
Abstract
Rift Valley fever virus (RVFV) is an emerging virus that causes serious illness in humans and livestock. There are no approved vaccines or treatments for humans. The purpose of the study was to identify inhibitory compounds of RVFV infection without any preconceived idea of the mechanism of action. A whole-cell-based high-throughput drug screening assay was developed to screen 28,437 small chemical compounds targeting RVFV infection. To accomplish both speed and robustness, a replication-competent NSs-deleted RVFV expressing a fluorescent reporter gene was developed. Inhibition of fluorescence intensity was quantified by spectrophotometry and related to virus infection in human lung epithelial cells (A549). Cell toxicity was assessed by the Resazurin cell viability assay. After primary screening, 641 compounds were identified that inhibited RVFV infection by ≥80%, with ≥50% cell viability at 50 µM concentration. These compounds were subjected to a second screening regarding dose–response profiles, and 63 compounds with ≥60% inhibition of RVFV infection at 3.12 µM compound concentration and ≥50% cell viability at 25 µM were considered hits. Of these, six compounds with high inhibitory activity were identified. In conclusion, the high-throughput assay could efficiently and safely identify several promising compounds that inhibited RVFV infection.
Introduction
Rift Valley fever virus (RVFV) belongs to the Phlebovirus genus of the Bunyaviridae family. It is an emerging virus that causes serious illness in humans and livestock in Africa and the Arabian Peninsula. RVFV is transmitted by mosquitoes and by direct contact with infected animals or animal products. 1 Human RVFV infection ranges from a mild illness associated with fever and liver abnormalities to much more severe, with symptoms such as retinitis, encephalitis, and hemorrhagic fever. The case fatality rate has varied between 2% and 30% in outbreaks. 2 In livestock, RVFV causes hepatitis, hemorrhagic fever, and abortion with a 100% mortality rate in newborn sheep, goats, and cattle. 3 The ability of RVFV to infect a broad range of mosquito species and its capability to cause major epidemics among humans and livestock demonstrate a serious public health concern all over the world. RVFV is classified as a category A pathogen by the National Institutes of Health in the United States. There are no approved therapeutic or prophylactic treatments for humans. Ribavirin has been recommended for treatment of patients; 4 however, it has only been shown to display partial protection in animal models. 5 The efficacy in humans remains to be proven and there are undesirable side effects. 6
RVFV is an enveloped, negatively stranded RNA virus composed of three RNA segments, complexed with the nucleocapsid (N) protein. Each segment is connected to an RVFV L-protein with RNA-dependent RNA polymerase activity. The first step of RVFV replication involves an interaction between the viral glycoproteins Gn and/or Gc and the cell surface receptors. After attachment, RVFV has been shown to use dynamin-dependent caveola-mediated endocytosis 7 for entry, and then negative-sense viral RNA is transcribed to mRNA by the L-protein. After translation of viral mRNA, RNA replication takes place, followed by budding into the Golgi cisternae, where Gn and Gc are located. Finally, cytoplasmic vesicles containing viruses fuse with the plasma membrane to release mature virions. NSs, the RVFV nonstructural protein encoded from the S-segment, disrupts the host antiviral interferon response and induces downregulation of the double-stranded RNA-dependent protein kinase (PKR).8,9 The other nonstructural virus protein, NSm, appears to have a functional role in the vector competence of mosquitoes for RVFV at the level of the midgut barrier, 10 and it is also reported to suppress apoptotic pathways in host animal cells after infection. 11
High-throughput screening (HTS) of large compound libraries to identify inhibitors of pathogen infection is now a relatively common method, and most HTSs have a preconceived idea of the mechanism of action. Cell-based approaches have less limitations, albeit they are usually more labor-intensive. Taking this into consideration, a whole-cell-based HTS assay was developed to screen a large chemical compound library with the aim to achieve both speed and robustness. The method allowed for identification of any compounds that had an overall phenotypic effect on RVFV infection, without having any aforementioned knowledge of its target or mechanism of actions. A recombinant replication-competent RVFV, where the NSs gene was replaced with a fluorescent reporter gene, was developed by reverse genetics. The recombinant virus was used to identify compounds that directly or indirectly inhibited the infection of RVFV. Here we describe HTS of approximately 28,000 compounds for antiviral activity against RVFV infection. Six compounds with low cell toxicity that inhibited RVFV infection at the micromolar concentration were identified.
Materials and Methods
Compound Collection
The 28,437 compounds in the collection used for the primary screen were from the Chemical Biology Consortium Sweden (CBCS) collection and represent the current primary screening set offered by CBCS. All compounds were dissolved in DMSO at 10 mM and were stored under appropriate conditions (REMP system, REMP, Oberdiessbach, Switzerland). Prior to HTS, the compounds were transferred by a Labcyte Echo 550 (Labcyte, Inc., Sunnyvale, CA) at the CBCS (Karolinska Institute, Stockholm, Sweden) into 384-well polypropylene plates and shipped to the CBCS node in Umeå.
