
Abstract
Autoimmune diseases arise from the loss of tolerance to endogenous self-antigens, resulting in a heterogeneous range of chronic conditions that cause considerable morbidity and mortality worldwide. In Western countries, over 5% of the population is affected by some form of autoimmune disease, with enhanced or inappropriate activation of nuclear factor (NF)–κB implicated in a number of these conditions. Although treatment strategies for autoimmunity have improved significantly in recent years, current therapeutics are still not capable of achieving satisfactory disease management in all patients, and as such, the therapeutic modulation of NF-κB is an attractive target in autoimmunity. To date, no NF-κB inhibitors have progressed to the clinic for the treatment of autoimmunity, but a variety of promising approaches targeting multiple stages of the NF-κB pathway are currently being explored. This review focuses on the current strategies being investigated for the inhibition of the NF-κB pathway in autoimmune diseases and considers potential future strategies for the therapeutic targeting of this crucial transcription factor.
Introduction
Nuclear factor (NF)–κB is an evolutionarily conserved, inducible transcription factor that is ubiquitously expressed in all mammalian cell types and has important roles in the coordination of cell cycle progression, differentiation, migration, and survival. 1 NF-κB is considered the master regulator of the immune response, with the majority of NF-κB target genes encoding proinflammatory proteins, such as cytokines,2–4 chemokines, 5 cell adhesion molecules, and proteins involved in antigen presentation (www.nf-kb.org).
NF-κB is a heterogeneous collection of homo- and heterodimers, the subunits of which comprise members of the Rel family of proteins. The five members of the Rel family (p50, p52, p65 [RelA], c-Rel, and RelB) all share a conserved 300–amino acid Rel homology domain (RHD), which confers the ability of NF-κB members to dimerize and bind DNA. 6 p65, RelB, and c-Rel also contain a C-terminal transactivation domain (TAD) necessary for transcriptional activity. p50 and p52, however, lack this domain and as such do not possess any intrinsic transactivational activity. The members of the Rel family can dimerize to make up to 15 different NF-κB dimers ( Fig. 1 ), with the p50/p65 heterodimer representing the most physiologically abundant of these. 7 The combination of subunits within NF-κB results in distinct DNA sequence specificities and transactivation properties, 8 directing a diverse range of outcomes through the control of the transcription of hundreds of different genes.
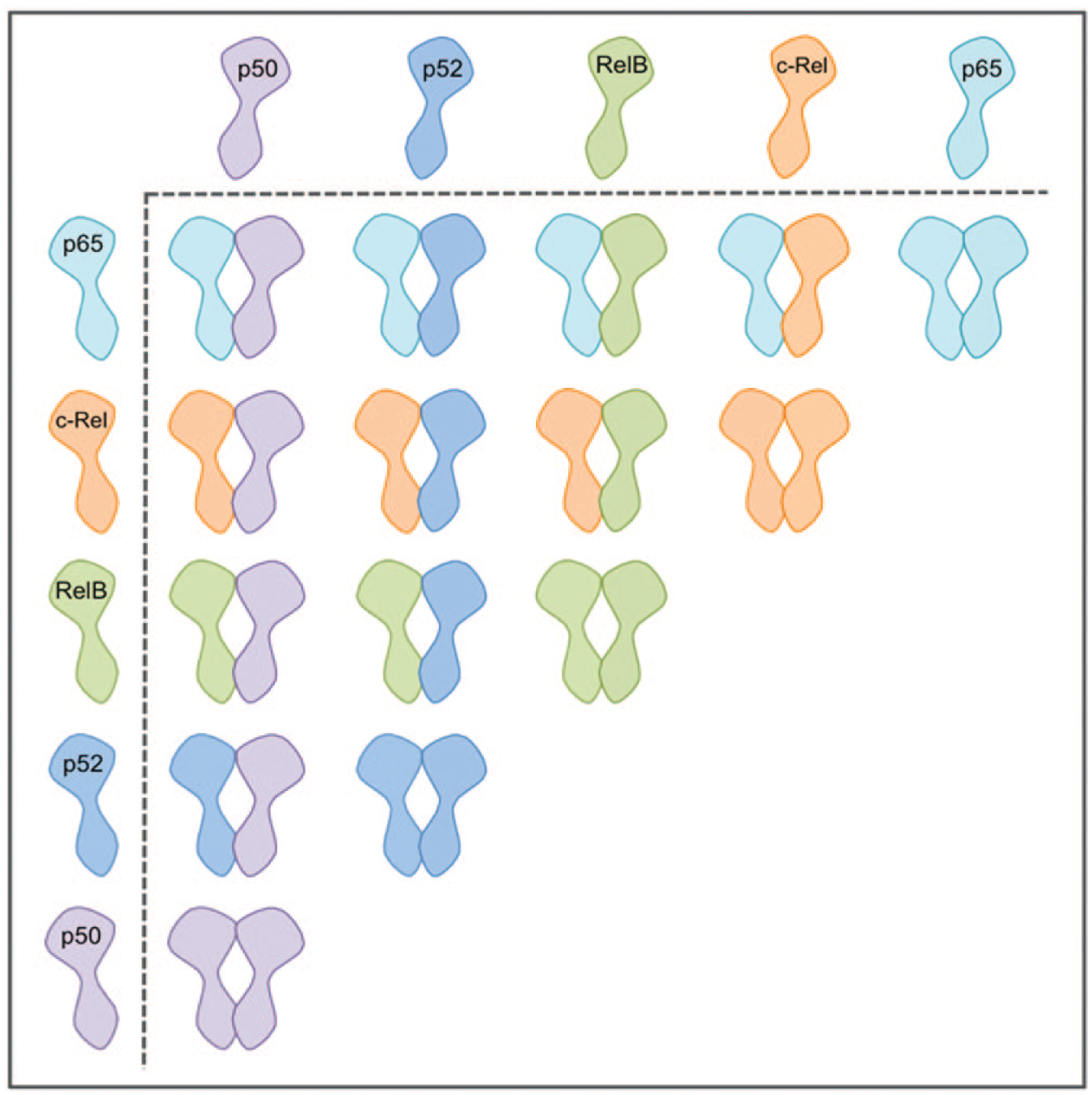
Nuclear factor (NF)–κB dimer pairs. NF-κB is a heterogeneous collection of homo- and heterodimers, comprising members of the Rel family, which can combine to make up to 15 different NF-κB dimers.
