
Abstract
Calcitonin gene-related peptide (CGRP) is a small neuropeptide and a potent vasodilator that is widely associated with chronic pain and migraine. An antibody that inhibits CGRP function would be a potential therapeutic for treatment of these disorders. Here we describe the isolation of highly potent antibodies to CGRP from phage and ribosome display libraries and characterization of their epitope, species cross-reactivity, kinetics, and functional activity. Homogenous time-resolved fluorescence (HTRF) binding assays identified antibodies with the desired species cross-reactivity from naïve libraries, and HTRF epitope competition assays were used to characterize and group scFv by epitope. The functional inhibition of CGRP and species cross-reactivity of purified scFv and antibodies were subsequently confirmed using cAMP assays. We show that epitope competition assays could be used as a surrogate for functional cell-based assays during affinity maturation, in combination with scFv off-rate ranking by biolayer interferometry (BLI). This is the first time it has been shown that off-rate ranking can be predictive of functional activity for anti-CGRP antibodies. Here we demonstrate how, by using just four simple assays, diverse panels of antibodies to CGRP can be identified. These assay formats have potential utility in the identification of antibodies to other therapeutic targets.
Keywords
Introduction
The small neuropeptide calcitonin gene-related peptide (CGRP) is a member of the calcitonin family and exists in two isoforms: CGRPα and CGRPβ. 1 CGRP is present in the central and peripheral nervous system. 2 It binds to a receptor complex consisting of the G protein–coupled receptor (GPCR) calcitonin receptor-like receptor (CALRLR), receptor activity-modifying protein 1 (RAMP1), 3 and receptor component protein (RCP). 4 CALRLR couples to the Gαs G protein, which signals predominantly through adenylate cyclase to increase cyclic adenosine monophosphate (cAMP) levels. 5 CGRP can also stimulate release of calcium from intracellular stores through the inositol phosphate pathway. 6 The activation of the CGRP receptor complex by CGRP causes vasodilation that can lead to chronic pain and migraine symptoms. 7 CGRP receptor antagonists such as Olcegapant (BIBN 4096BS) 8 and Telcegapant (MK0974) 9 are efficacious in treating acute migraine; however, the requirement for intravenous administration (Olcegapant) and toxicology issues (Telcegapant) have hindered further development of these molecules. 10 An antibody therapeutic to CGRP could potentially overcome the toxicology issues through its specificity and could be delivered subcutaneously.
Antibodies with therapeutic potential can be generated by in vivo or in vitro approaches. Phage and ribosome display are two powerful in vitro technologies that enable identification and generation of high-affinity antibodies from naïve libraries.11,12 In phage display, antibody single-chain variable fragments (scFv) are expressed in fusion with a phage minor coat protein that results in their display on the surface of phage particles. 13 Ribosome display is an alternative platform that uses in vitro prokaryotic or eukaryotic cell–free systems to display antibody fragments in libraries containing up to 1014 variable heavy- (VH) and variable light- (VL) chain combinations, linking phenotype to genotype through mRNA–ribosome–antibody complexes. 14 Populations of antibodies in these libraries that recognize a specific target antigen can be enriched through binding to decreasing concentrations of antigen in a selection process. 15 Antibodies that bind the target of interest and possess the required drug properties are identified by screening representative scFv from these selection outputs in a range of different assays that can measure binding, functional activity, species cross-reactivity, and selectivity. The scFv are expressed in unpurified Escherichia coli periplasmic extracts and routinely screened at a single dilution of unknown concentration. Functional cell-based assays would be the preferred choice for screening to identify antibodies with desired functional activities. However, in addition to scFv, these high-throughput E. coli extracts contain bacterial components and endotoxins in a hyperosmotic buffer, and are therefore not well tolerated in cell assays. 16 For this reason, biochemical assays using recombinant proteins are commonly used in primary screens for identifying antibodies to therapeutic targets. These assays are typically simple binding formats such as an enzyme-linked immunosorbent assay (ELISA). Additional biochemical binding assays that tolerate bacterially expressed samples include label-free formats such as surface plasmon resonance (SPR). These assays have the benefit of being able to determine kinetic parameters and measure the affinities of purified and unpurified antibody fragments, but are usually lower throughput. However, it is possible to measure off-rates of unpurified scFv in a relatively high-throughput manner using label-free biolayer interferometry (BLI) on systems such as ForteBio’s Octet (Menlo Park, CA), enabling the identification and ranking of scFv with reduced dissociation rates, and therefore potentially higher affinities, to the target of interest. 17
Antibodies that are identified from screening as unpurified scFv can be produced as purified scFv and immunoglobulin G (IgG) and profiled for desired characteristics, including affinity, functional potency, epitope, selectivity over related proteins, and species cross-reactivity. If improvements in antibody potency or affinity are required, libraries of antibody sequence variants can be generated by random or targeted mutagenesis of the antibody–antigen binding region, with the aim of identifying mutations that increase the affinity of the antibody. Antibodies with improved affinities can be enriched through further rounds of selection on decreasing concentrations of antigen and improved variants identified through single-point screening, as described earlier.
