
Abstract
Tau aggregation is the pathological hallmark that best correlates with the progression of Alzheimer’s disease (AD). The presence of neurofibrillary tangles (NFTs), formed of hyperphosphorylated tau, leads to neuronal dysfunction and loss, and is directly associated with the cognitive decline observed in AD patients. The limited success in targeting β-amyloid pathologies has reinforced the hypothesis of blocking tau phosphorylation, aggregation, and/or spreading as alternative therapeutic entry points to treat AD. Identification of novel therapies requires disease-relevant and scalable assays capable of reproducing key features of the pathology in an in vitro setting. Here we use induced pluripotent stem cells (iPSCs) as a virtually unlimited source of human cortical neurons to develop a robust and scalable tau aggregation model compatible with high-throughput screening (HTS). We downscaled cell culture conditions to 384-well plate format and used Matrigel to introduce an extra physical protection against cell detachment that reduces shearing stress and better recapitulates pathological conditions. We complemented the assay with AlphaLISA technology for the detection of tau aggregates in a high-throughput-compatible format. The assay is reproducible across users and works with different commercially available iPSC lines, representing a highly translational tool for the identification of novel treatments against tauopathies, including AD.
Introduction
Tauopathies are a group of neurodegenerative diseases characterized by the presence of neurofibrillary tangles (NFTs) in limbic and neocortical areas of the brain. NFTs are composed of insoluble and hyperphosphorylated microtubule-associated protein tau. More than 30 mutations in MAPT, the gene encoding tau, have been associated with hereditary forms of frontotemporal dementia. 1 Alzheimer’s disease (AD) is the most prevalent tauopathy and the most common neurodegenerative disorder. A strong correlation between NFT distribution and cognitive decline has been described in AD patients. 2 Although it is accepted that tau aggregates are rather a cause than a consequence of neurodegeneration, the precise molecular mechanisms by which tau pathology is triggered and progresses throughout brain areas are not yet well understood. Nevertheless, due to the correlation between tau histopathology and dementia, it is hypothesized that manipulating tau aggregation and/or clearance may be a potential therapeutic entry point to treat AD. 3
Identification of new therapies for treating neurological disorders requires translational and disease-relevant models. Tissue culture cell lines lack the physiological relevance and rodent primary cultures the scalability needed to test large compound libraries even in miniaturized format, in addition to the fact that they are nonhuman. In the preceding decade, the development of induced pluripotent stem cell (iPSC) technology has aided in overcoming historic limitations of in vitro models. 4 Stem cells can be differentiated into a virtually unlimited source of cortical neurons5–8 and have been already utilized in the in vitro modeling of a number of neurological disorders, including autism, schizophrenia, and AD.9–12 Despite the enormous potential of iPSCs in drug discovery, the current lack of standardized differentiation protocols has led to variability between laboratories and cell lines. In addition, scalability, long-term adherence to scaffolding/plating material, cell distribution, and slow maturation are among the biggest issues when adding stem cell technology to the drug development pipeline. Therefore, it is of general interest the establishment of standard operating procedures (SOPs) that allow maintenance of long-term cultures by reducing potential sources of variability. Ultimately, these SOPs can then be miniaturized and automated for their use in high-content imaging (HCI) or HTS platforms typically used for phenotypic screenings in drug discovery programs.
Here, we describe a highly reproducible tau aggregation assay suitable for drug discovery that is based on human iPSC–derived cortical neurons. We have adapted published neuronal differentiation protocols to miniaturized 384-well format that allows the maintenance of healthy mature cortical neurons for more than 5 weeks. In order to address challenges of cell handling typically experienced upon extended incubation periods impacting on cell distribution in wells, to protect neurons from mechanical stress introduced by, for example, medium exchange, and to ensure a consistent and supporting scaffolding environment, we used diluted Matrigel to create thin-layer three-dimensional cultures. 13 Moreover, we have applied previously described methods to trigger tau aggregation in vitro 14 and successfully reproduced pathophysiological features associated with tauopathy. Finally, we complemented the model using a highly sensitive nonwash “amplified luminescence proximity homogenous assay” (AlphaLISA) for the detection of tau aggregates. The robustness of the assay was confirmed across different cell lines and by multiple users. In conclusion, we have developed a human neuron–based model of tau aggregation that can be used in automated drug discovery platforms for the identification of novel therapies to treat neurodegeneration.
Material and Methods
iPSC Lines
Two commercially available cell lines generated from clinically unaffected donors were used in this study: IPSC0028 (Sigma, St. Louis, MO) and ChiPSC6b_m1 (Cellectis, Paris, France). IPSC0028 was generated by reprogramming epithelial cells from a 24-year-old female donor with the four Yamanaka factors (Oct3/4, Sox2, Klf4, and c-Myc) using retroviral vectors. Episomal technology for expressing human factors Sox-2, Klf4, Oct3/4, Lin28, L-myc, and glis-1 was used for generating ChiPSC6b_m1 from primary human fibroblasts derived from a male donor. Both cell lines have been characterized for expression of stem cell markers and have normal karyotype. Their capacity to differentiate into all three germ layers has also been tested by the manufacturer. Cells were cultured and maintained in feeder-free conditions using Matrigel (BD Biosciences, Franklin Lakes, NJ) and mTeSR media (STEMCELL Technologies, Vancouver, British Columbia).
Neuronal Differentiation
iPSCs were differentiated into cortical neuronal progenitor cells (NPCs) by Axol
Biosciences (Cambridgeshire, UK) adapting already published protocols.12,15 Briefly, iPSCs were dissociated into
single-cell suspension and neuronal induction was triggered by following classical dual
SMAD inhibition for a period of 12 days. After this time, NPCs were treated with dispase
and subplated for amplification three more times (at days 17, 20, and 25 approximately).
Between days 25 and 30, NPC frozen stocks were prepared and kept in liquid nitrogen for
subsequent experiments. Three days after thawing (considered experimental day 0), cells
were dissociated using accutase and subplated in a mixture of differentiation media and
Matrigel (1:15 v/v) for the final neural maturation. To block cell proliferation, cells
received two treatments with 10 µM
N-[N-(3,5-difluorophenacetyl)-
Cell Line Culture and Transfection
Human kidney–derived QBI-293 (QBiogene, Carlsbad, CA) were grown in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% (v/v) heat-inactivated fetal bovine serum (FBS), 1% pyruvate (10 mM), 1% penicillin-streptomycin (PenStrep), and L-glutamine (20 mM). Cells were maintained at 37 °C, in humidified atmosphere containing 5% CO2.