Cell and Culture Conditions
Human lung adenocarcinoma basal epithelial cells, A549, were cultured in cell culture medium (Dulbecco’s Modified Eagle’s Medium [DMEM], Sigma-Aldrich, St. Louis, MO) containing 0.75 g NaHCO3/L, 20 mM HEPES (4-[2-hydroxyethyl]-1-piperazineethanesulfonic acid) (EuroClone, Milan, Italy), penicillin G (100 IU/mL), and streptomycin sulfate (100 µg/mL) combined (1X PEST, Gibco, Carlsbad, CA), and 5% fetal bovine serum (FBS, Gibco) at 37 °C. For virus infection, cell maintenance medium was used containing the same components, except at a lower FBS concentration (2%).
Generation of Recombinant RVFV and Assay Optimization
Recombinant RVFV expressing the far-red fluorescent protein Katushka 12 instead of the NSs protein (rRVFVΔNSs::Katushka) was constructed and generated as described for the GFP-expressing virus rRVFVΔNSs::GFP. 13
To obtain a maximum and consistent fluorescent signal in the assays, the number of cells per well, the multiplicity of infection (MOI) of the recombinant virus, and the incubation time after infection were optimized for both the primary and secondary screening assay and the dose–response assay. For the primary and secondary screening optimization, the kinetic analysis was performed in a Tecan Infinite 200Pro Plate reader (Tecan Ltd., Männedorf, Switzerland) programmed to hold 37 °C and read fluorescence (excitation at 588 nm and emission at 635 nm) every hour for 40 h. For the dose–response optimization, it was important to generate a sufficient number of discernible fluorescent cells. Thus, different MOIs were experimentally analyzed using the Trophos plate runner HD (Trophos, Roche Group, Marseille, France), as described below. After optimization, the incubation time was set to 16 h and the virus MOI and cell number varied according to assay, as described below.
The primary screening was performed under BSL-2 conditions, approved by the department biosafety committee. The virulence gene RVFV NSs was deleted in the recombinant RVFV and replaced with the reporter gene. The modified virus cannot replicate in immunocompetent cells and was described to be nonpathogenic to humans and animals.8,14 However, results published just after the primary and secondary screenings were completed, showed that RVFV lacking NSs is 100% lethal in immunocompetent mice via aerosol exposure. 15 That led us to perform the remaining experiments in BSL-3 conditions (approved by the department biosafety committee and the Swedish Work Environment Authority).
Primary Screening of Compounds for an Antiviral Effect
The assay/cell plates were prepared by seeding 5000 A549 cells in 40 µL of cell culture medium to each well of a 384-well plate (3683 Corning CellBIND surface black plate with transparent bottom) using a Matrix WellMate multipipet dispenser (Thermo Fisher Scientific, Langenselbold, Germany) machine. The assay/cell plates were incubated for 24 h at 37 °C in a 5% CO2 incubator. We used a robust semiautomated approach to solve the automation needed to adapt the suggested assay for the HTS. To remove media from the assay/cell plate and add compounds to the cells, a Beckman Coulter (Brea, CA) NxP system was used with a 384-well pipetting head. Cell maintenance medium (20 µL) was added to each well of the compound plate, already containing 0.2 µL (10 mM) of library compound. After removing 35 µL of medium from the assay/cell plate, 20 µL of each compound dissolved in cell maintenance medium was added. Then, 15,000 plaque-forming units (PFU) of rRVFVΔNSs::Katushka virus (MOI 3), in a volume of 15 µL, were added to each well of the assay/cell plate with a Matrix WellMate dispenser, to give a final compound concentration of 50 µM. Sixteen negative control wells containing 5000 A549 cells in 40 µL of cell maintenance medium were included in each plate. In addition, 16 positive control wells containing 5000 A549 cells in 25 µL of cell maintenance medium, and 15,000 PFU of rRVFVΔNSs::Katushka virus (MOI 3) in a volume of 15 µL/well were included. The assay plates were incubated for 16 h at 37 °C in a 5% CO2 incubator. Katushka expression was assessed as the fluorescence intensity at 588 nm (emission spectra 635 nm) by using a Synergy H4 hybrid multimode microplate reader equipped with the BioStack microplate stacker (BioTek Instruments, Inc., Winooski, VT). As a positive control, ribavirin was added and virus infection was inhibited at 20 µM (74%) and 10 µM (43%).
Cell Toxicity Assay
After measuring the fluorescence intensity, the cellular toxicity of screened compounds was assessed by using the Resazurin cell viability assay (Sigma-Aldrich). This assay measures the metabolic activity of living cells and is based on the oxidoreduction of the nontoxic indicator blue dye resazurin. Viable cells with active metabolism can reduce resazurin into resorufin, which is pink and fluorescent. 16 To analyze cell survival/toxicity, 10 µL (40 µM final concentration) of resazurin was added per well, incubated for 3–4 h at 37 °C in a 5% CO2 incubator, and the fluorescent intensity was measured at 535 nm (emission spectra 590 nm). The Resazurin cell viability assay was also used to evaluate the cytotoxicity of DMSO. A549 cells were treated with DMSO at different concentrations, ranging from 0.2% up to 2%. DMSO cytotoxicity was then compared between cells treated with DMSO and the cells without any DMSO treatment.