Here we describe the application of four simple assay formats for the identification and characterization of anti-CGRP antibodies. Using a novel combination of homogeneous time-resolved fluorescence (HTRF), 18 BLI, and cell-based cAMP assays, we have identified a panel of potent species cross-reactive CGRP neutralizing antibodies. Interestingly, we demonstrate for the first time how biochemical competition and off-rate ranking assays can be used as surrogate assays for functional potency to CGRP. This combined use of a simple panel of assays could be applied to enable the identification and characterization of antibodies to other soluble therapeutic targets.
Materials and Methods
Materials
CGRP peptides were purchased from Bachem (Torrance, CA): biotinylated human CGRPα (H5688), biotinylated rodent CGRPα (H5684), human CGRPα (H1470), human CGRPβ (H6730), rodent CGRPα (H2265), mouse CGRPβ (custom-made), and rat CGRPβ (custom-made). HTRF reagents were purchased from Cisbio Assays (Codolet, France): streptavidin-cryptate (610SAKLB), anti-His-XL665 (61HISXLB), and cAMP dynamic 2 HTRF detection kit (62AM4PEC). IgG was labeled using a small-scale DyLight 649 (Thermo Scientific, Waltham, MA; 84536) or DyLight 650 (Thermo Scientific, 53051) antibody labeling kit according to the manufacturer’s instructions.
Isolation of Anti-CGRP Antibodies
Phage display technology was used to isolate scFv antibodies to CGRP from naïve human antibody libraries generated by MedImmune.11,19,20 CGRP-specific scFv were enriched through a series of selections on biotinylated human CGRPα. 15 Two rounds of solution-based selections were carried out using 1000 and 500 nM biotinylated human CGRPα. A deselection step was performed using a 10-fold molar excess of streptavidin and a structurally related protein to minimize the possibility of isolating scFv to these proteins. ScFv specific to CGRP were identified by screening unpurified scFv expressed in E. coli 21 in CGRP scFv binding HTRF assays. ScFv that bound human and rodent CGRPα and had unique VH CDR3 sequences were produced as purified scFv and IgG1 according to standard procedures. 21
Affinity Optimization of Clone 2
Diversity was introduced into the variable regions of clone 2 using error-prone PCR (error rate of 8.1 nucleotides per 1000 base pairs). ScFv libraries were constructed in ribosome display format,19,22 with an average mutational load of 3.6 or 5.7 amino acids per scFv. Antibodies with improved affinity for CGRP were enriched through a series of selection rounds, decreasing antigen concentration from 1 µM to 50 nM biotinylated CGRPα over three selection rounds. Individual scFv from selection outputs were prepared as unpurified bacterial periplasmic extracts according to standard protocols 23 in 50 mM MOPS, 0.5 mM EDTA, and 500 mM sucrose (pH 7.4) and screened in HTRF epitope competition assays to identify clones with improved affinity for CGRP. Clones were sequenced to assess diversity, and those with improved affinities differing by more than one amino acid were produced as purified scFv and IgG1 21 and profiled in the described assays. Beneficial “hot spot” mutations that were identified were introduced into the improved clones to rationally recombine beneficial mutations through standard site-directed mutagenesis. Further random mutagenesis libraries were constructed in ribosome display format on improved clones identified during the screening process, as described previously. In total, four rounds of selection were performed on second-generation libraries where antigen concentration was decreased from 50 to 1 nM biotinylated human CGRPα.