One day prior to transfection, 80% confluent cells were trypsinized and then seeded in 10 cm2 dishes at 1.5 × 106 cells per well. The growth medium was renewed directly before transfection. DNA mixture containing 2.5 µg pcDNA6-TR and 2.5 µg 2N4R-TauP301L-pcDNA4/TO was diluted in 500 µL OptiMEM and 15 µL FuGENE 6. Transfection reagent diluted in 500 µL OptiMEM was added. The mixture was incubated for 15 min at room temperature, and then added to the cells. After incubation for 24 h, the growth medium was removed and replaced with a new one containing 5 µg/mL blasticidin and 200 µg/mL Zeozin. The cells were cultured until selection was complete. Monoclonal lines were generated by limited dilution.
Preparation of K18 Fibrils and Seeding
Tau seeds were generated mixing 40 μM recombinant tau protein (Myc-K18-P301L) with 10 μM of low-molecular-weight heparin and 2 mM DTT in 100 mM sodium acetate buffer (pH = 7.0) and incubated 3 days at 37 °C. For the seeding step, Myc-K18-P301L fibrils were diluted in 100 nM sodium acetate buffer up to 10 μM and sonicated. Seeds were then added to each well according to the final concentration required and incubated for 3 days. For the doxycycline-inducible P301L, cells were treated 3 h before seeding with 100 µg/mL doxycycline at 300,000 cells per well in a 6-well plate.
Induction of Tau Aggregation in iPSC-Derived Neurons
Tau aggregation was carried out following previously described protocol 14 with slight modifications. Ten days after thawing, seeded NPCs were transduced at a multiplicity of infection (MOI) 100 with adeno-associated virus driving expression of the 2N4R full-length isoform of the human P301L mutant tau under the human synapsin promoter. Adeno-associated virus only expressing enhanced green fluorescent protein (EGFP) under the synapsin promoter was used as a control. Seeding with fibrils was performed 4 days later by adding recombinant truncated tau containing four microtubule-binding repeats with P301L mutation (K18) at a concentration of 50 nM per 96 wells and 12 wells, and 133 nM per 384 wells. Shortening of the protocol was performed by reducing the transducing and seeding times to days 4 and 7 postthawing, respectively.
Immunocytochemistry
Cells were fixed using 4% paraformaldehyde (PFA) diluted in phosphate-buffered saline (PBS) with 4% sucrose, pH 7.4, for 10 min at room temperature and permeabilized with 0.25% Triton X-100 (PBST) for 10 min. After three washes with PBS, 1% bovine serum albumin (BSA) in PBST was used for blocking for 1 h before incubation with primary antibody. Cells were incubated with primary antibodies diluted in 1% BSA in PBST in a humidified chamber for 1 h at room temperature or overnight at 4 °C. Detection of insoluble tau was done by fixing cells in a solution containing 4% paraformaldehyde, 4% sucrose, and 1% Triton X-100 in PBS for 10 min.
Images were acquired with a LSM510 confocal laser scanning microscope (Carl Zeiss, Oberkochen, Germany) equipped with an axiocam camera and processed using ImageJ. All antibodies used in this study are described in the supplementary material.
Protein Extraction and Western Blot Analysis
Protein extracts for Western blot (WB) were generated by differentiating and treating neurons in 12-well plates (300,000 NPCs were seeded per well). Cells were lysed and sonicated in RIPA buffer (Sigma) containing phosphatase and protease inhibitors. Lysate was cleared by 15 min centrifugation at 12,000g and supernatant was recovered for protein quantification. Twenty micrograms of protein extract was then loaded per lane and resolved on SDS-PAGE before transferring to nitrocellulose membranes. Blocking was done for 1 h with 5% nonfat milk solution in PBST before incubation with primary antibodies. For immunoblotting of insoluble tau, sequential extraction was performed as previously described 14 using Triton lysis buffer (1% Triton X-100 dissolved in 50 mM Tris, 150 mM NaCl at pH 7.6) containing protease and phosphatase inhibitors. Sarkosyl extracts were prepared following already described methods. 16
AlphaLISA Immunoassay
Cell lysis was performed in culture plates for 1 h at room temperature (RT) in an orbital
shaker using RIPA buffer–containing phosphatase (PhosSTOP, Roche, Mannheim, Germany) and
protease inhibitors (cOmplete, Roche). Lysis volume was optimized according to the plate
format: 20 µL per 384 wells and 40 µL for 96 wells. For hTAU10/hTAU10 and AT8/AT8 tau
aggregation assays, we performed measurements in duplicates using 5 µL of lysate each
time. Five microliters of the remaining lysate was used for the relative quantification of
total tau and for measuring cell viability, respectively. Each sample was transferred to a
384-well assay plate for AlphaLISA reaction, in which 5 µL of cell extracts were incubated
for 2 h at RT with a mixture of biotinilated antibody and acceptor beads at the
concentration described in
Cell Viability
Cell viability was measured using the CellTiter-Glo luminescent cell viability assay. Five microliters of lysate from each well was used and incubated for 10 min before measuring emission at 560 nm on the EnVision plate reader.
Data Analysis
Unless otherwise indicated, one-way ANOVA followed by Dunnett’s multiple comparison test was performed using GraphPad Prism version 6.00 for Windows (GraphPad Software, La Jolla, CA, www.graphpad.com).
Results
Scalable Format for Differentiation of iPSC Cortical Neurons
We first generated neuronal progenitor cells following an already published protocol 15 and prepared NPC frozen stocks following 30 days of amplification. Thawing day was considered day 0, and after 3 days NPCs were resuspended in a 1:15 dilution of differentiation media–Matrigel (v/v) and seeded in 384-well plates to test their neuronal differentiation capacity over a 40-day period ( Fig. 1A ). As already described, 12 diluted Matrigel allows for cell sedimentation and forms a thin film of gel that embeds cells, providing a scaffold and permitting long-term adherence. Furthermore, Matrigel introduces an extra physical protection barrier that reduces considerably the shearing stress linked to media change and plate handling. This is particularly important, as these issues partially explain the limitations of scaling up, long-term incubation, and automation of iPSC-derived neuronal assays in drug development in neuroscience.
