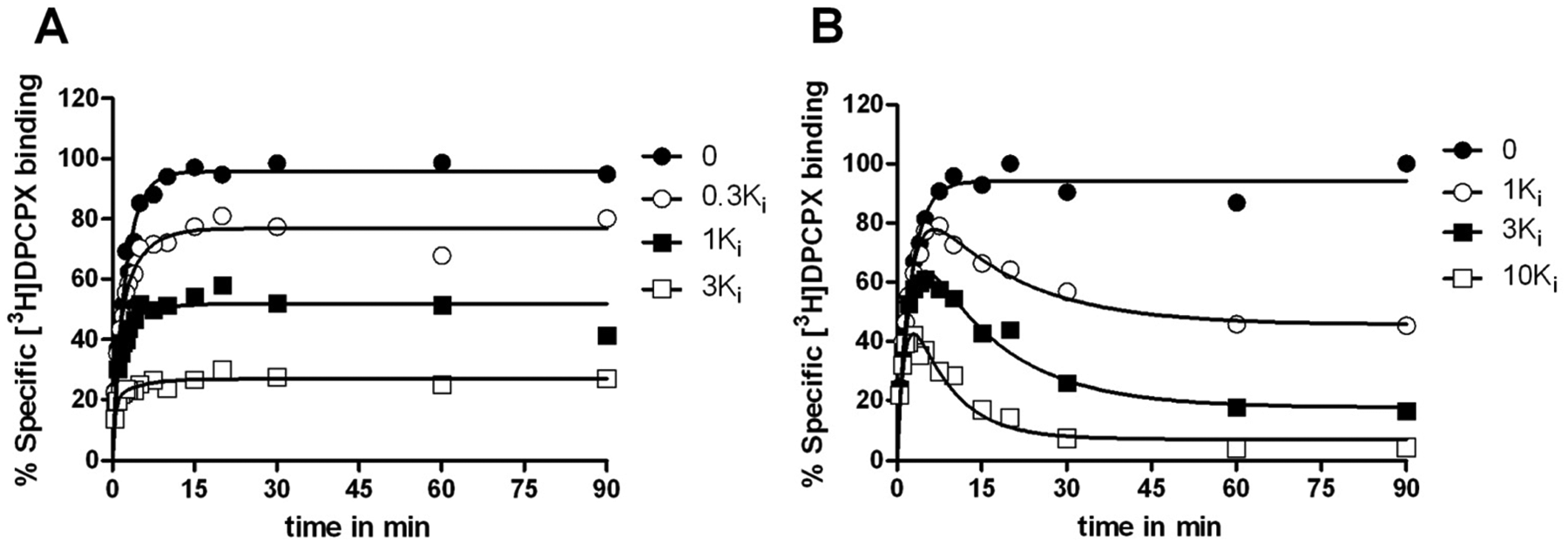
Competition association assay with [3H]DPCPX at 25 °C from human adenosine A1 receptors stably expressed on Chinese hamster ovary (CHO) membranes in the absence or presence of 0.3-fold, 1-fold, and 3-fold Ki value of unlabeled DPCPX (
Optimization of the Dual-Point Competition Association Assay at hA1R
For the dual-point competition association assay, it was necessary to decide on the optimal compound concentration and the two time points (t1 and t2) to obtain a robust kinetic screening assay. Therefore, we decided to optimize the assay using the reference antagonist FSCPX as a positive control, which is known to bind irreversibly to the hA1R. 16
First, we used concentrations that represented 3-, 10-, and 30-fold Ki values of unlabeled FSCPX to study the influence of different concentrations on the kinetic assay. It follows from Figure 4A that a long incubation time (i.e., 180 min) is needed for FSCPX to abrogate all [3H]DPCPX binding at a low concentration (i.e., 3-fold its Ki value). The co-application of 30-fold its Ki value resulted in a faster knockdown of [3H]DPCPX binding sites (i.e., after approximately 60 min). Such a significant decrease of [3H]DPCPX binding reduced the KRI window. Incubation of the membranes with an FSCPX concentration equaling 10 times its Ki value led to an intermediate situation ( Fig. 4A ). Notably, the set of three FSCPX concentrations are higher than the concentrations used in experiments with LUF5962 and DPCPX ( Fig. 3 ). To achieve radioligand equilibrium binding as much as possible with slowly dissociating competitors (or irreversibly binding competitors like FSCPX) in the same experimental incubation time, higher concentrations need to be used than for competitors with relatively faster or equal dissociation rates. The latter has also been described for the muscarinic M3 and histamine H3 receptor.9,24 In the present study, we observed that 10-fold Ki of the competitor gave reasonably large assay windows for all three ligands displaying divergent off-rates (i.e., DPCPX, LUF5962, and FSCPX). Hence, for screening purposes, it was decided to use concentrations equal to 10-fold the Ki values of the respective unlabeled ligands. It is important to note that 10-fold Ki is optimal for A1R in the current protocol, yet this concentration may not be suitable on other drug targets, especially when divergent concentrations of radioligand are used in the competition association assay. For instance, Dowling and Charlton 9 used approximately 25-fold KD of the radioligand in their competition association assay on the muscarinic M3 receptor. This requires higher concentrations of