Over 150 distinct stimuli induce NF-κB activity, 9 including proinflammatory cytokines, viral and bacterial components, and oxidative stress (www.nf-kb.org). The NF-κB–dependent transcriptional programs induced by these stimuli have the potential to inflict significant damage to healthy host tissue, and as a consequence, NF-κB activity is tightly controlled. 10 The principal means of control of NF-κB activity is through retention of inactive dimers in the cell’s cytoplasm by inhibitor of κB (IκB) molecules. 11 The IκB molecules are a family of related proteins that share a homologous central ankyrin repeat domain, composed of six to seven ankyrin repeats, which mediates their interaction with NF-κB dimers. 12 The cytoplasmic inhibitory IκB molecules include the prototypical IκB molecules (IκBα, IκBβ, and IκBε), as well as the p50 and p52 precursors, p105 and p100. Although subunit precursors, p105 and p100 are classified as members of the IκB family due to the C-terminal ankyrin repeat domain present in these proteins. Activation of NF-κB requires the phosphorylation-dependent ubiquitination and proteasomal degradation of IκB proteins, with this degradation leading to the liberation of active NF-κB dimers, enabling them to translocate into the nucleus. 13 In the case of p105 and p100, the C-terminal IκB portion of these proteins is degraded upon ubiquitination, releasing functional p50 and p52 subunits that are able to bind DNA target sequences in the nucleus. 14 p105 and p100 can also inhibit preformed NF-κB dimers,15,16 in a similar way to the prototypical IκB molecules. Phosphorylation of the IκB proteins is carried out by the IκB kinase (IKK) complex, which is composed of two related IκB kinases, IKKα and IKKβ, and the scaffold protein NF-κB essential modifier (NEMO, also known as IKKγ). Phosphorylation of IκB proteins by the IKK complex leads to their recognition and polyubiquitination by the ubiquitin E3 ligase β-transducin repeat–containing protein (βTrCP), targeting the IκB molecules for proteasomal degradation.17,18
Two distinct signaling pathways are involved in NF-κB activation, the classical (or canonical) pathway and the noncanonical pathway, each resulting in diverse biological outcomes. The classical pathway involves NF-κB dimers composed of the p65, c-Rel, and p50 subunits and requires the degradation of the prototypical IκB proteins IκBα, IκBβ, and IκBε, as well as the NF-κB p50 precursor p105. As well as regulating NF-κB activity, IκBα is also an NF-κB target gene, creating an autoregulatory feedback loop. Following NF-κB activation, newly synthesized IκBα protein moves to the nucleus where it binds NF-κB dimers, blocking their DNA binding ability and helping limit the duration of NF-κB–dependent responses. 6 The noncanonical NF-κB pathway is highly evolutionarily conserved and controls genes important in the regulation of homeostatic processes, such as lymphoid organogenesis, bone metabolism, and B-cell survival. 19 This pathway is triggered by a limited number of members of the tumor necrosis factor receptor (TNFR) superfamily, including the lymphotoxin-β receptor (LTβR), B-cell activating factor (BAFF), and CD40. This pathway centers on the activation of the RelB subunit, which is bound to p100 in the cytoplasm of unstimulated cells. 20 Activation of the noncanonical pathway is generally slower than that of the classical pathway and begins with the activation of the NF-κB inducing kinase (NIK) in response to receptor ligation. NIK-mediated phosphorylation of IKKα leads to the subsequent processing of p100 to p52 and the liberation of RelB and p52 containing dimers, which are then able to undergo nuclear translocation.
In addition to the cytoplasmic IκB proteins, a number of atypical IκB proteins (IκBδ,
21
B-cell lymphoma 3-encoded protein
Enhanced or inappropriate activation of the NF-κB pathway has been implicated in numerous pathological conditions, including cancer, septic shock, and multiple autoimmune diseases ( Table 1 ). NF-κB activation has been detected in the synovium of patients with rheumatoid arthritis (RA)27,28 and plays a key role in the expression of proinflammatory genes associated with lymphocyte recruitment, cartilage destruction, and pannus formation. 29 Inappropriate NF-κB activation is also seen in mucosal biopsy specimens from patients with active Crohn’s disease and ulcerative colitis (collectively termed inflammatory bowel diseases [IBDs]).30–32 This NF-κB activation leads to the production of proinflammatory mediators, with some of these mediators further activating NF-κB, leading to a positive autoregulatory loop and the continuation of pathology. Importantly, aberrant expression of NF-κB does not always result in enhanced activity. The abnormal expression of NF-κB subunits has been described in T cells from patients with systemic lupus erythematosus (SLE),33,34 with reduced levels of NF-κB DNA binding detected in peripheral blood lymphocytes. 35
Autoimmune Diseases Associated with Dysregulated Nuclear Factor (NF)–κB Activation.

The involvement of aberrant NF-κB activity in numerous autoimmune pathologies, as well as the ever-expanding understanding of NF-κB regulation, has generated a great deal of interest in the therapeutic targeting of this signaling pathway. NF-κB inhibition has already been shown to be efficacious in a variety of cancers36,37 and has the potential to become an important component of the therapeutic arsenal against autoimmunity.
Current Treatments That Modulate NF-κB
Although very few specific NF-κB inhibitors are currently in use in the clinic, a variety of commonly used anti-inflammatory agents modulate the NF-κB pathway as part of their mechanism of action, including glucocorticoids, nonsteroidal anti-inflammatory drugs (NSAIDs), disease-modifying antirheumatic drugs (DMARDs), and certain immunosuppressive agents. Although these drugs do not show specificity for NF-κB, they act on varying stages of the NF-κB signaling pathway, and their immunomodulatory properties, at least in part, can be ascribed to the inhibition of NF-κB.
For example, the efficacy of sulfasalazine, a DMARD used in the treatment of autoimmune diseases such as RA and IBD, has been shown to be partially dependent on the prevention of NF-κB nuclear translocation. Sulfasalazine administration interferes with IκBα phosphorylation and degradation, inhibiting NF-κB activation in colon cells in response to tumor necrosis factor α (TNFα), LPS, or phorbol ester stimulation. 38 Another DMARD, mesalamine, also demonstrates activity against NF-κB, inhibiting posttranslational modifications of p65 that are required for the initiation of gene transcription. 39 NSAIDs are commonly used in the treatment of chronic inflammatory conditions, with aspirin and sodium salicylate shown to inhibit the activity of IKKβ, preventing the degradation of IκB and blocking NF-κB nuclear translocation.40,41 Glucocorticoids are highly effective in the treatment of active IBD and have been shown to inhibit NF-κB through both indirect and direct mechanisms. Indirectly, glucocorticoids induce the transcription and synthesis of IκBα, enhancing the retention of NF-κB in the cytoplasm and effectively inhibiting its activation.42,43 However, under certain conditions, glucocorticoids can directly inhibit activated NF-κB via competition between p65 and the glucocorticoid receptor for limited nuclear supplies of coactivator proteins. Both receptors require these coactivators for maximal activity, and by sequestering the available stores, glucocorticoids interfere with p65-dependent gene activation.44,45
The anti-cytokine biologics anakinra (IL-1 receptor antagonist), infliximab, and adalimumab (both anti-TNFα monoclonal antibodies) are valuable tools in the treatment of autoimmune diseases such as arthritis and Crohn’s disease and can also be said to mediate their beneficial effects through the modulation of NF-κB activity. NF-κB can be induced by proinflammatory cytokines, leading to further transcription of proinflammatory mediators, amplifying already present inflammation and contributing to disease pathogenesis. In RA, activated synovial cells produce mediators involved in joint destruction and are responsible for the invasion and degradation of cartilage and bone within the joint. 46 Stimulation of human synovial cells with TNFα results in NF-κB activation and subsequent cell proliferation, with NF-κB blockade able to activity inhibit this TNFα-induced proliferation. 4 Anti-cytokine biologics effectively remove these upstream activatory stimuli, and as such, the inhibition of NF-κB activation can be considered part of their mechanism of action.
Given the variety of stimuli involved in the activation of the NF-κB pathway and the crucial role of NF-κB–dependent genes in multiple biological processes, it is perhaps not surprising that so many drugs currently used in the treatment of autoimmune diseases have been shown to modulate NF-κB signaling. The success of these drugs in helping to ameliorate autoimmune pathologies suggests that specific NF-κB inhibitors would be a useful addition to the currently available therapeutics.
Strategies for Specific NF-κB Inhibition
The development of inhibitors specifically targeted to the NF-κB signaling pathway is a more recent endeavor. Studies in NF-κB knockout mice in the mid-1990s supplied evidence that the NF-κB pathway could be selectively targeted and that individual subunits play distinct roles that cannot be fully compensated by other Rel-family members.47,48 So far, over 780 inhibitors of the NF-κB pathway have been experimentally identified. 49 These vary from broad-range inhibitors of NF-κB to those with more selective inhibition that target the later stages of the pathway, and include both natural and synthetic molecules ( Fig. 2 ). Therapeutics targeting the NF-κB pathway are now available in the clinic36,37 and have become a mainstay in the treatment of certain malignancies. The use of pharmacological NF-κB inhibitors for the treatment of autoimmunity, however, is less advanced. Currently, there are no NF-κB inhibitors approved for use in autoimmune diseases, but this is a highly active research area. To date, many promising preclinical studies have been published targeting multiple stages of the NF-κB pathway using a variety of strategies able to prevent or diminish the activation of NF-κB.