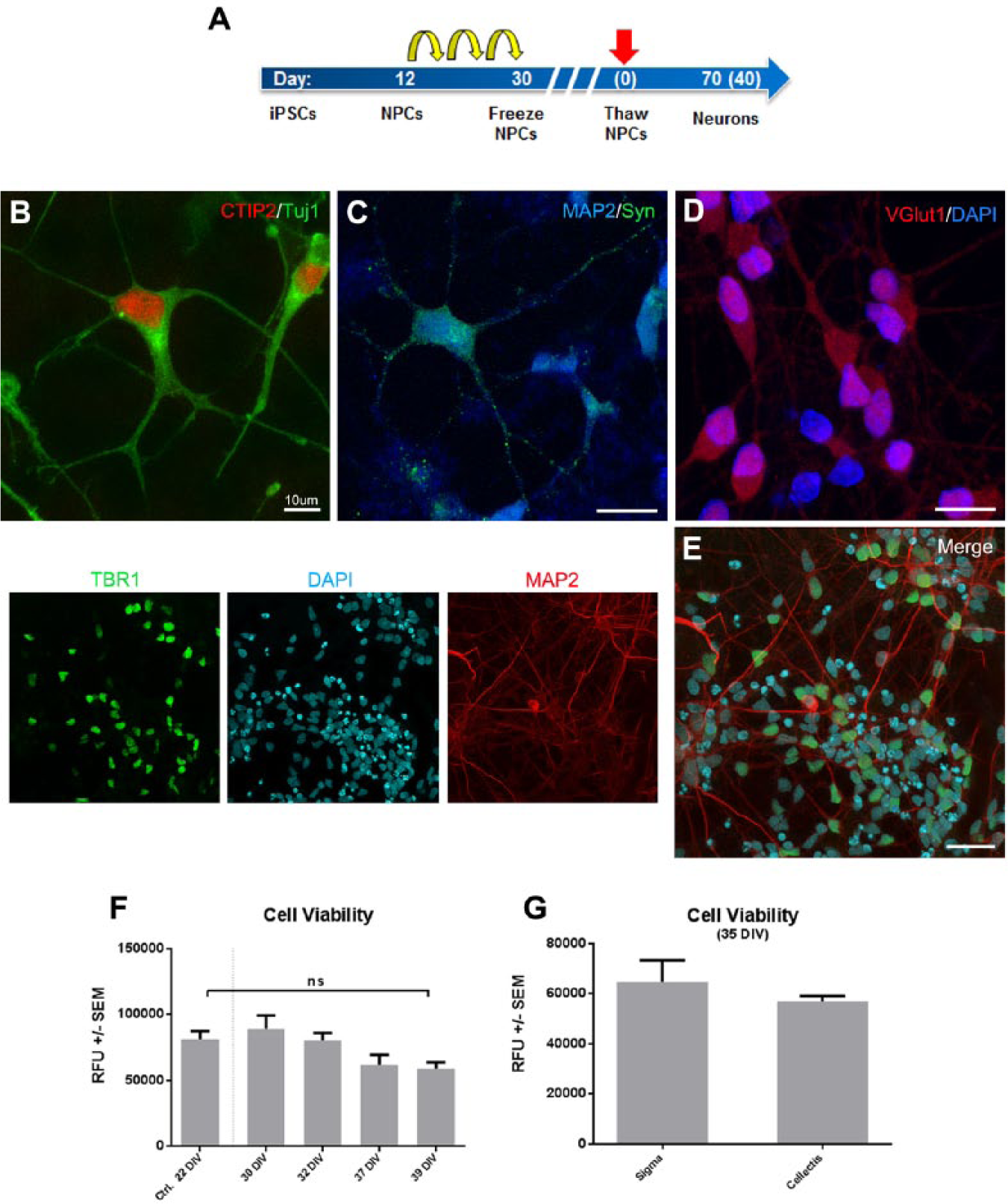
Healthy cortical neurons can be differentiated in 384-well plates and maintained for
at least 5 weeks. (
One week after seeding, NPCs display pyramidal morphology and express key cortical neuronal identity markers as detected by antibodies for beta-III tubulin (TUJ1) and CTIP2 ( Fig. 1B ). Thirty days after seeding cortical neurons show increased morphological complexity and the presence of apical and basal dendrites that are positive for maturity markers, including MAP2 and TBR1 ( Fig. 1C–E ), and synaptic proteins such as synapsin and VGlut1 ( Fig. 1C , D ). Using ATP as surrogate marker for cell viability, we observed that there is a progressive process of cell death that is intrinsic to the in vitro cell culture conditions. However, neurons cultured for 39 days are as viable as control cells kept in culture for 22 days ( Fig. 1F ). We also compared ATP values in two different commercially available cell lines, ChiPSC6b and IPSC0028, after 35 days in culture and found no significant differences in cell viability ( Fig. 1G ), confirming that culture conditions do not impair cell survival among the cell lines used. Therefore, final differentiation of NPCs in diluted Matrigel/384-well plates allows for the maintenance of mature and healthy cortical neurons that can be maintained in culture for more than 5 weeks.
Downscaling a Tau Aggregation Assay Using Human Neurons
We have recently demonstrated that tau aggregation can be triggered in human iPSC–derived cortical neurons 17 in a way similar to that described using primary rodent cultures. 14 We now wanted to validate the model using different culture conditions and formats in order to increase its scalability and potential automation. Due to the low amount of endogenous tau expression, current models require tau overexpression and the addition of recombinant truncated tau aggregates formed in vitro, such as the 4-repeat tau-repeat domain K18 harboring a P301L mutation to trigger aggregate formation in cells. 18 P301L is a tau mutation identified in frontotemporal dementia (FTD) and widely used in these aggregation models. The mutation lies within tau’s 4-repeat microtubule binding (4R) domain and facilitates the aggregation process in vitro. 19 Using WB analysis, we first studied the isoforms of tau endogenously expressed in our model. Using an isoform-specific antibody (RD3, Millipore, Billerica, MA), we confirmed the predominant expression of the “fetal” 3-repeat (3R) form of tau and the absence of 4R forms ( Fig. 2B ), indicating that tau overexpression may be necessary to promote aggregation in human iPSC–derived neurons. Therefore, we performed adenoviral-mediated overexpression of mutant tau P301L and subsequently seeded cells with recombinant K18 fibrils consisting of the tau P301L 4-repeat domain. The human synapsin promoter was used to ensure that expression of P301L was restricted solely to postmitotic mature neurons. Cells were transduced 10 days postthawing and seeded with fibrils 4 days after transduction ( Fig. 2A ).