unlabeled ligands (from 10- to 1000-fold their respective Ki values) to achieve full resolution of radioligand equilibrium binding within the frame of incubation time. Similarly, Slack et al. 24 used higher concentrations of unlabeled ligands (from less than 15-fold to over 200-fold Ki) in their competition association assays on histamine H1 and H3 receptors. Therefore, for a general optimization of the dual-point assay on other drug targets, it is suggested that one should take into account the radioligand’s concentration, assay incubation time, and expected kinetic profiles of the competitors for optimization of the competitors’ concentration, since all these parameters influence the resolution of the assay.
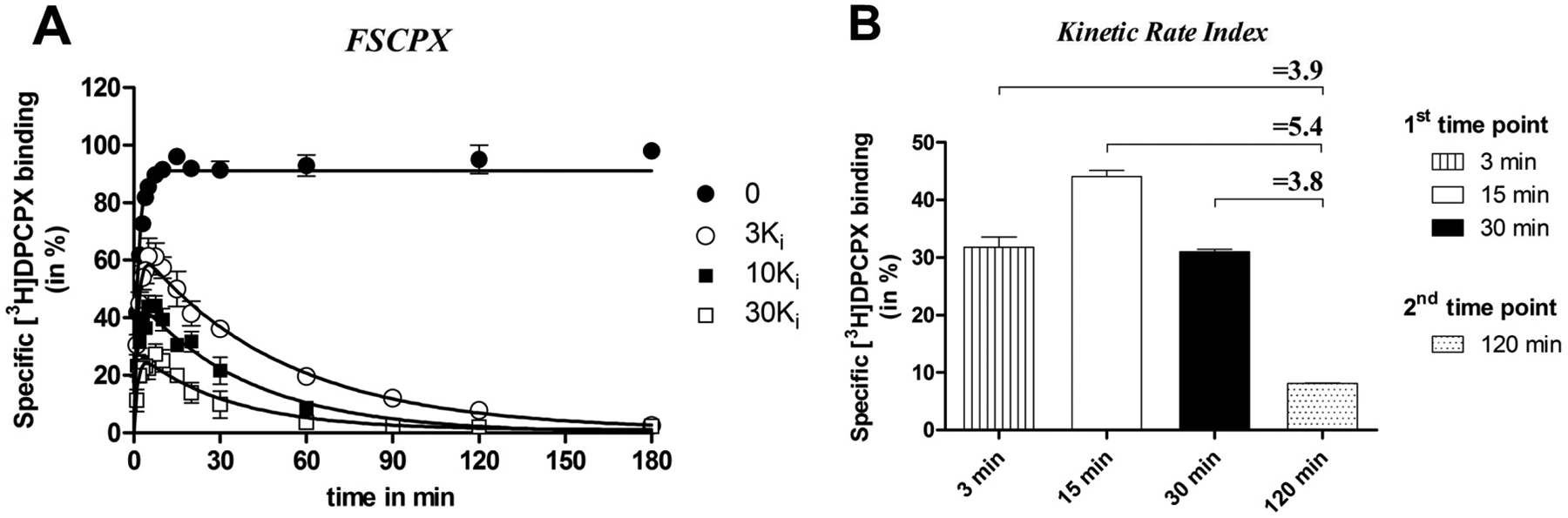
Competition association assay with [3H]DPCPX at 25 °C from human adenosine A1 receptors stably expressed on Chinese hamster ovary (CHO) membranes in the absence or presence of 3-, 10-, and 30-fold Ki value of FSCPX (
Second, we optimized the two time points (t1 and t2). As for t2, the incubation time was set at 120 min, which was based on the observation in Figure 4A that in the presence of an FSCPX concentration of 10-fold its Ki value, [3H]DPCPX binding was negligible after a 120-min incubation. As mentioned above, we assumed that within this time frame, most compounds would equilibrate with the target, even if they were kinetically different, as further corroborated by Figure 3 . As for t1, the incubation time was set at 15 min since under control conditions (i.e., absence of unlabeled ligand), [3H]DPCPX binding reached equilibrium after 15 minutes at 25 °C according to equation (5) and Figure 2A . We also tried to set t1 at a much longer (i.e., 30 min) or shorter incubation time (i.e., 3 min). From Figure 4B , it follows that the selection of t1 and t2 clearly affects the KRI values obtained. In other words, a shorter or longer incubation time than 15 min for t1 reduced its margin over the control (KRI = 1.0)—that is, KRI values = 3.9 or 3.8, respectively—whereas t1 = 15 min provided a more significant KRI value of 5.4 for FSCPX. Therefore, incubation times of 15 min (t1) and 120 min (t2) were used for the dual-point competition association assay with [3H]DPCPX at the hA1R. Notably, it is impossible to define an absolute value or cut-criterion of t1 and t2 since the two time point setup is dependent on several factors (i.e., radioligand concentration, temperature). For a general application on other targets, we suggest that one can start with setting t1 at a time when the radioligand just reaches equilibrium by using equation (5), whereas for t2, a later time point is chosen long enough for the binding of the unlabeled ligand to reach a plateau. A late t2 of course compromises the convenience of this screening method, as the total assay time increases. Therefore, it may be necessary to perform similar “tailored” optimizations of time points and concentrations, and even the choice of the radioligand, to obtain robust results on other drug targets.