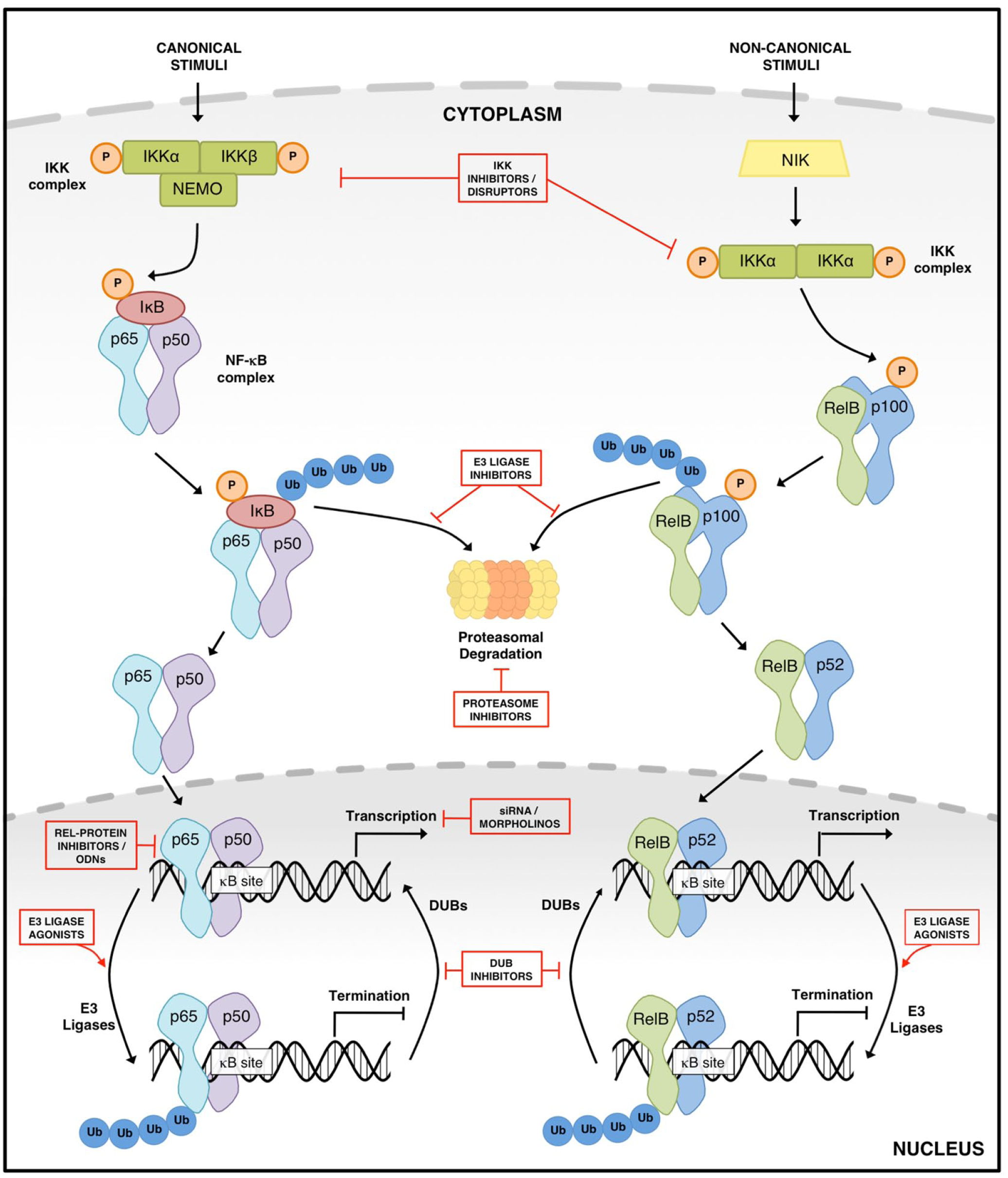
Schematic of the nuclear factor (NF)–κB pathways of activation, indicating current and potential targets for inhibition. Various stimuli result in the activation of NF-κB through either the canonical or noncanonical signaling pathways. Numerous inhibitors of the NF-κB pathway have been identified, with these able to target both the canonical (classical) and noncanonical signaling pathways. Inhibitors vary from broad-range inhibitors to those with a more precise inhibition, which target the later stages of the pathway.
Targeting the IKK Complex
The IKK complex plays a central role in integrating upstream signals from multiple signaling pathways into the NF-κB activation pathway, and increased IKK activity is well recognized in numerous autoimmune diseases.50,51 Consequently, this kinase complex has been a major target for the development of NF-κB signaling inhibitors. The clinical success of imatinib, a tyrosine kinase inhibitor (TKI) that blocks the catalytic activity of the deregulated BCR-ABL tyrosine kinase found in chronic myeloid leukemia, has confirmed the druggability of kinases, encouraging this area of investigation and resulting in the development of a number of IKK inhibitors. Because IKK is essential for the activation of NF-κB, an IKK-specific inhibitor could be expected to have potent anti-inflammatory activity in autoimmunity due to effects on a variety of inflammatory mediators. Several studies indicate that targeting IKK may be of particular benefit in chronic inflammatory conditions52–56 and as such may also be particularly efficacious in the treatment of autoimmune disorders.
The classic IKK complex comprises two catalytically active kinases, IKKα and IKKβ, which can form homo- and heterodimers, and the regulatory scaffold subunit NEMO. NEMO is also an ubiquitin-binding protein, and on receipt of an NF-κB–activating stimulus, polyubiquitin chains are formed within the cytosol that bind to NEMO, as well as specialized kinase complexes. The binding of NEMO indirectly recruits IKKα and IKKβ to the kinase complexes, resulting in the phosphorylation and activation of IKK. Activated IKK then phosphorylates IκB molecules, liberating NF-κB dimers. However, numerous non-IκB targets of IKK have also been identified, including Histone H3, 57 cyclin D1, 58 interferon regulatory factor 7 (IRF7), 59 the signaling molecules B-cell lymphoma/leukemia 10 (Bcl10) 60 and β-catenin, 61 the nuclear coactivators steroid receptor coactivator 3 (SRC3) 62 and CREB-binding protein (CBP), 63 the kinase Aurora A, 64 and p65. 65 The outcome of IKK-mediated phosphorylation is different for every protein. For example, posttranslational modification of p65 by IKK enhances its transactivational activity by inducing the interaction of p65 with nuclear coactivator proteins. 66
Most IKK-selective inhibitors developed so far have been identified through a combination of high-throughput screening (HTS), in silico methods, and rational design. IKKα and IKKβ have different affinities for specific NF-κB dimers, and historically IKKβ has been pursued as the prime IKK target isoform for inhibition due to its role in the activation of the canonical NF-κB pathway, resulting in the production of proinflammatory mediators. As such, the majority of existing IKK inhibitors are either directed solely at IKKβ or show dual activity against both the IKKα and IKKβ isoforms. However, the essential role for IKKα in canonical pathway activation, 19 as well as the elucidation of a potential inflammatory role for IKKα in noncanonical NF-κB activation, 20 means that interest in the specific targeting of this IKK isoform is likely to grow.