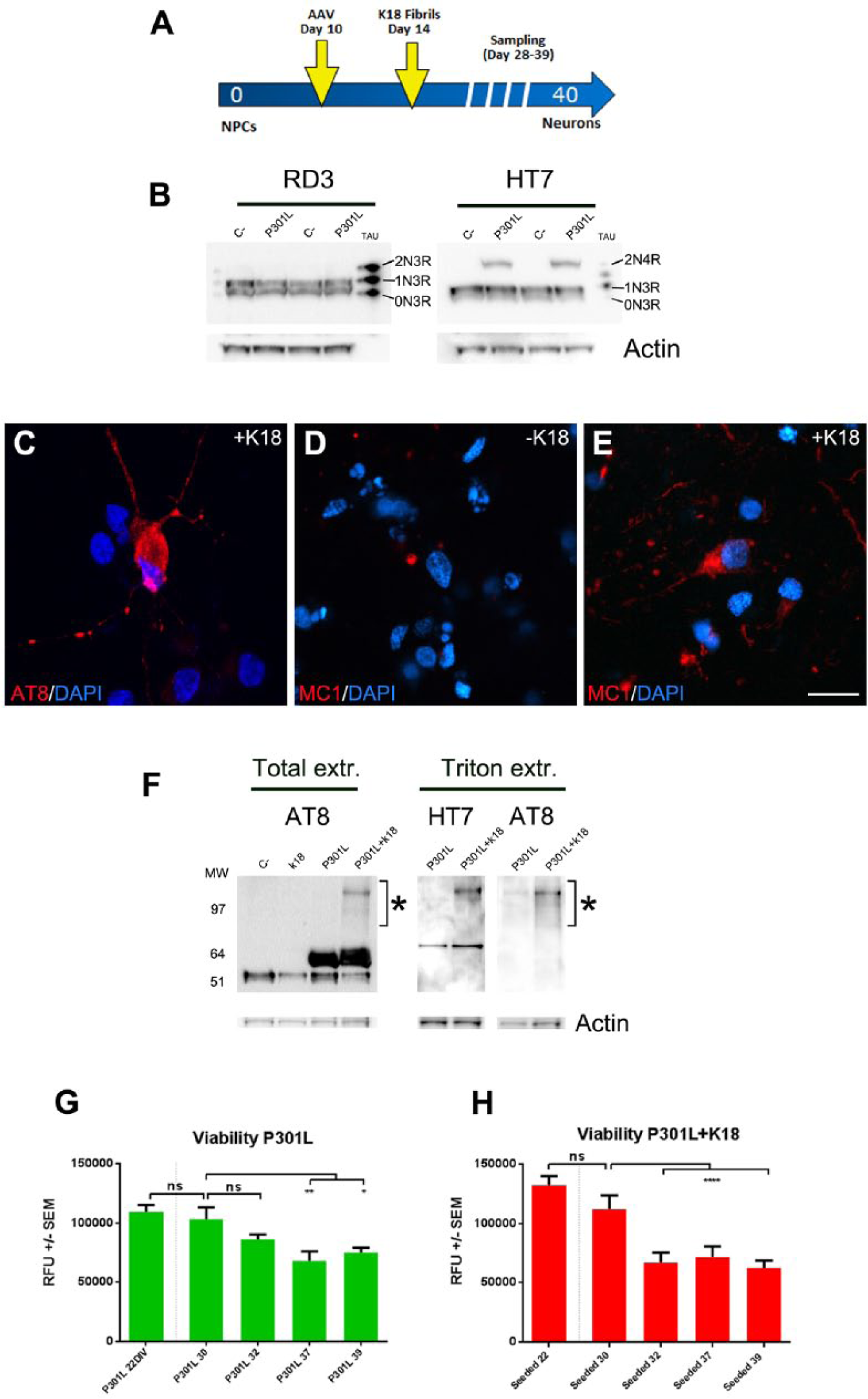
Tau aggregation in human cortical neurons cultured in Matrigel. (
At 28 days, neurofibrillary tangle-like inclusions of insoluble hyperphosphorylated tau were detected by immunofluorescence (IF) with AT8 antibody (which specifically detects hyperphosphorylated tau typically present in NFTs) after treatment with 1% Triton diluted in 4% PFA as previously described14,20 ( Fig. 2C ). AT8 positive tau aggregates were mostly detected in transduced and seeded cells, but not in control samples (not shown). We also confirmed the pathological nature of the aggregates by immunofluorescence with the antibody MC1, which has been described to specifically detect disease-specific tau conformation. 21 MC1 positivity was only found after 32 DIV in seeded and transduced cells ( Fig. 2E ), but not in only transduced cells ( Fig. 2D ).
The 4R forms were only detected in transduced cells by using anti-pan-tau antibody HT7 ( Fig. 2B ). Interestingly, in transduced and seeded samples we also observed an extra band/smear that is indicative of the presence of tau fibrils or oligomers. In order to define the presence of aggregates, we performed sequential extraction of Triton-soluble and nonsoluble tau following already described methodology. 14 Only transduced and K18-treated samples displayed the presence of a Triton-insoluble protein of high molecular weight that was detected by both HT7 and AT8 antibodies, suggesting that it might correspond to aggregated forms of tau ( Fig. 2F ). Interestingly, the presence of AT8 positive aggregates correlated with a 50% decrease in cell viability occurring between 30 and 32 days in culture ( Fig. 2H ), suggesting a toxic effect due to the seeding-induced intracellular tau aggregation. Therefore, the human tau aggregation model that we describe is able to reproduce in vitro prototypical tangle-like AT8 positive inclusions present in AD brain.
Validation of AlphaLISA Technology to Detect Tau Aggregation In Vitro
The most commonly used methodology to detect aggregated tau requires multiple centrifugation steps and subsequent immunoblot analysis of the sarkosyl/Triton insoluble fraction. The development of a rapid and scalable assay to perform relative quantification of tau aggregates represents a valuable tool in the establishment of robust assays oriented to detect compounds targeting tau aggregation. AlphaLISA is a “no wash” fluorescence-based high-throughput-compatible technology that has been previously described for the detection of total and aggregated tau in cell lines and primary cultures, respectively.20,22 We have recently described the proof of principle for its applicability to detect tau aggregates in human neurons. 17 We now wanted to demonstrate its compatibility with the new culture format using in vitro models as cell lines and primary cultures as validation tools.