Kinetic Screening of hA1R Antagonists Using Dual-Point Competition Association Assay
In the present study, 35 hA1R antagonists synthesized in house17,18 were screened in the newly developed dual-point competition association assay ( Fig. 5 ). The specific binding of [3H]DPCPX was measured after 15 min and 120 min and for [3H]LUF5962 after 30 min and 120 min in the absence and presence of a single concentration of unlabeled competitive ligand (10-fold Ki, in both [3H]DPCPX and [3H]LUF5962 assays). This allowed us to calculate the KRI values for each compound by using equation (6). The KRI cutoff value for kinetically interesting compounds was arbitrarily set at 1.2 ( Fig. 5 , the stippled line) rather than 1.0 to avoid false positives. It is worth mentioning that the cutoff value for the KRI can be flexible based on a different screening purpose. In other words, one can specifically set it higher to select only longer residence time compounds or lower to obtain more hits for future chemical modifications. In the present study, most compounds were found to have KRI values equal to or less than 1 ( Fig. 5 ) with both radioligands, which were thus considered to have similar or faster off-rates from the hA1R compared with the radioligands used, that is, [3H]DPCPX or [3H]LUF5962. Seven compounds, excluding FSCPX, had KRI values ≥1.2 with [3H]DPCPX as the radioligand, which indicated that these compounds dissociated more slowly from the receptor than [3H]DPCPX ( Fig. 5 , white bars). KRI values for the same panel of unlabeled ligands against [3H]LUF5962 were also determined ( Fig. 5 , gray bars). In the latter situation, all compounds displayed a KRI <1.2, except FSCPX, since the radioligand used ([3H]LUF5962) has a relatively slow off-rate (0.021 min−1). Interestingly, most of the compounds adopted a concerted trend with a decreased KRI value compared with that obtained with [3H]DPCPX. This is in accordance with the observation that indeed most compounds have faster dissociation rates than LUF5962, as determined by full kinetic experiments with [3H]DPCPX ( Table 2 ). For validation of the dual-point assay, four compounds with lower or similar KRI values (compared with DPCPX, KRI ≤1) and four compounds with higher KRI values (≥1.2) were further characterized in follow-up kinetic assays ( Table 2 ).