IKK inhibitors generally exert their activity via interaction with the conserved adenosine triphosphate (ATP)–binding site of IKK molecules. ATP-competitive IKK inhibitors are of low molecular weight, are orally bioavailable, and have been repeatedly shown to successfully inhibit target proteins, with this class of inhibitor more readily identifiable by HTS than those targeting allosteric sites. One of the first IKK inhibitors described as a treatment for inflammatory and autoimmune diseases was the quinazoline analogue SPC-839 (AS602868), 67 with SPC-839 shown to reduce disease severity in a preclinical model of RA. 68 This kinase inhibitor progressed to phase I trials, although in the context of hematological malignancies rather than autoimmunity. Unfortunately, clinical development was later stopped by Merck Sorono for reasons unconnected to drug performance. 69
Several other ATP-competitive IKKβ-specific inhibitors have since been reported. The β-carboline compound ML120B blocks IKKβ-mediated IκBα phosphorylation, reducing proinflammatory cytokine production by human fibroblast-like synoviocytes, chondrocytes, and mast cells in vitro. 70 In a rat model of adjuvant-induced arthritis, ML120B treatment resulted in significantly reduced inflammation and a decrease in both cartilage and bone destruction. 71 The recently developed IKKβ inhibitor [5-(p-fluorophenyl)-2-ureido] thiophene-3-carboxamide (TPCA1) has also shown promise in early stage development. TCPA1 inhibits the production of TNFα, IL-6, and IL-8 by LPS-stimulated human monocytes, with nanomolar potency. 72 Prophylactic or therapeutic administration of TCPA1 reduced disease severity in a murine collagen-induced arthritis (CIA) model, significantly decreasing nuclear localization of p65, as well as tissue levels of IL-1, IL-6, TNFα, and interferon γ (IFNγ) in treated mice. 72 More recently, TCPA1 has been shown to reduce the severity of type 1 helper T-cell (Th1)–mediated local immune responses. 73 Multiple autoimmune disease are considered to be broadly Th1-mediated, including multiple sclerosis (MS), type 1 insulin-dependent diabetes mellitus, and posterior uveitis, and although a variety of factors contribute to the complex symptoms of each disease, the abrogation of Th1-type responses via NF-κB modulation could potentially help to alleviate such conditions. Another novel IKK inhibitor, 3-[(dodecylthiocarbonyl)methyl]glutarimide (DTCM-glutarimide), has been found to have anti-inflammatory activity both in vitro and in vivo. A synthetic analogue of the natural compound 9-methylstreptimidone, DTCM-glutarimide is able to inhibit LPS-induced nitric oxide production in RAW264.7 cells, 74 as well as inhibit receptor activator of nuclear factor κB ligand (RANKL)– and LPS-induced osteoclast differentiation in primary mouse bone marrow–derived macrophages. 75 DTCM-glutarimide treatment was also able to ameliorate disease in a preclinical model of IBD, 76 indicating its potential as an autoimmune therapeutic.
Although promising, ATP-competitive IKK inhibitors have the potential to generate considerable “off-target” effects as a result of cross-reactivity with other kinases as ATP-binding sites are highly conserved between kinases. 77 A number of these inhibitors have demonstrated unfavorable outcomes, 78 and as such, more recent efforts to develop IKK inhibitors have moved away from ATP-competitive inhibitors. 4-(2′-Aminoethyl)amino-1,8-dimethylimidazo[1,2-a]quinoxaline (BMS-345541) is a ATP-noncompetitive inhibitor of IKKβ, targeting allosteric sites on the kinase, 79 with an approximately 10-fold greater inhibitory effect on IKKβ compared with IKKα. Prophylactic administration of BMS-345541 is able to prevent joint inflammation and destruction in a murine model of CIA, reducing the levels of IL-1β transcript present in the joints and reducing both the incidence and clinical signs of disease. 54 Administration of BMS-345541 is also effective in blocking both clinical and histological end points of inflammation and injury in murine dextran sodium sulfate (DSS)–induced colitis, 80 a preclinical model used to study both ulcerative colitis and Crohn’s disease.
An additional strategy for the targeting of IKK has been the development of IKK complex “disruptors”: small peptides that disrupt the protein-protein interactions within the IKK complex, preventing the phosphorylation of IκB and subsequent NF-κB nuclear translocation. This disruption is mediated by small cell-permeable peptides targeted at the NEMO-binding domain (NBD) of IKK, effectively blocking the association of NEMO with the catalytic IKK proteins. 81 Multiple studies have shown that inhibition of the IKK complex by NBD peptides is efficacious in preclinical models of autoimmunity. NBD peptide treatment of 2,4,6-trinitrobenzenesulfonic acid (TNBS)– and DSS-induced colitis, as well as spontaneous chronic colitis, ameliorated colonic inflammation and downregulated the production of proinflammatory cytokines mediated by NF-κB.82,83 In a preclinical model of MS, treatment with the NBD peptide reduced spinal cord invasion by mononuclear cells, resulting in an overall shift from a Th1 to a type 2 helper T-cell (Th2) immune response and a reduction in clinical disease symptoms. 84 NF-κB plays a crucial role in osteoclast differentiation, and inhibiting the NF-κB signaling pathway with NBD peptides has shown to be an effective strategy for the treatment of diseases in which bone resorption plays a pathological role. IKK complex blockade by the NBD inhibited RANKL-stimulated osteoclastogenesis in vitro and in vivo 52 and prevented both bone erosion and inflammatory responses in the joints of mice with experimental arthritis.52,53 The anti-inflammatory effects of the NBD peptide have been confirmed in human cells in vitro. Treatment of human monocyte–derived dendritic cells with the NBD peptide rendered them resistant to LPS-induced maturation and inhibited their production of the proinflammatory cytokines IL-6, IL-12, and TNFα. 85
Targeting the IKK-NEMO interaction, rather than the catalytic activity of IKK, blocks the activation of NF-κB in response to proinflammatory stimuli without affecting basal NF-κB activity. 81 This selective blockade of NF-κB induction may help to reduce possible side effects, such as the induction of undesired apoptosis and the toxicity issues that can be associated with direct IKK inhibition. However, before the progression of the NBD peptide into a clinically usable treatment can occur, further development will need to be undertaken to introduce the desired drug-like characteristics and appropriate pharmacology to this inhibitor.
Gene therapy targeting the IKK complex is also being pursued as a strategy for effective NF-κB blockade. Local gene therapy is an attractive alternative to systemic protein delivery, and an anticipated benefit of this type of intervention compared with pharmacological approaches is that it has the potential for a sustained beneficial effect to patients without the need to readminister a given treatment—a particular advantage in the treatment of chronic conditions such as autoimmunity. Although a promising approach, it should be noted that the long-term nature of gene therapy also has an important potential drawback. As opposed to pharmacological approaches, gene therapy cannot be stopped or treatment regimens easily altered once the therapeutic is administered. This feature could therefore cause problems if the patient experiences unwanted side effects or encounters circumstances in which he or she needs to mount a robust immune response.
Vector-mediated gene transfer is a highly efficient system for the delivery of genes, with the choice of vectors and promoters used directing the resultant protein expression profile, allowing maximal therapeutic effects to be achieved. Adenoviral vectors carrying a dominant-negative mutant IKKβ construct (IKKβdn) have been used to target NF-κB both in vitro and in vivo. Overexpression of the catalytically inactive IKKβdn results in inhibition of endogenous IKKβ activity, and in vitro, this has been shown to inhibit maturation and effective antigen presentation by human dendritic cells (DCs), 86 prevent RANKL-induced osteoclast formation by RAW264.7 cells, 87 and block endothelial cell expression of markers involved in the recruitment of lymphocytes during inflammation. 88 Intra-articular injection of IKKβdn in rats with adjuvant-induced arthritis resulted in significant amelioration of disease severity, accompanied by a substantial decrease in NF-κB DNA expression and reduced proinflammatory cytokine expression in the synovium without altering expression of the anti-inflammatory cytokine IL-10.56,89
IKK inhibitors have shown promise in preclinical studies, but to date, relatively few IKK inhibitors have made it to clinical trials ( Table 2 ) despite the tremendous investments made by both the pharmaceutical industry and the wider research community. Importantly, it should be noted that there are considerable toxicity concerns associated with the interruption of IKK function, regardless of the method used, with the toxicity of IKK inhibition first predicted by genetic models of IKK deficiency. Both IKKβ knockout and NEMO knockout mice are embryonically lethal due to increased hepatocyte apoptosis, 90 while IKKα knockout mice show a number of severe developmental abnormalities and do not survive after birth.91–93 Interestingly, these outcomes are not related to IKK kinase activity: transgenic mice expressing a kinase-dead mutant of IKKα are phenotypically normal. 94 Conditional IKK knockout mice also develop defects in multiple tissue compartments. Hepatocyte-restricted NEMO deletion results in the development of spontaneous chronic hepatitis in adult mice, which ultimately progresses to hepatocellular carcinoma, 95 indicating NEMO is involved in tumor suppression in the liver. IKK also plays a key role in gut homeostasis, with specific deletion of NEMO or IKKα and IKKβ in murine intestinal-epithelial cells causing spontaneous inflammation of the colon, characterized by immune cell infiltrates and enhanced cytokine expression. 96 IKKα-null bone marrow chimeras also demonstrate immune system abnormalities, with altered NF-κB activation in macrophages 97 and defective B-cell function. 98 IKK signaling is also involved in the maintenance of mature B cells, with the disruption of either IKKβ or NEMO resulting in the disappearance of mature B cells from the periphery. 99 Mature T cells also depend on IKK signaling. T-cell–specific deletion of NEMO or the replacement of IKKβ with a dominant-negative mutant prevents the development of mature peripheral T cells in a mouse model. 100 These wide-ranging side effects must be considered if IKK inhibitors are to be used clinically, especially where treatment is likely to be long term, such as in cases of autoimmunity.