To develop a tau aggregate–specific AlphaLISA assay, we verified the suitability of several antibodies specific for multiple epitopes and posttranslational modifications of tau. As a first step in assay validation, we used samples generated from cell lines (HEK-293/QBI) and primary neuronal cultures in which tau aggregation had been induced following already established protocols.14,23 Based on the high signal, anti-human tau antibody hTAU10/hTAU10 combination was selected due to its high affinity for aggregated tau. 24 As shown in Figure 3A , B , even in 100 times diluted samples (final dilution) significant differences between K18-seeded and nonseeded conditions in both HEK cells and primary cultures were detectable ( Fig. 3A , 3B , p < 0.0001). We also validated the combination of AT8/AT8 and HT7/hTAU10 antibodies to detect phosphorylated aggregates and total tau, respectively. AT8/AT8 AlphaLISA confirmed the specificity of our assay as it exclusively detected tau aggregates only in seeded samples in both cellular models ( Fig. 3C , D , p < 0.0001). Finally, anti-human tau HT7 antibody in combination with hTAU10 was used for the detection of total tau. The HT7/hTAU10 combination detected overexpressed tau in both cell models and in seeded and nonseeded cells, making it more suitable for the detection of total rather than aggregated tau ( Fig. 3E , F ).
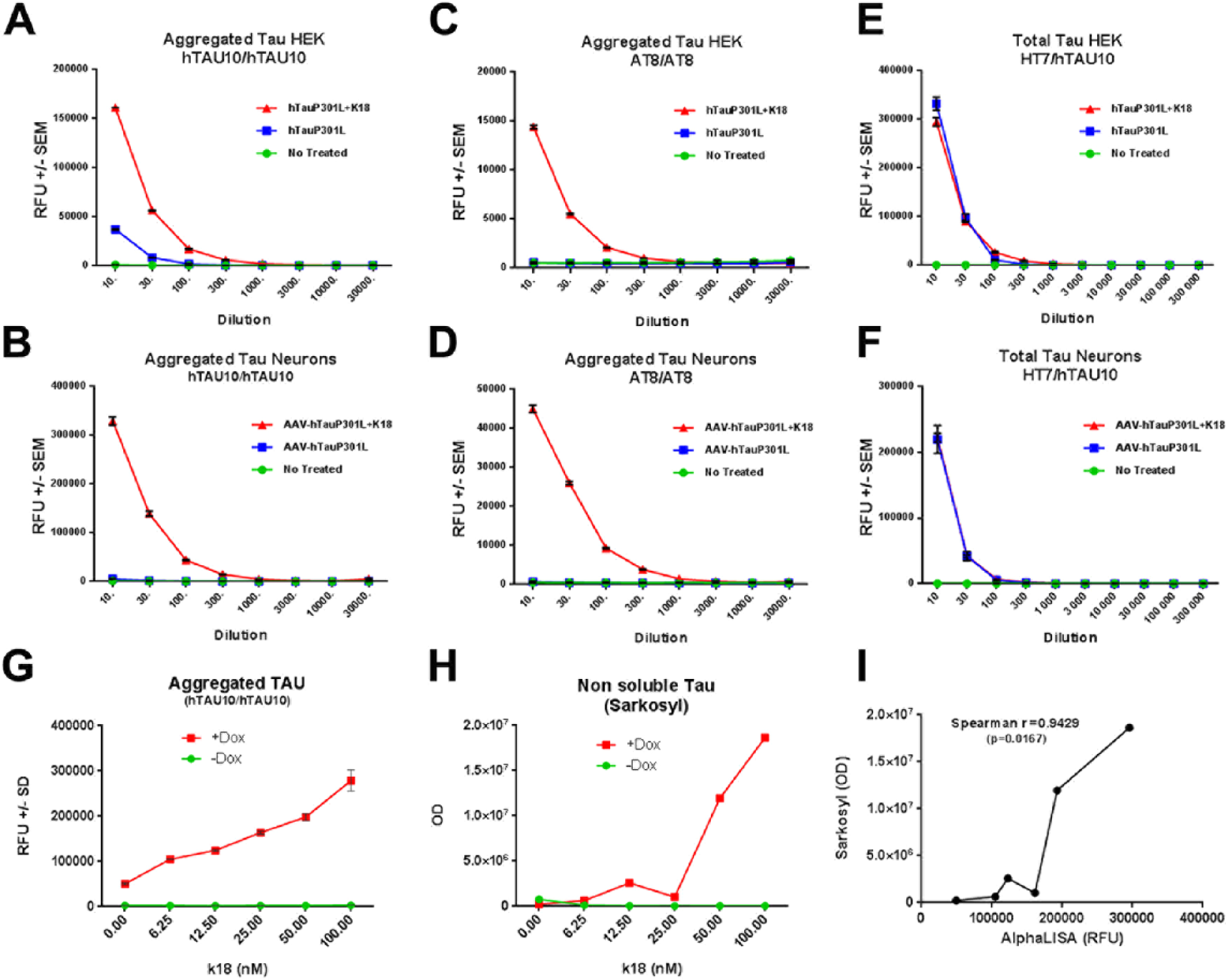
AlphaLISA technology for the relative quantification of tau aggregates in vitro.
Antibody validation to quantify aggregated and total tau using two different models,
HEK cells (
One of the most accepted methods to detect insoluble tau is by immunoblots of sarkosyl
extracts.
16
In
order to define the specificity of our assay to detect tau aggregates, we computed the
signal obtained from the hTAU10/hTAU10 AlphaLISA assay with the densitometric values
obtained from sarkosyl extracts. To this end, we performed a K18 dose–response experiment
seeding HEK cells expressing human P301L mutant tau under the doxycycline-inducible
promoter (
Fig. 3G–I
).