Data Analysis and Hit Scoring
The primary screen had two readouts for each assay plate. The definition for a compound to be active was the ability to reduce the expression of Katushka far-red fluorescence by 80% and produce more than 50% fluorescence intensity from the cell viability assay. Chemical structure and assay readout were matched using the MScreen informatics system (University of Michigan), 17 in addition to Excel with the definition of active compound as threshold. The average Z′ (calculated as defined by Zhang et al.) 18 was 0.54 and the median was 0.55. Factors responsible for low Z′ values (9 of 56 plates had a Z′ below 0.4) were identified. In a few cases, cells were limited for a given set of plates, resulting in a characteristic pattern in the final wells (<6) dispensed into. In some cases, clogging of pipette tips due to long intervals between tip-box changes was a factor. Liquid handling and logistics improved along the screening campaign. Consequently, in the final days of the campaign, the plates had an average Z′ of 0.82 (CV = 6%). Thus, the factors contributing to the poor Z′ were due to technical origin and did not reflect the biology and assay design.
Secondary Screening
Compounds that fulfilled the selection criteria to be active in the primary screening were further analyzed in a dose-dependent manner to validate the hits. The selected compounds were prepared in 384-well plates at the CBCS node at Karolinska Institutet (Stockholm). The compounds were plated at four different concentrations (25, 12.5, 6.25, and 3.12 µM), enabled by using an Echo 550 (Labcyte). The same procedure, methods, and protocols as for the primary screen were then used.
Dose–Response Analysis
The hit compounds identified from the secondary screening were further analyzed with an assay based on fluorescent intensity of individual infectious foci. The day before infection, 10,000 A549 cells per well were seeded in 96-well black plates with transparent bottoms (Corning). The next day, compounds were twofold serially diluted in six steps from 12.5 µM to 0.39 µM and mixed with 1000 PFU of rRVFVΔNSs::Katushka virus in a total volume of 100 µL. Then, the growth medium was removed from the cell plate and 100 µL of compound/virus was added to the cells for each compound concentration (MOI = 0.1). The plates were incubated for 16 h at 37 °C in a 5% CO2 incubator. After incubation, the medium was removed and cells were fixed with 1% paraformaldehyde for 1 h at room temperature and washed with phosphate-buffered saline (PBS). The Trophos plate runner HD (Trophos, Roche Group) identified all individual Katushka-expressing cells and quantified the fluorescence intensity in each well. The intensity corresponded to the infection rate, and from that the IC50 value was calculated.
Translational Inhibition Analysis
Hek293t cells (10,000 per well) were seeded in 96-well plates 1 day before transfection. An SV40 expression plasmid, encoding the Renilla luciferase gene (30 ng/well), was transfected by using the Genejuice transfection protocol (Merck Millipore, Merck KGaA, Darmstadt, Germany). Plasmid and transfection reagent were removed after 2.5 h, and 100 µL of cell culture medium containing hit compounds of different concentrations (10, 5, 2.5, and 1.25 µM) was added. Cycloheximide was used as the control. After 24 h at 37 °C, the luciferase expression was analyzed by the Renilla luciferase assay system (Promega Corporation, Madison, WI) in a Tecan Infinite 200Pro Plate reader (Tecan). All experiments were performed in triplicate.
Plaque Reduction Neutralization Test (PRNT)
Vero B4 cells (120,000 per well) were seeded in 12-well plates 1 day before infection. Cells were infected in triplicate with RVFV clone 13 (40 PFU/well) 19 and incubated for 1.5 h at 37 °C. Virus was removed, 600 µL of Avicel overlay (300 µL of 2 × cell culture medium + 300 µL of 2.4% Avicel [FMC BioPolymer, Philadelphia, PA]) containing hit compounds (12.5 and 3.12 µM) was added, and the plate was incubated at 37 °C for 72 h. Then, the overlay was removed and the wells were washed with PBS. To count the plaques, cells were fixed with 4% paraformaldehyde for 1 h at 20 °C and then stained with 1% crystal violet.
Results
DMSO Tolerability Analysis
The compounds analyzed by HTS were dissolved in DMSO. It has been reported that DMSO causes cell cycle arrest at the G1 phase of lymphoid cell lines, having an adverse effect on the cell viability of lung adenocarcinoma CL1–5 cells and membrane permeability in MDCK cells.20,21 Therefore, it was important to measure the effect of DMSO on the viability of A549 cells. The analysis showed that DMSO reduced A549 cell viability in a dose-dependent manner. Cells treated with 2% DMSO showed 53% viability, which gradually increased upon reducing the DMSO concentration. Almost 100% viability was observed when cells were treated with ≤0.4% DMSO. Thus, for the primary HTS assay, 0.1% DMSO was used and the DMSO concentration was never above 0.4% for any experiment including a compound.
Development and Validation of the HTS Assay
For the HTS assay to be robust, several conditions were optimized; cells were infected with rRVFVΔNSs::Katushka at different MOIs, cell densities, and incubation times. The expression of the fluorescent reporter gene increased with time until 18 h after infection, where it reached a plateau, and then slowly increased again, most probably because of newly produced virus ( Fig. 1 ). At 16 h the increase in fluorescence was still linear and the signal was high; thus, it was decided to use 16 h as the time of infection. The amount of cells and virus (MOI) in each well is important to avoid cell overgrowth and to get a consistent virus infection. To obtain a maximum and consistent fluorescent signal, A549 cells were seeded in 384- and 96-well plates at different densities and infected with rRVFVΔNSs::Katushka at different MOIs. The optimization led to an adequate number of cells per well and an MOI that fulfilled the requirements of the assay.