IKKβ Inhibitors Involved in Clinical Trials.
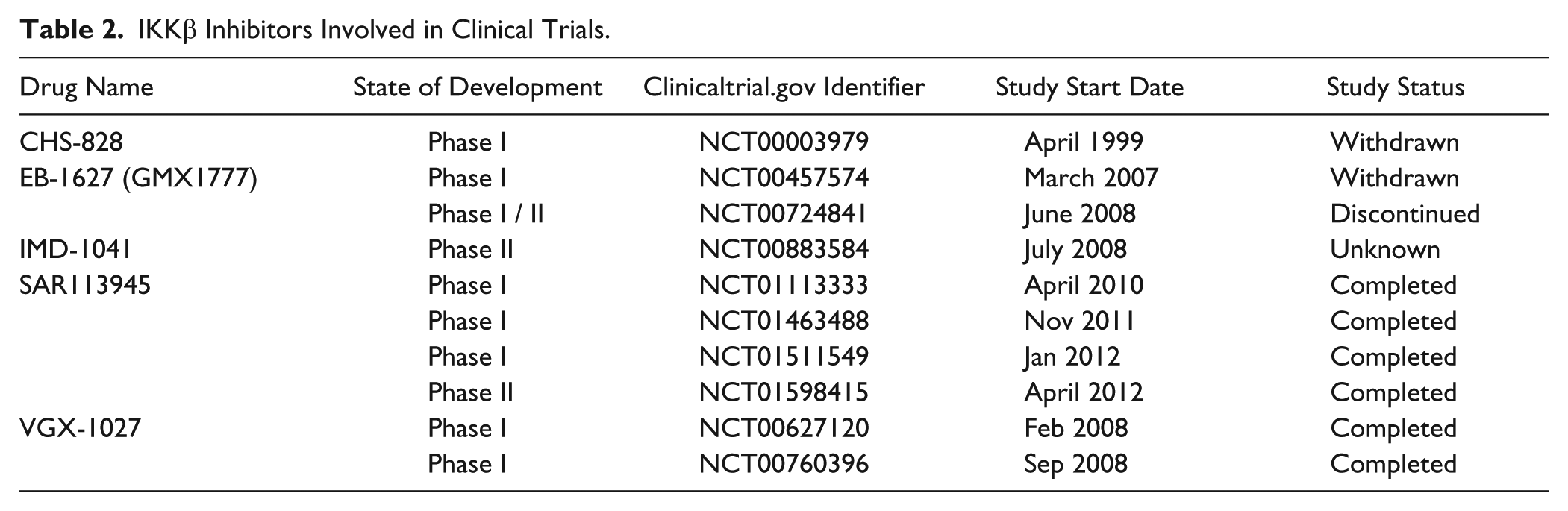
Due to the numerous adverse effect of IKK deletion, along with concerns regarding the side effects of potential IKK inhibitors, the development of NF-κB signaling inhibitors has been moving away from IKK and instead focusing on the development of inhibitors directed at components further downstream in the NF-κB signaling pathway. This strategy allows a more selective approach, reducing the likelihood of extensive adverse outcomes and toxicities that can result from broad-spectrum IKK inhibition.
Targeting IκB Proteins
The IκB proteins are of great interest as targets for therapeutic intervention due to their key roles in the regulation of NF-κB complexes. Maintaining a high level of IκB proteins in the cytoplasm effectively prevents NF-κB induction. Gene constructs that overexpress endogenous IκBα, or the IκBα super repressor (IκBαSR), a mutated form of IκBα lacking the phosphorylation sites required for its degradation, lead to NF-κB sequestration in the cytoplasm of cells and inhibition of its transcriptional activity. IκBα does not interact with all NF-κB subunits, however. As such, the overexpression of IκBα or introduction of an IκBαSR into cells does not generate total NF-κB inhibition, allowing selective targeting of specific NF-κB proteins, rather than blanket inhibition of all NF-κB cellular activity. Adenoviral vectors carrying IκBα or IκBαSR have been used to successfully block NF-κB activation in multiple in vitro systems,101–104 indicating the possibilities of this potential therapeutic strategy. Overexpression of the inhibitory subunit IκBα in primary human macrophages, fibroblasts, and T cells from RA patient synovial explants significantly inhibits the spontaneous production of proinflammatory cytokines, as well as the matrix metalloproteases (MMPs) MMP-1 and MMP-3.2,105 Intestinal epithelial cells (IECs) play a crucial role in the maintenance of homeostasis in gut, and dysregulation of these processes can lead to IBD, characterized by abnormal immune responses and inflammation at the intestinal mucosa. Delivery of the IκBαSR into IECs has been shown to effectively block induction of inducible nitric oxide synthase, IL-1β, and IL-8 genes by proinflammatory stimuli, suggesting the IκBα super-repressor may be an exciting approach for in vivo intestinal gene therapy. 106
It should be appreciated that although adenoviral vectors have been used extensively for NF-κB inhibition both in vitro and in vivo, current adenoviral vectors are too immunogenic for use in the treatment of chronic conditions. 107 Stimulation of potent antiviral host immunity in response to adenoviral administration results in transient gene expression, which is short-lived and, consequently, not therapeutically beneficial. To overcome this, alternate viral vectors can be employed to mediate gene transfer, such as adeno-associated virus (AAV). AAV is unable to replicate, lacks many of the immunogenic characteristics of adenoviral vectors, and has been successfully used in multiple clinical trials to date. 108 Furthermore, several naturally occurring AAV serotypes exists, each of which displays a different tropism, potentially allowing vector selection to be tailored to target expression in particular tissues in vivo.109,110 As NF-κB is ubiquitously expressed in human cells, this characteristic could help to reduce off-target effects and unwanted side effects. Another option is ex vivo gene transfer, which could remove the need for viral vectors altogether. In ex vivo gene transfer, primary autologous cells are isolated, transfected in culture, untransduced cells selected out, and the cells that express the desired gene injected into the target tissue. This method of gene transfer has proven useful in several preclinical models of RA,111–113 but this approach has not yet been used to inhibit the NF-κB pathway, and so it remains to be seen whether it would be successful in this setting.
The atypical IκB proteins are also an attractive target for NF-κB inhibition. Bcl-3 is a nuclear regulator of NF-κB that stabilizes homodimers of p50, promoting the formation of a stable p50 repressor complex at NF-κB DNA binding sites. Homodimers of p50 are critical negative regulators of NF-κB–mediated inflammatory gene expression,26,114 and the activation of p50 homodimers is associated with the resolution phase of inflammation. 115 As such, Bcl-3 is critical in limiting the expression of proinflammatory cytokines. 26 A recently developed Bcl-3 mimetic peptide, BDP2, is able to inhibit LPS-induced cytokine expression in vitro and has potent anti-inflammatory activity in vivo. Preclinical studies in mice show BDP2 significantly inhibits carrageenan-induced paw inflammation, reducing local expression of the proinflammatory cytokines TNFα and IL-6 and inhibiting edema. 116 Although based on a model of acute inflammation, these findings indicate that mimicking Bcl-3 function may be a promising strategy for the inhibition of inflammation in a clinical setting. However, a better understanding of this class of NF-κB regulator is needed to allow its full potential as a therapeutic target to be explored.