As shown in
Figure
3G
,
H
,
the increase in aggregates is detected by both AlphaLISA (
Fig. 3G
) and WB in a dose-dependent
manner. The correlation between the two signals is significant as measured by the
Spearman’s coefficient (r = 0.9429, p = 0.0167;
Fig. 3I
). Importantly,
the total levels of tau expressed stay relatively constant despite the increase of K18
(
Assay Optimization: Defining Noise Signal Values, Sampling Time, and Protocol Length
Next, we designed a set of experiments to define the baseline values for AlphaLISA assays when using human cortical neurons in which endogenous tau is being expressed. All control conditions, consisting of seeding but nontransduced cells, or only cells transduced with viral vectors overexpressing either EGFP or P301L but without K18 seeding, provided baseline values of approximately 1300 counts (p < 0.0001; Fig. 4A ) for hTAU10/hTAU10 AlphaLISA. Detection of total tau using HT7/hTAU10 AlphaLISA confirmed that baseline values of tau increased by more than 50% only in cells that had been transduced with P301L overexpressing viruses (p < 0.0001; Fig. 4B ). After defining negative values of the assay, we then performed time course experiments, which would additionally provide us with an optimal sampling time point to detect tau aggregation. The optimal sampling time point was established at 35 days in vitro (DIV), corresponding to the maximum signal obtained in the shortest time for hTAU10/hTAU10 AlphaLISA ( Fig. 4C ). The time-dependent increase in signal for aggregated tau does not correlate with the increase in total tau, as the values for HT7/hTAU10 do not change significantly after 28 DIV ( Fig. 4D ). Figure 4E , F shows dose–response experiments when using different K18 seed concentrations in 96-well plates. Importantly, this increase in signal is detected both with hTAU10 ( Fig. 4E ) and with AT8 ( Fig. 4F ) antibodies, indicating that the aggregates are also hyperphosphorylated. Combining the optimal sampling time (35 DIV) with a seed concentration of 133 nM in 384-well plates, the assay has a Z′ factor of 0.52 in the hTAU10/hTAU10 ( Fig. 4G ). The final step for assay optimization involved a reduction in duration of incubation by changing the time point of transduction and seeding. We therefore transduced cells 4 days after plating and seeded them 3 days later. As shown in Figure 4H , our results demonstrate that by changing these parameters, we are able to reduce the sampling point by more than 12 days to 22 days after thawing.
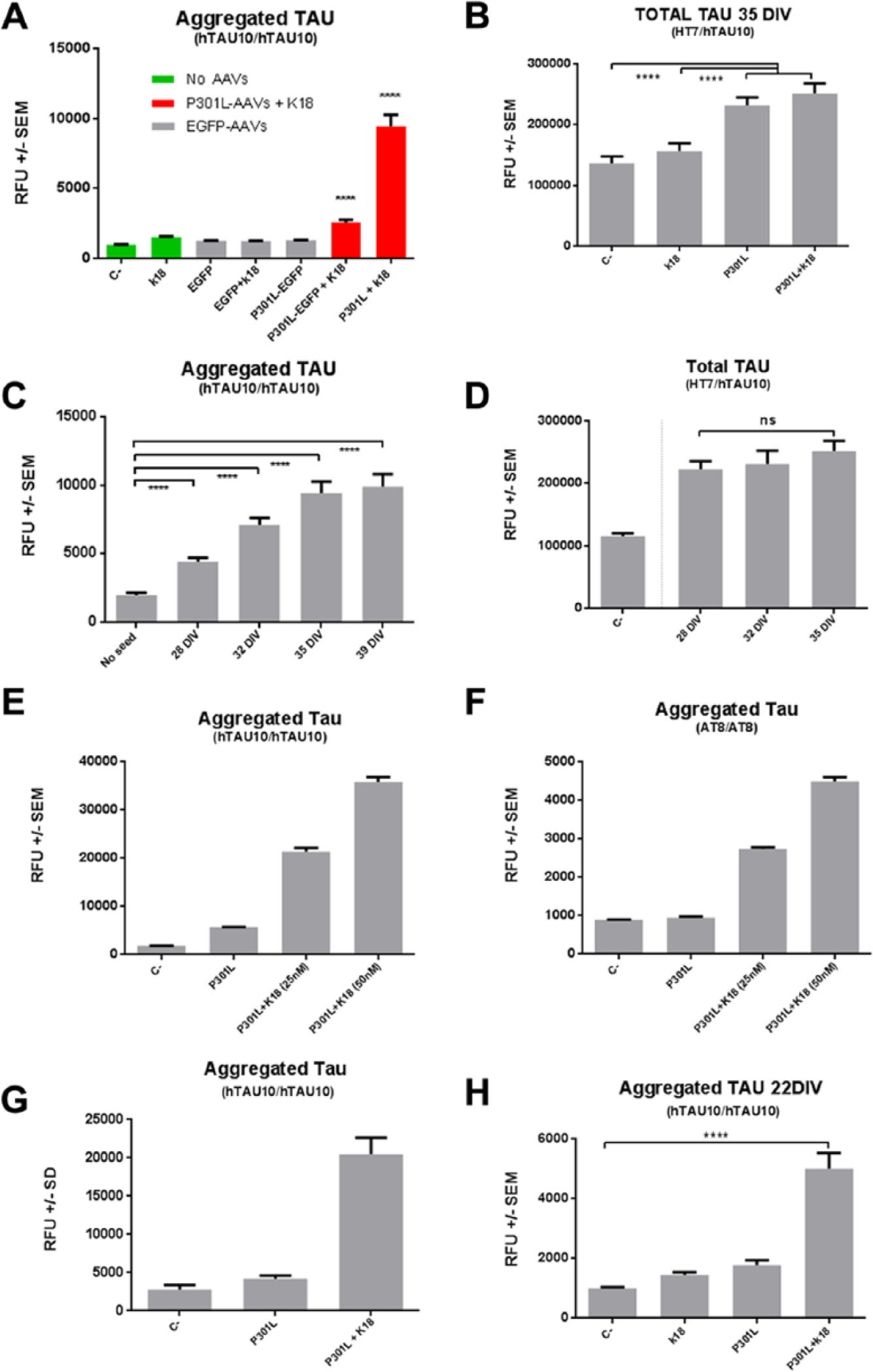
Validation of AlphaLISA technology for the relative quantification of tau aggregates
in human neurons cultured in Matrigel. (
Taken together, our results indicate that high levels of aggregated tau are only detected in cells that have been previously transduced and treated with K18 seeds, suggesting that (1) endogenous levels of wild-type tau and (2) overexpression of mutant P301L tau alone are not sufficient to trigger aggregation under these culture conditions.