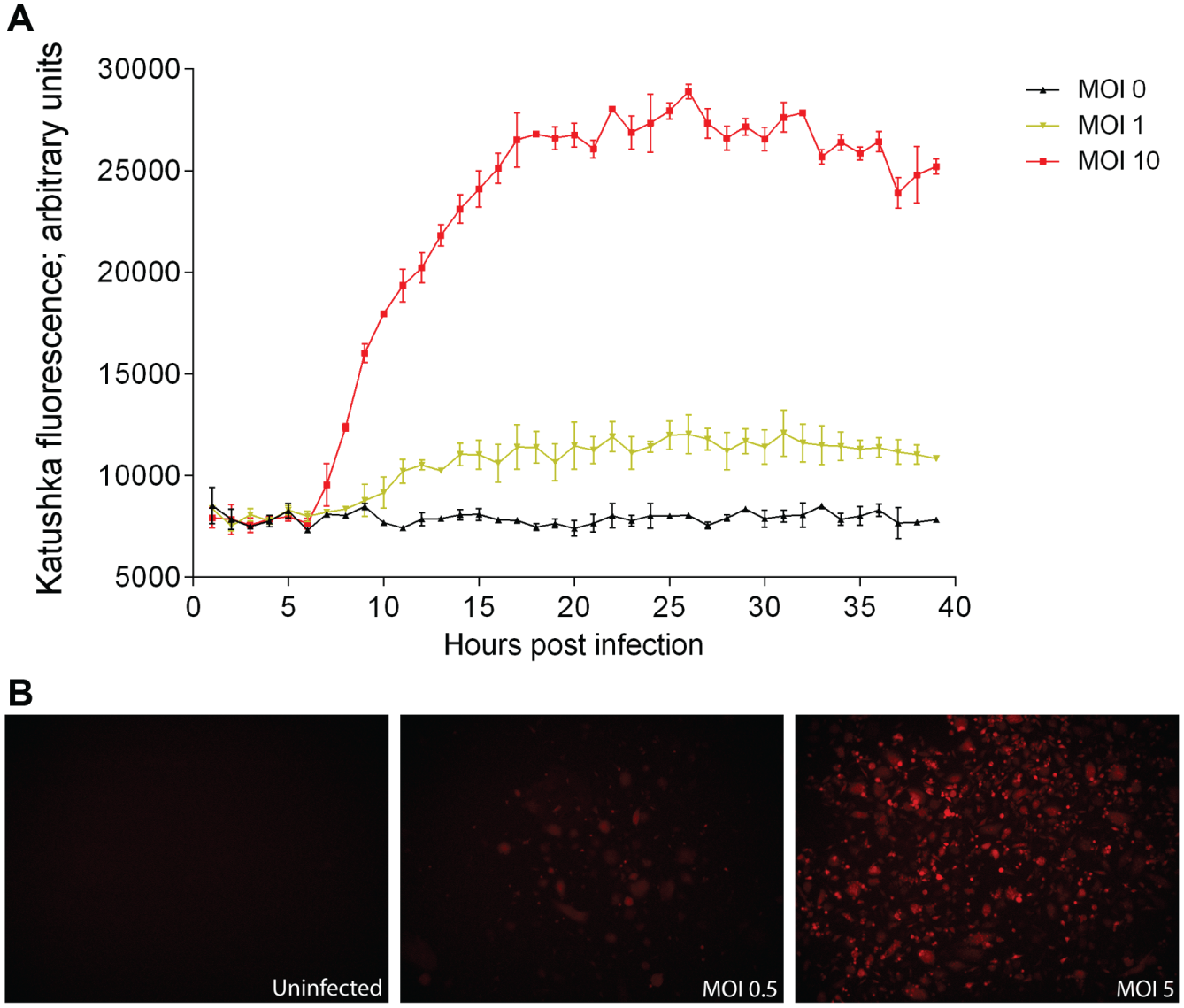
Assay development. (
Primary and Secondary Screenings of the Compound Collection
After assay optimization and validation, we screened the 28,437 compounds at 50 µM for their ability to inhibit far-red fluorescence after infection of A549 cells with rRVFVΔNSs::Katushka. The threshold for a hit compound was a decrease of fluorescent intensity by more than 80% and >50% cell viability. The selection process combined the mentioned threshold and the data visualization (heat map function), all integrated within the MScreen informatics system, in addition to Excel. These criteria were fulfilled for 641 compounds that were identified as primary hits, representing a hit rate of 2.25%. The principle is shown in Figure 2 , where 13 of the 320 compounds tested in the plate showed >80% inhibition of virus infection and 2 of these had <50% cell viability. Thus, for this specific plate, the hit rate was 3.4% (11/320) ( Fig. 2 ).