Targeting the Ubiquitin-Proteasome System
The ubiquitin-proteasome system (UPS) regulates the synthesis and degradation of proteins within the cellular environment 117 and is crucial not only to activate NF-κB but also to terminate the transcriptional responses in the nucleus. Once activated, NF-κB translocates to the nucleus, where it binds specific sequences in the promoter-regulatory regions of target genes. 118 This DNA binding event triggers the ubiquitination and subsequent proteasomal degradation of NF-κB, thereby terminating transcriptional activity. 119 E3 ubiquitin ligases conjugate ubiquitin to ε-amino group lysine residues on target proteins, such as NF-κB, tagging them for destruction by the proteasome. 120 Prior to interaction with the proteasome, deubiquitylating enzymes can mediate the removal and processing of ubiquitin chains, rescuing certain proteins from degradation, and altering their location and activation. 121 It is a balance of this ubiquitination and deubiquitination that regulates the cell’s protein content, maintaining cellular homeostasis. The deregulation of this system is involved in multiple pathological conditions, including cardiovascular disease, 122 a number of cancers, 123 and several autoimmune diseases.124,125 Targeting specific elements of the UPS has the potential to interfere with the process of NF-κB activation, as well as limiting the duration of inappropriate NF-κB responses.
The Proteasome
Proteasome inhibitors (PIs) were originally developed as anti-inflammatory agents 126 before gaining recognition as valuable anticancer therapeutics. PIs are structurally heterogeneous and can be broadly divided into two groups depending on whether they covalently bond with the proteasome’s threonine active site.
The potential activity of PIs in autoimmunity has been demonstrated in multiple preclinical animal models. Treatment with bortezomib has been shown to be efficacious in murine models of lupus nephritis, 127 myasthenia gravis, 128 MS, 129 and colitis.130,131 Bortezomib forms covalent adducts with active site threonines on the proteasome, while coinhibiting the chymotrypsin-like and caspase-like active sites, resulting in the disruption of the proteasome’s proteolytic function. This synthetic inhibitor is currently used clinically in the treatment of several malignancies, including multiple myeloma (MM),132–135 and in the treatment of antibody-mediated allograft rejection in posttransplant patients.136,137 Bortezomib’s major mechanism of action has been identified as the selective destruction of antibody-producing plasma cells, and as such, it may also be efficacious in the treatment of antibody-mediated autoimmune diseases. Extensive protein synthesis by a cell results in the formation of unfolded proteins, and inhibition of the proteasome in these cells leads to the accumulation of misfolded proteins, activating the terminal unfolded protein response. 138 Due to their high rate of antibody production, plasma cells are particularly sensitive to proteasome inhibition. 139 Interestingly, bortezomib treatment is also associated with osteoblast activation in multiple myeloma patients. 140 Proteasome inhibitors have been shown to promote bone formation in in vitro assays and in mice,141,142 through the inhibition of osteoclastogenesis 143 and stimulation of osteoblastogenesis. 144 Osteopenia is a well-known feature of established RA. As such, the bone-anabolic activity demonstrated by proteasome inhibitors indicates that RA patients treated with these agents may benefit not only from the impact on the immune system but also from the effects on the skeletal system.
Based on these findings, recruitment is currently under way for a clinical pilot study investigating the efficacy of bortezomib treatment in SLE, myasthenia gravis, and RA. The TAVAB study (ClinicalTrials.gov Identifier: NCT02102594) is set to run until late 2016, with the prediction that treatment will lead to reduced autoantibody titers and improved clinical outcome in participants, and results are eagerly awaited. Despite these advancements, the future of bortezomib as a treatment for human autoimmune diseases is unsure, as a considerable proportion of patients treated with this PI have gone on to present with some form of peripheral neuropathy in response to therapy.145,146
In an attempt to improve on the results seen with bortezomib, several second-generation proteasome inhibitors have now been developed. These novel PIs belong to a range of distinct structural classes (β-lactones, salinosporamides, peptidyl epoxyketones, and peptidyl boronic acids) and have varying pharmacological and pharmacodynamic activity profiles. Carfilzomib (PR-171), 147 marizomib (NPI-0052), 148 and oporozomib (PR-047/ONX 0912) 149 bind irreversibly to the proteasome, producing a more sustained inhibition of proteolytic activity. Lower incidences of peripheral neuropathy have been observed in MM patients treated with carfilzomib, 150 indicating it may be of more use than bortezomib in the long-term treatment of autoimmune disease. Marizomib and oporozomib, along with the reversible inhibitors delanzomib (CEP-18870) 151 and ixazomib (MLN-9708), are orally active PIs, allowing for greater flexibility and convenience for patient dosing, with marizomib, delanzomib, and ixazomib all having reached the stage of phase I/II clinical trials.152–154 The different structures, drug kinetics, and administration routes of these novel PIs may influence the tissue distribution of these inhibitors and consequently the levels of proteasome inhibition within biological niches, something that may prove to be useful in the targeting of specific pathogenic cells while reducing off-target systemic effects.
Formerly, most PIs have been “dual-target agents”: they act indiscriminately on both the constitutive proteasome and the immunoproteasome, a variant proteasome composed of distinct proteolytic subunits. The immunoproteasome is expressed in cells of hematopoietic origin and induced in nonhematopoietic cells during inflammation and after exposure to certain proinflammatory cytokines.155–157 A subset of second-generation inhibitors is able to selectively target the chymotrypsin-like subunit of the immunoproteasome, such as the immunoproteasome-specific inhibitors (IPSIs) PR-957 (ONX 0914) 158 and IPSI-100. 159 Studies in murine lupus using PR-957 have demonstrated that targeting the immunoproteasome is equally as efficacious as treatment with dual-target agents. This novel inhibitor prevented disease progression and treated nephritis in established lupus, reducing serum tires of both anti–double-stranded DNA and total immunoglobulin G (IgG) levels in treated animals. 160 PR-957 is also able to ameliorate disease in experimental arthritis models, with treatment resulting in enhanced disease inhibition compared with mice treated with either the dual-target agent carfilzomib or the constitutive proteasome-specific PR-825. 158 In these animals, reduced joint cellular infiltration and subsequent bone erosion were observed, as well as diminished expression of proinflammatory cytokines and autoantibody levels. 158
Although the therapeutic potential of proteasome inhibition has been successfully demonstrated with the use of bortezomib and carfilzomib in the clinics, these agents offer broad-spectrum inhibition. This means the shared final step involved in all degradation mediated by the UPS is blocked, and as such, these agents are not entirely NF-κB specific. The hope is that the use of selective IPSIs may reduce the occurrence of toxicities that can arise from the blanket inhibition of all cellular proteasomal activity seen with dual-target agents.146,161
DUBs and E3 Ligases
Another angle from which the actions of the UPS can be modulated is through the development of small-molecule inhibitors of individual enzymes involved in this system. This approach promises increasingly specific therapeutic agents with the capability to treat autoimmune disorders.