Reproducibility of the Assay
After validating iPSC-derived neurons cultured in Matrigel as a suitable model of tau aggregation, we next challenged the robustness of the assay by changing key parameters. We first analyzed the “line-to-line” variability by testing an additional commercially available cell line provided by a different supplier (ChiPSC6b, Cellectis). We differentiated, transduced, and seeded NPCs according to our initial experimental design and sampled after 35 DIV. As shown in Figure 5A , we confirmed that only transduced and seeded neurons display a significant increase in tau aggregation as quantified by hTAU10/hTAU10 AlphaLISA (p < 0.0001). Finally, we further verified the robustness of the assay by testing it in a different cell culture format and multiple users. First, the experimental setup was adapted to 96-well plates in which 20,000 or 30,000 cells were plated per well. The transduction and tau aggregate seeding time point was following the 384-well format procedure and the assay performed by different users. hTAU10/hTAU10 AlphaLISA results demonstrate that we retain tau aggregation in a 96-well plate format, which is reproduced across users ( Fig. 5B ). Importantly, AlphaLISA positive values raised up to 60-fold when increasing the number of plated cells, providing key information about how to increase the dynamic range of the assay. Although there is a degree of variability in the total values obtained across the different users, a minimal difference of 10-fold is detected between positive values and the negative controls. Thus, we demonstrate that it is possible to trigger tau aggregation in cell lines generated in different laboratories regardless of genetic background or reprogramming method. Furthermore, we demonstrate that the high dynamic range of the assay can be reproduced across users, underlining the robustness and reproducibility of this miniaturized tau aggregation model.
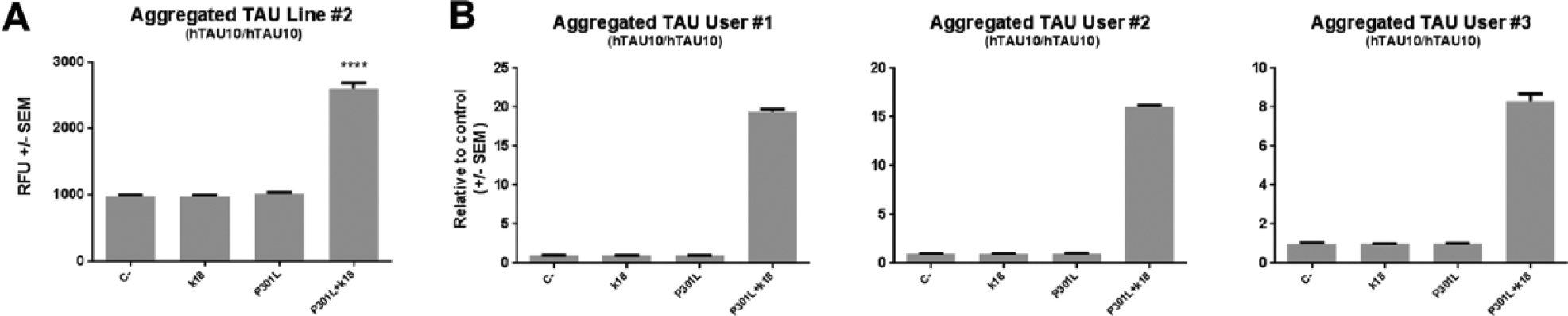
Tau aggregation assay is reproducible among different lines and across users.
(
Discussion
Adapting Stem Cell Technology to Drug Development in Neuroscience
iPSCs can be differentiated into cortical neurons, providing a unique biological substrate for in vitro disease modeling.7,8 However, one of the major limitations of this technology is the lack of a robust standard procedure that allows data reproducibility among laboratories. Dual SMAD inhibition is a highly reproducible method used to trigger rostralization and dorsalization of neuronal progenitors derived from stem cells. 25 Most of the published protocols combine SMAD inhibition with a variety of growth factors and supplements, such as BDNF, GDNF, or NT3, necessary for the final differentiation process. Despite sharing key basic similarities, the wide variety of differentiation protocols used among laboratories makes comparing and reproducing data difficult. In addition to media optimization, different plate coating methods have also been applied, with polyornithine/laminin (POL) being the most widely used. The POL coating method has been highly valuable when using mouse primary cultures, and has also been translated to iPSC neuronal differentiation protocols. However, batch-to-batch variability and the degradation of laminin during the long period of time required for differentiating human cortical neurons (50–70 days) raise concerns about its suitability for use at the industrial scale. Both media composition and cell culture format require an assay-by-assay optimization that needs to be standardized and validated for the scientific community. Recently, multiple laboratories have explored new approaches to overcome these limitations. Interestingly, the use of biomaterials such as hyaluronic acid, 26 Matrigel, 12 or Geltrex 27 offers new options for culture conditions that could help overcome these limitations. Biomaterials provide not only additional adhesion proteins and growth factors that accelerate maturation of functional neurons, but also mechanical support (as scaffold) that crucially contributes to keep culture stability and synaptic connectivity between cells. 28 A particular extra advantage of the use of embedding materials to study tau pathology is that dying cells in the process of releasing pro-aggregant tau species (or toxic insults accelerating neuronal death) will remain in the vicinity of neighboring cells, better recapitulating what occurs in the disease brain. Importantly, from the perspective of scalability and automation, embedding materials also provide protection to the shearing stress caused by media change and plate handling, which might eventually contribute to reduce potential sources of variability.