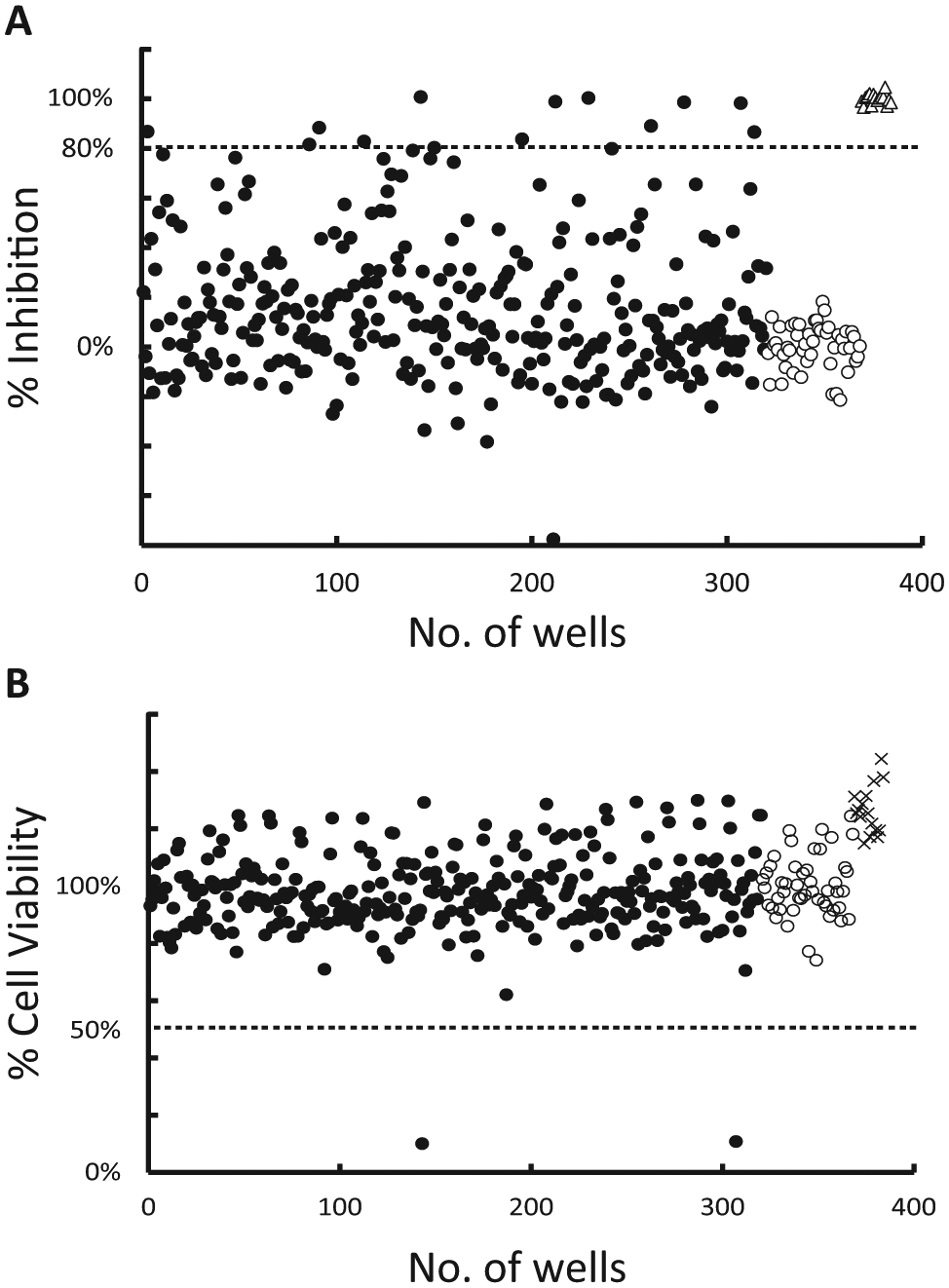
Selection of compounds from primary screening. The analyzed data from a 384-well plate illustrates the principle. The threshold was ≥80% inhibition of Katushka fluorescence intensity and ≥50% cell viability. (
To verify the hits, we performed a secondary screen of the 641 compounds at four different compound concentrations. The selection was based on a dose–response and a cutoff at >60% decrease of fluorescence intensity at the lowest concentration (3.12 µM). This resulted in 63 compounds that passed the threshold of the secondary screen.