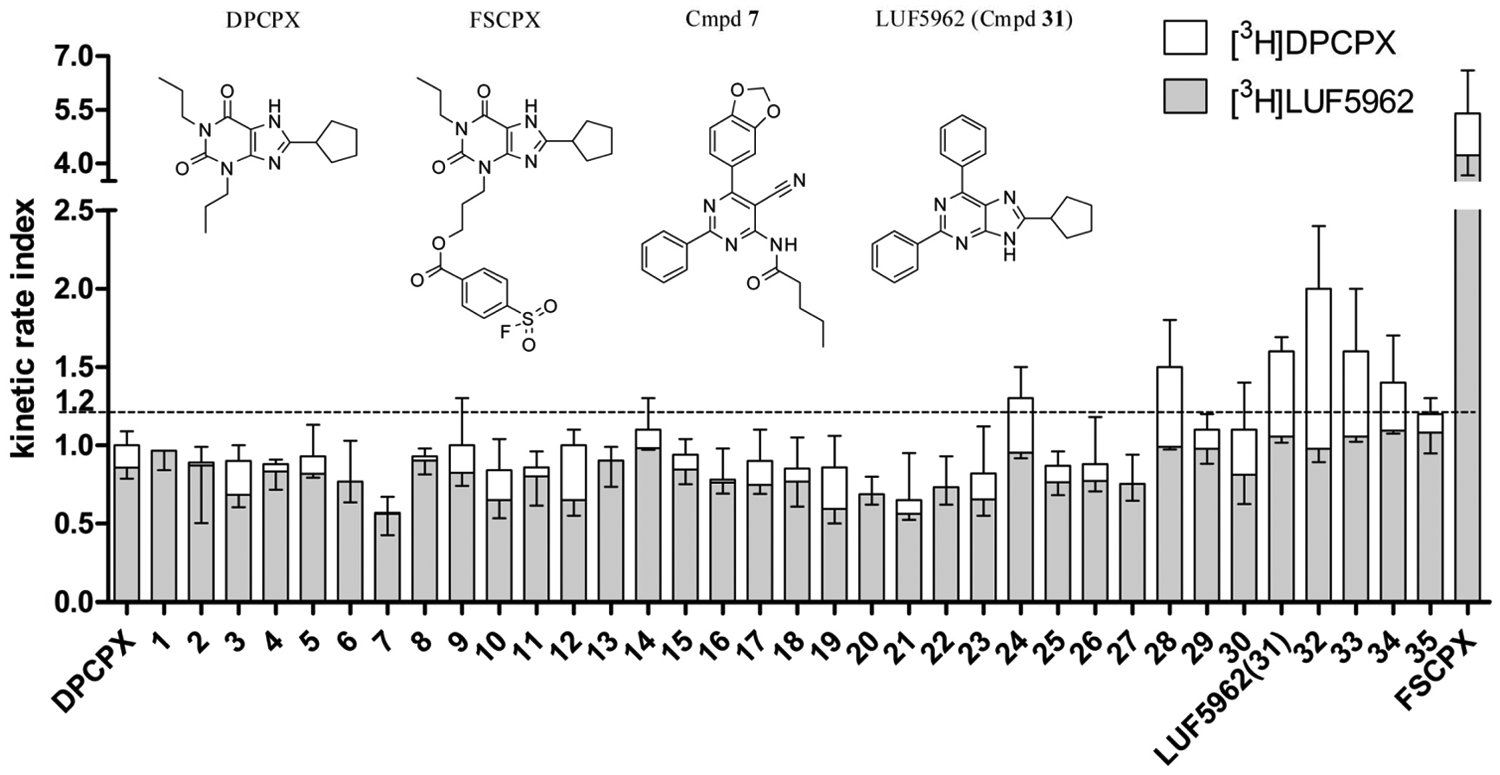
Kinetic rate indexes (KRIs) of 35 in-house hA1R antagonists, DPCPX and FSCPX.16–18 The KRI values of the same panel of unlabeled ligands against [3H]LUF5962 (gray bars) are integrated with the results obtained with [3H]DPCPX (white bars). Values were obtained from the dual-point competition association assays and calculated by using equation (6). The kinetic rate indexes of the reference compounds, unlabeled DPCPX or LUF5962 (compound
Subsequently, competition association assays in the presence of [3H]DPCPX were performed to determine the binding kinetics of the eight compounds that had divergent KRI values (
Table 2
). In the present study, we used FSCPX as a tool compound, which had a KRI value of 5.4 ± 1.2 at the hA1R, indicative of negligible dissociation (
Table 2
and
Fig. 4A
). Similar to FSCPX, the specific binding of [3H]DPCPX in the presence of unlabeled LUF5962 (compound
(
Advantages of the Dual-Point Competition Association Assay and Its Future Application in Kinetic Binding Screening
Currently, there are several assays available developed for kinetics screening. For instance, Heise et al.