The human genome encodes approximately 95 deubiquitylating enzymes (DUBs), with around 80 of these identified as functional proteases. 121 DUBs can be grouped into two broad classes, based on their mechanism of catalysis (metallo or cysteine proteases), with these further subdivided based on their ubiquitin-protease domains. The ubiquitin-specific proteases (USPs), a class of cysteine protease, constitute the largest subfamily of DUBs, containing more than 60 human members. Each of these DUBs possesses varied specificities, with sequences both inside and outside the catalytic domain contributing to DUB target selectivity. Most research focused on the modulation of DUB activity has focused on the use of drug-like deubiquitylase inhibitors, and initial problems with the specificity of DUB inhibitors 162 have been overcome in recent years with the development of small-molecule inhibitors able to selectively target specific DUBs. 163 Multiple deubiquitylase inhibitors are currently involved in preclinical studies investigating their therapeutic capacity in a range of pathologies.164–166
Ubiquitin-specific-processing protease 7 (USP7), previously named herpesvirus-associated ubiquitin-specific protease (HAUSP), is one of the most extensively studied DUBs. Multiple targets of USP7 have been identified, including phosphatase and tensin homolog (PTEN), 167 Ataxin-1, 168 claspin, 169 the tumor suppressor protein p53 170 and its regulator mouse double minute 2 homolog (mdm2), 171 and multiple transcription factors.172,173 Several small-molecule inhibitors of USP7 have been developed,162,174–176 with some of these demonstrating activity both in vitro and in animal models of multiple myeloma. 176 USP7 has recently been shown to mediate the deubiquitination of the NF-κB p65 subunit, 177 identifying it as an important regulator of inflammation. Inhibition of USP7 activity in murine macrophages with the small-molecule inhibitor HBX41,108 prevents the production of proinflammatory cytokines in response to TLR and TNFR activation. 177 These findings demonstrate that the deubiquitination of NF-κB by USP7 is important for target gene transcription and that blocking this interaction has the potential to ameliorate inflammatory processes crucial to the pathology of multiple autoimmune disorders. It should be noted, however, that the current anti-USP7 small-molecule inhibitors target the catalytic domain of UPS7 and as such inhibit the deubiquitination of all of USP7’s substrates, not solely p65. This universal blockade of USP7 function has the potential to cause detrimental off-target effects in vivo, suggesting that a novel approach, which is able to specifically target the USP7-p65 interaction, would hold more promise as a future therapeutic.
E3 ubiquitin ligases work alongside DUBs as key members of the UPS: they are responsible for the posttranslational ubiquitination of proteins, targeting them for degradation by the proteasome. Over 600 different E3 ligases are encoded by the human genome, 178 and protein ubiquitination occurs with a high level of substrate specificity. The E3 ubiquitin ligases Mindbomb E3 Ubiquitin Protein Ligase 1 (MIB1), Ring Finger Protein 121 (RNF121), and βTrCP have all been identified as enhancers of NF-κB activation through the mediation of IκB ubiquitination, and findings indicate that IκB-specific E3 ligases are promising therapeutic targets for autoimmunity. MIB1-deficient cells have enhanced basal levels of IκBα compared with wild-type cells, 179 while the knockdown of RNF121 results in impaired IκBα degradation in response to a range of stimuli, including the ligation of TLRs, Nod-like receptors (NLRs), RIG-I–like receptors (RLRs), or after DNA damage. 180 βTrCP has been shown to mediate the ubiquitination of both IκBα 17 and IκBβ, 18 and a dominant negative form of the ligase prevents localization of NF-κB to the nucleus of human embryonic kidney (HEK) cells in response to TNFα stimulation. 17 Inhibition of these ligases in a targeted manner could result in reduced IκB degradation and consequently the diminished translocation of active NF-κB to the nucleus, regulating NF-κB–dependent gene transcription and the production of proinflammatory mediators.
Another E3 ligase, F-box/WD repeat-containing protein 7 (FBXW7), has been shown to constitutively target the noncanonical inhibitory IκB molecule p100. 181 Accumulation of p100, either through enhanced expression or inhibition of its degradation, has been shown to limit TNFα-mediated bone resorption in mice via the inhibition of osteoclastogenesis. 182 This model reproduces conditions seen in common erosive bone diseases such as RA in which TNF is highly expressed, with some evidence that inflammation-induced low-grade bone loss may also occur in patients with diabetes mellitus. 183 Together, these data suggest that inhibition of specific E3 ligases, such as FBXW7, may also have potential for the treatment of bone loss resulting from autoimmune responses.
Three E3 ligases have been identified that target NF-κB subunits directly: PDZ and LIM Domain Protein 2 (PDLIM2), 184 Suppressor of Cytokine Signaling 1 (SOCS1), 185 and Inhibitor of Growth Protein 4 (ING4). 186 These ligases show some selectivity in the NF-κB subunits they ubiquitinate and as such may perform nonredundant roles in the regulation of NF-κB–dependent responses. Full characterization of these ubiquitinating enzymes is lacking, and it is likely that further NF-κB–specific ligases will also be described. It is not implausible to assume that these E3 ligases will have differing selectivities for the different NF-κB dimers ( Fig. 1 ), adding an additional layer of control to NF-κB responses. Understanding the detail of these interactions would open up the possibility of targeting the destruction/stabilization of specific Rel proteins. For instance, the ability to stabilize repressor p50 homodimers through the inhibition of a putative p50-specific E3 ligase would likely shift the balance in autoimmunity toward a more anti-inflammatory environment, helping to regain control of an overactive system. To date, however, the identity of an E3 ligase for p50 homodimers remains unknown.
Targeting NF-κB DNA Binding
Dehydroxymethylepoxyquinomicin (DHMEQ) is a low-molecular-weight inhibitor based on the structure of the antibiotic epoxyquinomicin C that directly targets NF-κB subunits. 187 DHMEQ binds to specific cysteine residues of Rel family proteins to inhibit their DNA-binding activity and subsequent gene transactivation, and under certain conditions, it can block the nuclear translocation of NF-κB. 188 This novel pharmacologic can inhibit both canonical and noncanonical NF-κB signaling, 189 and its therapeutic efficacy has been demonstrated in preclinical models of multiple autoimmune diseases, including RA, 190 SLE, 191 and colitis.76,188 The success of DHMEQ treatment in these models and the absence of apparent toxicity in animal studies indicate DHMEQ may be an attractive agent for the clinical treatment of autoimmune diseases, although further studies are needed to confirm this.
Direct targeting of NF-κB by gene therapy has also been explored with the use of synthetic double-stranded DNA oligodeoxynucleotides (ODNs). ODNs act as “decoy” cis elements and bind directly to NF-κB dimers, inhibiting their NF-κB DNA-binding activity. It was originally hypothesized that the large and polar nature of NF-κB ODN may hinder their cellular uptake and bioavailability, affecting their therapeutic efficacy. However, this does not seem to be the case. NF-κB ODNs have now been used successfully in the clinic to suppress restenosis after nonsurgical interventions for the treatment of coronary disease,192,193 demonstrating the safety and clinical value of NF-κB ODN treatment. Blockade of NF-κB with decoy ODN has not yet reached the clinic for the treatment of autoimmunity, but it has shown success in several preclinical models. Intrarectal administration of decoy DNA encapsulated in a viral envelope is able to prevent and treat multiple murine colitis models, with no NF-κB inhibition observed in extra-intestinal organs. 194 Intracolonic delivery of “nonviral” NF-κB ODN is also able to ameliorate murine IBD, with ODN primarily localized in the cell nuclei after 6 h, reducing proinflammatory NF-κB heterodimers in inflamed cells. 195 NF-κB ODNs have also been shown to ameliorate disease pathology and joint destruction in animal models of arthritis. Intra-articular administration of decoy ODN to rats with CIA markedly suppresses joint destruction of treated joints and blocks production of IL-1 and TNFα in the synovium. 196 Osteoclast activity is also suppressed by intra-articular injection of NF-κB ODN, with significantly fewer osteoclasts present in the joints of treated rats. 197 Interestingly, in a model of streptococcal cell wall arthritis, ODN treatment resulted in significant inhibition of disease severity in the contralateral, untreated joints, indicating local NF-κB inhibition may have beneficial systemic effects. 198 NF-κB ODN has also been shown to inhibit NF-κB activity in human RA synovial explants. 199 Transfection of decoy ODN resulted in reduced proinflammatory cytokine and adhesion molecule expression, as well as the significant inhibition of synovial cell proliferation. 199 These findings indicate that local administration of ODN quickly targets NF-κB activity without the need for systemic administration or viral vectors. However, these molecules have a short half-life and need to be administered frequently, which may diminish their therapeutic potential for the treatment of chronic diseases such as autoimmunity.