Dose–Response, Hit Validation, and Selection
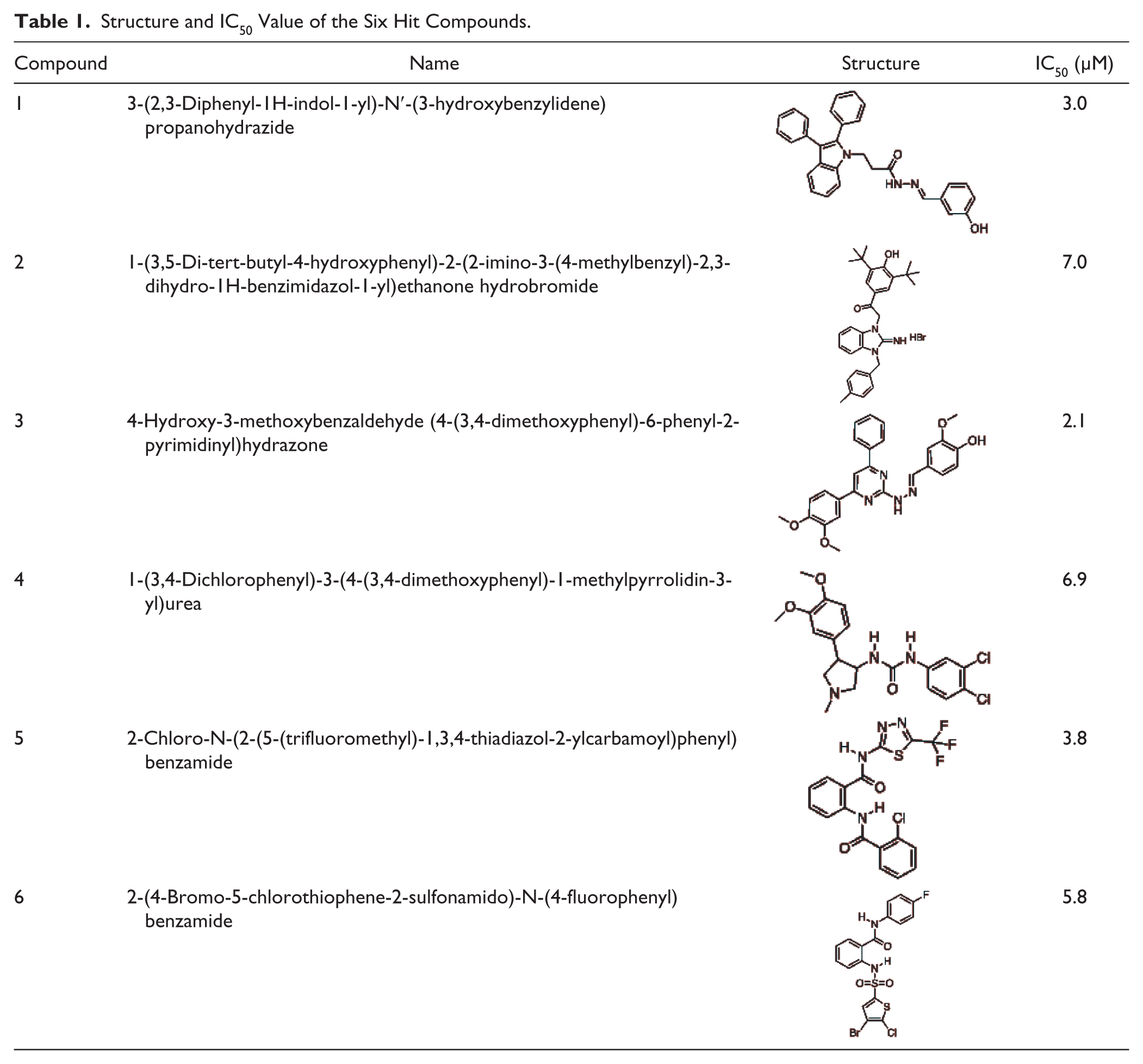
Next, a dose–response validation assay was performed where all individual Katushka-expressing cells in each well were first identified by the imaging instrument and then the fluorescence intensity of the foci was summarized and quantified in contrast to the primary and secondary screenings, where the total fluorescence in the well was measured. The 63 compounds were serially diluted in six steps and the IC50 values were calculated, and from this, 6 compounds with high inhibitory activity (IC50 2.1–7.0 µM) and low cytotoxicity were selected ( Fig. 3 , Table 1 ). To determine whether the decrease in Katushka expression could be due to translational inhibition, we performed a translational inhibition analysis using a transfected expression plasmid. For this, we selected two compounds with the lowest IC50 (compounds 1 and 3, Table 1 ) and one with the highest IC50 (compound 2, Table 1 ). No translational inhibition of Renilla luciferase expression was detected for any of the compounds ( Fig. 4 ). These three compounds were further investigated in an orthogonal assay, where the ability of the compounds to reduce virus plaques in a PRNT assay was evaluated. Compounds 1 and 2 showed 100% reduction at 12.5 µM and 75% and 10% reduction at 3.12 µM, respectively. Compound 3 showed 35% reduction at 12.5 µM and 25% at 3.12 µM.
Structure and IC50 Value of the Six Hit Compounds.