25
developed a scintillation proximity assay (SPA) to measure Ki values for gonadotropin-releasing hormone receptor (GnRH receptor) antagonists after a 30-min and 10-h incubation. This assay is based on the observation that a slowly dissociating compound will display a decrease of apparent Ki value over time, since it requires a longer incubation time to reach equilibrium.11,25 Another methodology is a functional assay described by Morriello et al.,
26
where hNK1R overexpressing CHO cells were pretreated with neurokinin 1 receptor (NK1R) antagonists before a measurement of the remaining activity induced by subsequently added substance P (endogenous agonist of hNK1R). A complete reversal of the hNK1R antagonism indicated a short receptor residence time of the compound under evaluation, whereas an insurmountable effect of the tested ligand suggested longer receptor occupancy. In such a way, the recovery of the response to substance P gave an indication of an antagonist’s receptor off
As represented in Figure 5 , the same set of unlabeled A1 receptor ligands have different KRI values if tested against two kinetically different radioligands. Indeed, since the dual-point approach and its derived KRI are mainly based on whether an unlabeled competitor dissociates faster or slower from its target than a particular radioligand, the choice of radioligand becomes the key for a reliable kinetic screening. Here we propose that the choice for an “ideal” radioligand (if one has the luxury of a number of radioligands) should take into account the drug target and the particular screening purpose. More specifically, if one searches for slowly dissociating compounds, a relatively long– residence time radioligand would be preferred, whereas for short–residence time compounds, a relatively fastly dissociating radioligand may be needed. By following this principle, the load of follow-up kinetic determinations for selected compounds can be reduced. However, it is also necessary to take the assay practicability into consideration. In the present study, for instance, LUF5962 has a slow off-rate. According to equation (5) regarding the t1 time point setup, it would require nearly 2 h for [3H]LUF5962 to reach equilibrium when a concentration of 1-fold its Ki value (0.24 nM) is used. Logically, this results in an even later t2 time point. Thus, we applied 1.0 nM [3H]LUF5962 (approximately four times its Ki) in our additional KRI experiments, enabling a relatively quick t1 at approximately 30 min and the same t2 as for [3H]DPCPX (120 min). The robustness of the dual-point competition assay is underscored by the fact that even a less than ideal radioligand, such as [3H]LUF5962, can still be used for screening purposes.
In the present study, we have merely described the application of the dual-point assay for a prototypical GPCR target—namely, the adenosine A1R. We believe that this assay can have a more general application for other drug targets as well. This consideration is supported by an increasing amount of studies that characterized receptor binding kinetics, such as for the histamine H1 receptor, the corticotropin releasing factor type–1 receptor (CRF1), and, more recently, the adenosine A2A receptor, where Motulsky and Mahan’s theory and their competition association assay were used.24,31,32 Similar patterns of competition association curves were observed in these studies—that is, in the presence of a fastly dissociating competitor, the specific radioligand binding approached equilibrium slowly, whereas in the presence of a slowly dissociating compound, it showed first the typical “overshoot,” followed by a constant decline to a plateau, as in Figure 3B . This, as a proof of concept, implies the feasibility of a dual-point kinetic screening and the use of KRI for searching desired leads on other drug targets as well.
In summary, the dual-point competition association assay is a valid tool to perform fast and high-throughput screening for compounds having either long or short residence times. The KRI value, which we coined in the present study, is an effective predictor of a compound’s dissociation rate from its target. We believe that this approach can be of general use at other drug targets (e.g., other GPCRs) after assay optimization for individual receptors.
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This project was financially supported by the Innovational Research Incentive Scheme of the Netherlands Research Organization (NWO; VENI-Grant 11188 to L.H.H.).