Targeting Posttranslational Modification of NF-κB
NF-κB subunits undergo extensive posttranslational modification (PTM), including phosphorylation, acetylation, nitrosylation, glycosylation, ubiquitination, and sumoylation, 200 with many of these modifications altering both the strength and duration of NF-κB activity and adding yet another level of specificity to the signaling pathway. Methylation of p65 in the nucleus negatively regulates NF-κB function by promoting degradation of DNA-bound active NF-κB. 201 Acetylation of p65 can result in either enhanced activation or reduced DNA binding specificity and nuclear export, depending on the specific lysine residues targeted.202,203 One approach for the direct targeting of NF-κB is the use of small-molecule compounds to alter these modifications. A number of agents have been shown to block NF-κB activation by inhibiting p65 acetylation in vitro,204–207 and the acetylation inhibitors Vorinostat and Romidepsin have recently been approved for use in T-cell lymphomas. The predominant PTM of NF-κB is phosphorylation, with a significant number of phosphorylation sites identified in all NF-κB subunits. A complete discussion of NF-κB phosphorylation is beyond the scope of this review, but it is worth noting that in many cases, site-specific phosphorylation contributes to the regulation of a select group of target genes. This suggests that modulation of NF-κB phosphorylation may be key to targeting precise transcriptional effects of therapeutic importance, without the consequences of a broad blockade of NF-κB activity. Although not yet investigated in the context of autoimmunity, the data indicate that the modulation of NF-κB posttranslational modification, whether methylation, acetylation, or phosphorylation, may also prove to be a valuable therapeutic target for the treatment of autoimmune disease. The primary potential benefit of targeting these modifications would be the ability to mediate gene-specific effects, altering only a small subset of NF-κB responses, while leaving the remaining responses intact.
Posttranscriptional Gene Silencing
An alternative approach to NF-κB inhibition is the use of RNA interference (RNAi) to block the production of NF-κB–dependent proteins. Small interfering RNA (siRNA), small hairpin RNA (shRNA), and morpholinos are all able to modulate and disrupt the functions of target proteins effectively, resulting in posttranscription gene silencing, with these antisense agents shown to effectively block the expression of NF-κB target genes.
siRNAs are small double-stranded RNA fragments, typically 20 to 25 base pairs long, which induce the formation of a RNAi silencing complex in the cytosol of target cells that mediates messenger RNA (mRNA) cleavage, resulting in the degradation of specific mRNA. 208 siRNA sequences can also be designed into shRNA, which are stem-loop RNA structures, and packaged in viral vectors and delivered easily into target tissues in vivo or in vitro, allowing more stable expression of siRNA constructs.
In recent years, a great deal of investment into the development of therapeutics using siRNA has been made, with numerous clinical trials undertaken targeting a wide variety of pathologies, including cancers, viral infection, renal failure, and transplant complications. 209 RNAi directed against NF-κB or other components of the NF-κB signaling pathway demonstrates potential as future therapeutics, and NF-κB silencing has been studied in a variety of experimental systems. siRNA directed against NF-κB p65 has been shown to inhibit the expression of mediators involved in inflammation and cartilage/bone destruction by rat chondrocytes in response to IL-1β and TNFα. 210 In primary human synoviocytes, siRNA-mediated knockdown of p65 and p50 inhibits inflammatory cytokine production and enhanced apoptosis in response to TNFα stimulation. 211 In addition, the feasibility of local delivery of siRNA directly into cartilage and synovium in vivo has been demonstrated in a rat model of osteoarthritis. 212 This is an approach that could be anticipated to also show efficacy in autoimmune-mediated arthritis.
Although promising, siRNA use has some limitations. The half-life of unmodified “naked” siRNA in vivo is short due to rapid degradation by endogenous serum RNases and elimination by the kidneys. To overcome this, a variety of delivery methods have been pursued to help stabilize siRNA and facilitate uptake, including encasement in liposomes, the conjugation of siRNA to targeting moieties, and inclusion in polyplexes to form nanoparticles, 209 but an optimal delivery system has yet to be determined. Off-target activity can be observed with siRNA administration, such as the regulation of unintended transcripts 213 and triggering of inflammatory responses,214–216 with the potential for these to result in broad toxicity issues.
NF-κB morpholinos are ODNs that have been modified to increase their efficacy and stability.217,218 In these antisense agents, the deoxyribose sugar of natural nucleic acids has been removed and replaced with a six-membered morpholino ring, and internal internucleotide linkages are also altered. These DNA analogues block mRNA translation in a sequence-specific manner comparable to siRNA. Morpholinos, however, require greater complementarity with their target mRNA (about 15 bases) to silence gene expression, and this enhanced specificity contributes to the lack of off-target effects seen with morpholinos, making them an attractive approach for targeting posttranscriptional gene silencing.
Considerations of Therapeutic Targeting NF-kB Signaling Pathway
The therapeutic targeting of NF-κB for the treatment of autoimmune diseases is a promising therapeutic strategy, but a number of considerations must be taken into account. Primary among these is the toxicity of systemic and indiscriminate blockade of NF-κB signaling. NF-κB is involved in numerous homeostatic and developmental pathways, as well as the beneficial inflammation involved in pathogen clearance, and so a delicate balance must be struck between suppressing undesired inflammation and interfering with normal cellular functions. As such, potential agents with the ability to target NF-κB signaling in a tissue- or cell-specific manner may demonstrate better therapeutic efficacy and reduce systemic toxicity. This could be achieved either through their mode of action, such as the plasma cell–specific effects resulting from proteasome inhibition, or by the local delivery of therapeutic agents, such as intra-articular injection of gene therapy constructs.
Another consideration is the propensity for NF-κB inhibition to cause significant apoptosis in target and nontarget cells due to NF-κB’s critical role as a regulator of apoptosis. In its role as an anticancer therapeutic, the apoptosis of malignant cells is a desired outcome of NF-κB inhibition, but this is not necessarily the case in the treatment of autoimmunity. For the majority of autoimmune diseases, the ability to selectively prevent the production of proinflammatory mediators by immune cells, while leaving these cellular compartments able to mount anti-inflammatory and antimicrobial responses, is the desired outcome. For example, it has been shown in a number of studies that malignant cells and highly active plasma cells are more susceptible to the cytotoxic effects of proteasome inhibition compared with other cells of the body, indicating low-dose treatment may not cause widespread issue. However, further work has raised concerns for the survival of normal epithelial 219 and cardiac cells 220 in response to other NF-κB modulating agents. As yet, it remains to be seen whether the widespread induction of apoptosis proves to be a major concern in the treatment of autoimmunity.
The immense complexity of the NF-κB pathway is both a help and a hindrance for the development of NF-κB–specific agents. The number of proteins involved in this signaling cascade allows a multitude of levels of NF-κB regulation to be targeted therapeutically, each with slightly varying extents of inhibition. Individual IκB and IKK regulatory proteins play distinct roles in the canonical and noncanonical activation pathways and in the activation of particular NF-κB dimers within these distinct pathways. Targeting of individual protein isoforms may therefore enable precise control of NF-κB modulation, with only the pathways or dimers involved in detrimental pathological effects targeted, while other NF-κB activities are untouched.
New screening methodologies are essential for the development of novel therapeutics. HTS has revolutionized the identification of new biologically active small molecules in drug discovery. However, currently available HTS methods do not capture all relevant processes, and many molecular changes cannot accurately be detected by available HTS methods. As such, continued investment in the methods used for initial detection of potential drug targets will enable more effective identification of compounds for clinical study, hopefully translating to the successful production of novel therapeutics for the treatment of autoimmunity.
In conclusion, NF-κB inhibitors are already successfully used for the treatment of a variety of malignancies, and the therapeutic modulation of NF-κB is an attractive target in autoimmunity. Numerous treatments are currently in use in the clinic for the treatment of autoimmune disease, but many are associated with adverse side effects and a subset of patients does not respond to available therapies. As such, novel therapeutics with different mechanisms of action are required. What will future NF-κB targeting therapies look like? Given the toxicity associated with IKK inhibitors, it is likely that any future therapy will target a postactivation step of NF-κB activity. In this respect, perhaps one of the more promising approaches may be the modulation of the ubiquitination and degradation of individual NF-κB subunits. Whatever the therapies may ultimately look like, it is clear that a NF-κB–based approach holds enormous potential for the treatment of a wide range of autoimmune and inflammatory diseases.
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.