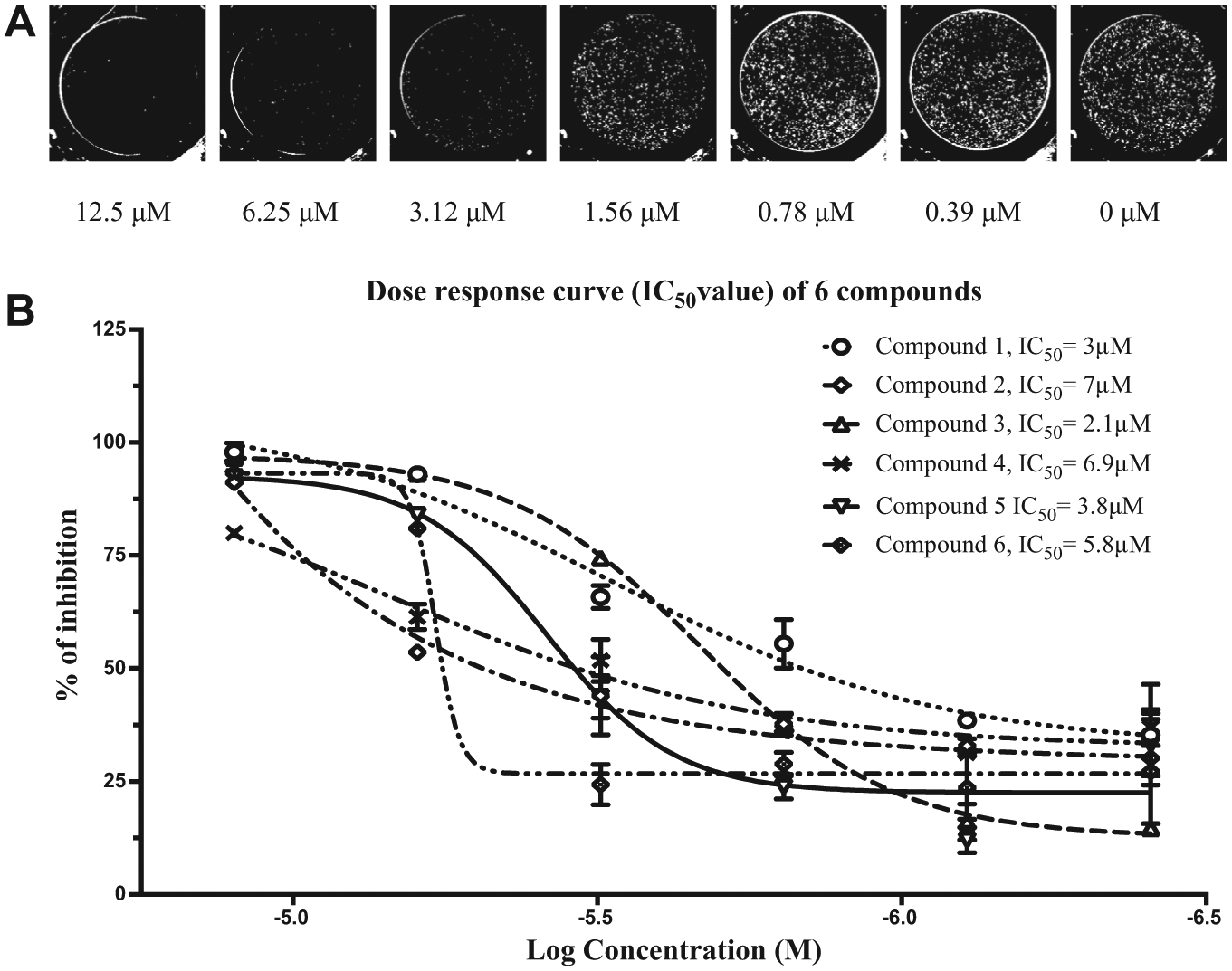
Dose–response assay. (
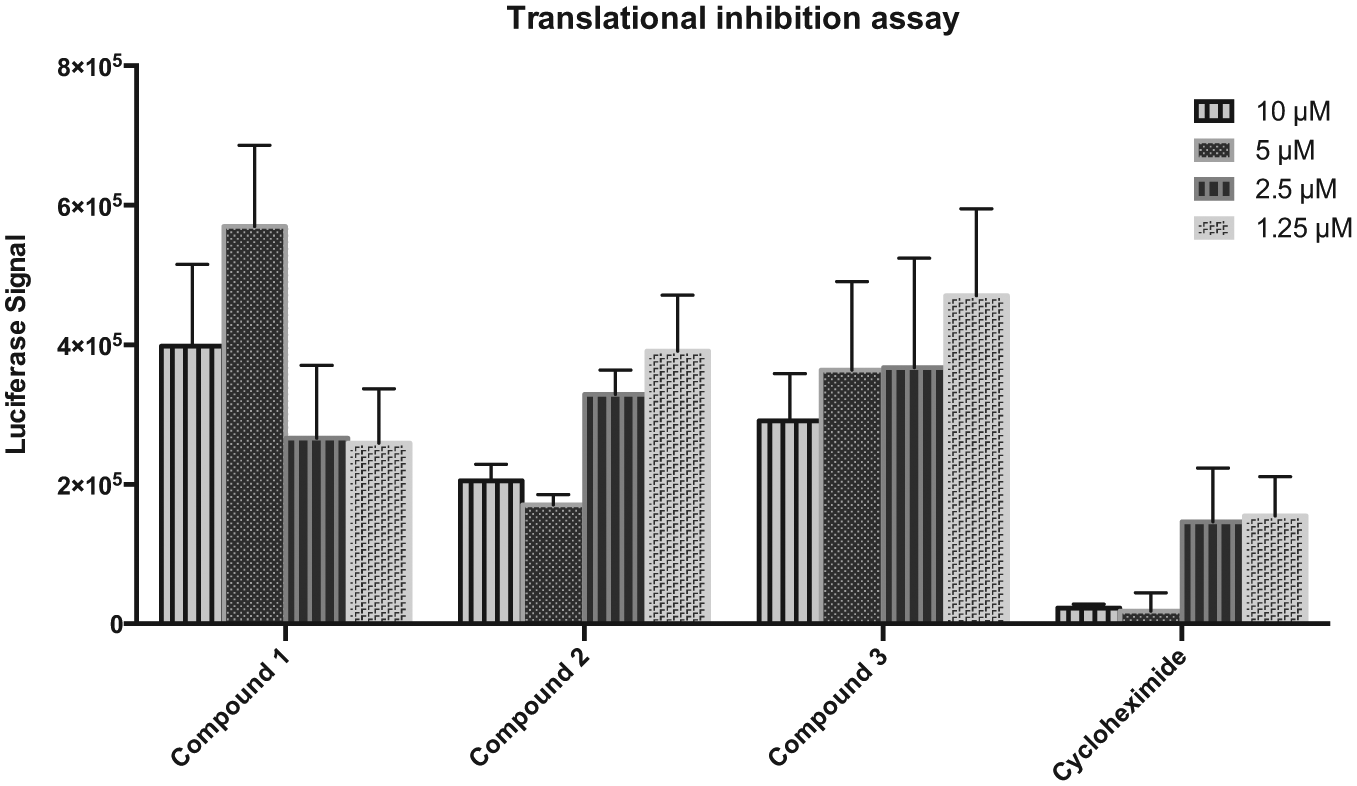
Translational inhibition assay. Compounds and cycloheximide were added at different concentrations to Hek293t cells, followed by transfection with an expression plasmid encoding the Renilla luciferase gene. Luciferase expression was analyzed after 24 h. All data points are the average of the signals from triplicate wells.