In this study, we have combined the classical dual SMAD inhibition protocol alongside Matrigel embedding with the objective of generating a standard procedure for the differentiation of cortical neurons that can be reproduced among different users, across laboratories, and using different iPSC lines. In addition, we provide evidence that this protocol works under different cell culture formats (12-, 96-, and 384-well plates) at different cell densities (300,000, 20,000, and 10,000 cells, respectively). Although we have demonstrated the reproducibility of the assay when using Matrigel, the eventual variability among batches and the elevated production costs might represent potential hurdles in the assay. Matrigel is a protein mixture obtained from Engelbreth–Holm–Swarm (EHS) mouse sarcoma cells, and the cell culture conditions of source cells may alter protein composition and thus introduce batch-to-batch variability. We have addressed these potential variability issues by using a type of Matrigel that has reduced growth factor content and is regularly used for the culture and maintenance of iPSCs. At least five different batches of Matrigel were used in this study, all of them yielding comparable results regardless of the user or the cell line used. By optimizing the working dilution to 1:15 and the culture format (384-well plates/10,000 cells per well), we have considerably reduced the amount of Matrigel and material required per assay. Currently, the priority in the neuroscience/stem cell research field is to minimize the sources of variability in the differentiation media composition and to accelerate the neuronal maturation process. Although we successfully reproduce tau aggregation in human neurons, we have not been able to generate a substantial number of cortical neurons displaying spontaneous activity at the latest time point analyzed (39 days postthawing). We are currently optimizing culture conditions, as the physiological relevance of the model would greatly benefit from assaying synaptically active and synchronized neuronal cultures. As recently described, new formulations of the neuronal differentiation media may help to adapt the assay accordingly. 29 Similarly, the optimization of chemically defined biomaterials will undoubtedly contribute to improve the technology in the same way, reducing further the eventual sources of variability.
New Tau Aggregation Model Using Human Cortical Neurons
A high-throughput-compatible assay to detect and quantify tau aggregation in human neurons does not exist yet, and current methods used (such as WB of sarkosyl/Triton-insoluble extracts) are tedious and require large amounts of material. Intracellular tau aggregation has traditionally been triggered by combining overexpression of mutant forms of human tau with the addition of paired helical filaments (PHFs) derived from AD brain extracts or tau aggregates produced in vitro. Few publications have shown the presence of aggregates in stem cell–derived neurons.12,17,30 Contrary to what has been described, 13 neurons cultured in Matrigel using our experimental conditions do not express 4R, but only 3R tau forms. 3R forms are less prone to form aggregates than the 4R variants that carry mutations associated with familial tauopathies. 31 We provide evidence that K18 seeding of 3R tau-expressing cells cultured in Matrigel does not trigger tau aggregation. Due to the low expression levels of endogenous 4R tau, we induced neuronal-specific overexpression of the human 4R mutant tau form P301L using adenoviral vectors under the control of the human synapsin promoter. Under these conditions, we observed a significant increase in the levels of phosphorylated mutant protein in our cultures, as detected by AT8 immunobloting. Accordingly, our data reproduce previously described effects observed in mouse primary cultures. 14 Only K18-seeded and transduced cells displayed significant values of tau aggregation, suggesting that (1) a sufficiently high and mutant protein expression is needed before aggregation can be observed and (2) P301L tau does not aggregate without adding tau seeds. Future experiments using more specific antibodies at different time points after neuronal induction may be performed to further characterize the different tau isoforms endogenously expressed by human cortical neurons in vitro. As for the development of alternative tau aggregation assay, cells carrying a combination of mutations known to alter the ratio and expression levels of 4R and 3R tau in addition to the pro-aggregant mutations would be a potential alternative to the described overexpression system.32,33
Robust Assay for the Relative Quantification of Tau Aggregates in a Scalable/Automatable Culture Format
It has been recently shown that it is possible to reproduce in vitro the key hallmarks of AD pathology using iPSC-derived neurons cultured in Matrigel (i.e., amyloid deposition and MC-1 positive immunoblots).12,13 Although highly innovative and disease relevant, the model proposed is rather complex (several fluorescence-activated cell [FAC] sorting and subplating steps) and requires 10 weeks for the detection of these pathological features. While an important proof of concept, the lack of scalability and the excessive length of the protocol make it difficult to adapt to drug discovery programs or automated platforms.
Here we provide a robust, scalable, and simple high-throughput-compatible assay that recapitulates several features already described, 13 reducing the time required for the formation of intracellular tau aggregates and allowing its relative quantification. Our assay requires only one transduction and one subplating step coupled to an in-well solubilization and readout with no washing steps necessary, which reduces potential loss of material/tau aggregates during the handling process. First, we demonstrate that our model works in a wide variety of formats ranging from 384- and 96- to 12-well plates. In addition, by modifying the time of transduction and seeding, we also demonstrate that tau aggregation can be triggered in human neurons as early as 22 days after subplating neuronal progenitor cells. Under these different conditions, we confirmed that the assay is reproducible at different cell densities, across users, and using different iPSC lines providing a consistent and ample dynamic range that reaches up to 60-fold. We did not perform dose–response experiments using inhibitors of tau aggregation, as reference compounds known to modulate tau aggregation/clearance do not exist yet. We performed, however, K18 dose–response experiments that demonstrate the sensitivity of the assay to the aggregation triggered by seeds, and allowed the identification of optimal seed concentration (50 and 133 nM for 96- and 384-well plates, respectively). Based on the signal-to-basal ratio values obtained in the small data set presented in this study, the proposed assay meets the criteria to be considered compatible for high throughput, as the Z′ factor was 0.52 in the hTAU10/hTAU10 assay in 384-well plates. 34 We hypothesize that the Z′ factor of the assay will certainly improve if larger data sets are used. Finally, the variability detected in AlphaLISA values between users can be explained by differences in cell number and reactive mixture preparations, and the batch of antibodies or components of the buffers. Nevertheless, the consistency of the dynamic range obtained on each user’s data set confirms the robustness of the model.
In conclusion, we have established a human neuron–specific tau aggregation model using Matrigel as embedding material that is suitable for both basic research and drug discovery purposes and that may overcome challenges related to standard differentiation protocols by providing the neurons extra physical support and protection. Using tau as an example, we demonstrate that AlphaLISA technology is a reliable method for the detection and relative quantification of protein aggregates in a fast and scalable manner that is not impacted by the use of scaffolding materials. Our proof-of-concept study allows us to suggest that this technology will be extremely useful for studying other protein aggregation diseases such as Huntington’s disease, ALS, or Parkinson’s disease, provided the availability of high-affinity antibodies.
Footnotes
Acknowledgements
We would like to acknowledge I. Powley, S. Fernandez, and J. Talpos for their critical comments when preparing this manuscript. We also thank members of Janssen NS department in Beerse for the technical support and helpful discussions, especially F. Pestana, M. Mercken, and K. Van Kolen. The research leading to these results has received funding from the Innovative Medicines Initiative Joint Undertaking under grant agreement number 115582 (EBiSC), the resources of which are composed of financial contribution from the European Union’s Seventh Framework Programme (FP7/2007-2013) and EFPIA companies’ in-kind contribution.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.