Discussion
Rift Valley fever virus is an emerging virus causing severe infection in humans and livestock. There is a need for a safe antiviral treatment that hitherto is lacking. Ribavirin, a nucleoside analog only approved for compassionate use treatment of RVF,4,22 has shown some activity against RVFV in vitro and in vivo.23,24 However, in animal models, ribavirin has caused anemia and teratogenic effects, which has limited the application to compassionate use under investigational new drug protocols.5,6 There are also other potential drugs in the development stage.24,25 Therefore, there is a great need to find new drugs to treat human infection with better efficacy.
Screening strategies are well suited for identification of compounds with potential antiviral activities. Our unique assay is based on a replication-competent RVFV vector where the detection of fluorescence by Katushka expression is directly related to RVFV genome expression. The robustness, simplicity, and direct measurement of inhibition of RVFV gene expression makes it a versatile assay. Replication-competent viruses represent powerful tools for drug discovery since the complete virus replication allows for identification of any compounds with an overall phenotypic effect on virus infection, without knowledge of its target or mechanism of actions. For RVFV there are, as far as we know, no HTSs that have used our strategy. Others have described HTS for RVFV inhibitory compounds with specific targets, such as nucleocapsid protein RNA binding, protease inhibitors, or signal transduction pathways.23,26,27 Moreover, viral constructs modified through reversed genetics enable antiviral HTS for highly pathogenic viruses.28,29
In the present study, we have used our assay to efficiently screen around 28,000 compounds against RVFV infection and several promising inhibitory compounds were identified. The primary screening identified 641 hits (>80% inhibition at 50 µM), and they were further analyzed in a concentration–response assay where 63 candidates (9.8%) fulfilled our stricter threshold criteria of >60% decrease of fluorescence intensity at 3.12 µM. The 63 compounds were further analyzed in another assay where the fluorescence of each infectious focus in the cell layer was quantified. From this assay, we identified six compounds with low cytotoxicity and high inhibitory activity with IC50 at micromolar levels, lower than or similar to those of other suggested inhibitory RVFV compounds. In addition, we performed an orthogonal PRNT assay with three selected compounds. All inhibited RVFV infection, although with different efficiencies, with compound 1 showing the highest inhibition.
None of the six lead compounds belonged in the same compound class. However, there were a few common fragments. Compounds 1 and 3 carry a hydrazone/acyl hydrazone fragment. Compounds 3 and 4 both carry cathecol-related fragments. Compounds shared definite similarities in both number and type of aryl groups as well as linkages, albeit the linkages are reversed. The overall 2D structures of the compounds are not obviously similar. However, one could view all as similar-sized compounds of joined aryl/heteroaryl groups, which makes it conceivable that some, or even all of the compounds, may present a similar 3D motif to a putative target, but this is not known at present. The compounds could potentially affect any step of the RVFV infectious cycle—binding, entry, primary transcription, replication, assembly, egress, and potential reinfection—since the incubation time was set to 16 h for the recombinant RVFV, in accordance with the time course of RVFV clone 13 infection in A549 cells. 30 When we analyzed translational inhibition in three selected compounds, none of them inhibited translation. Further studies are needed to determine the mode of action of the compounds, for example, time of addition assays.
In conclusion, the study demonstrated the application of HTS using a unique whole-cell virus replication reporter gene assay as an effective method to identify novel compounds with potential RVFV antiviral activities. The studies were performed in a 384-well plate format, rendering it possible to apply automation to screen large small-molecule libraries against the important, highly pathogenic RVFV.
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The study was supported by the Medical Faculty, Umeå University; the County Council of Västerbotten; the Laboratory for Molecular Infection Medicine Sweden (MIMS); and the SFB 1021 of the Deutsche Forschungsgemeinschaft.