HTRF Assays
HTRF assays were performed in 384-well white ShallowWell nonbinding plates (Corning Incorporated, Corning, NY; 3673) in assay buffer containing phosphate-buffered saline (PBS) (Life Technologies, Carlsbad, CA; 14190), 0.1% v/v bovine serum albumin (BSA) (Sigma, St. Louis, MO; A9576), and 0.4 M potassium fluoride ((VWR International, Radnor, PA; 26820). Time-resolved fluorescence at 590 and 665 nm was measured following excitation at 320 nm on an Envision (PerkinElmer, Waltham, MA) plate reader after the indicated incubation periods. Ratio values of (665 nm emission/590 nm emission) × 10,000 were used to calculate % Delta F according to the following equation:
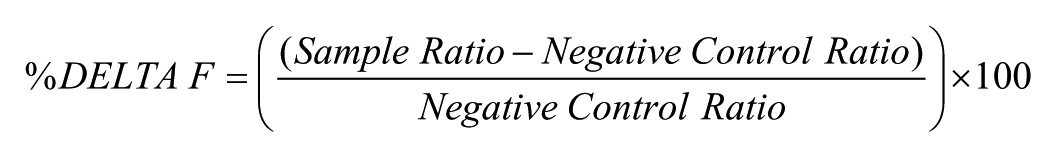
The negative control ratio was derived from nonspecific binding (NSB) control wells. Curves were analyzed using GraphPad Prism software using a four-parameter logistic curve-fitting equation.
CGRP scFv Binding HTRF Assays
ScFv were incubated with 1 nM biotinylated human CGRPα or rodent CGRPα, 0.5 nM streptavidin-cryptate, and 5 nM anti-His-XL665 to give a final assay volume of 20 µL, for 3 h at room temperature, prior to measurement of time-resolved fluorescent emission.
CGRP cAMP HTRF Assays
The CGRP-induced cAMP response was measured using SK-N-MC cells (ECACC, 90022302). All reagents were prepared in minimal essential media (Life Technologies, 51200) containing 10% v/v fetal bovine serum (SAFC Biosciences, St Louis, MO; 19106C), 1% v/v nonessential amino acids (Life Technologies, 11140), 1 mM sodium pyruvate (Sigma, S8636), and 0.5 mM 3- isobutyl- 1- methylxanthine (Sigma, I7018). Purified scFv and IgG were preincubated with EC50–80 CGRP (100 pM human CGRPα, 45 pM human CGRPβ, 30 pM rodent CGRPα, 60 pM rat CGRPβ, or 25 pM mouse CGRPβ) for 1 h at room temperature in HTRF assay plates. SK-N-MC cells (1500 cells per well) were then added to give a final assay volume of 10 μL, and the reaction was incubated for 20 min at room temperature. cAMP levels were determined using a cAMP HTRF detection kit (Cisbio, 62AM4PEC) according to the manufacturer’s instructions.
CGRP Epitope Competition HTRF Assays
For clone 2 IgG and clone 4 IgG epitope competition assays, 2.5 μL test scFv or IgG was incubated with 2.5 nM biotinylated human CGRPα, 1.25 nM streptavidin-cryptate, and 1.25 nM DyLight 650–labeled IgG 2 or IgG 4 in a total assay volume of 10 μL.
During clone 2 affinity optimization, a second, more sensitive epitope competition assay was established using a higher-affinity antibody that had been identified during the optimization process. Less biotinylated CGRP was required to generate a sufficient assay signal using this higher-affinity-optimized antibody variant, resulting in a more sensitive assay that could resolve the potencies of higher-affinity antibodies. This assay used 0.7 nM biotinylated human CGRPα or 2 nM biotinylated rodent CGRPα, 1.25 nM streptavidin-cryptate, and 3 nM affinity-optimized IgG labeled with DyLight 649. Assay plates were incubated for 3–4 h at room temperature prior to fluorescence measurement.
For this assay, the assumptions are made that it is performed at equilibrium and all antibody variants tested bind an overlapping epitope to that of clone 2. If these conditions are met, then an antibody with improved affinity for CGRP will have a higher potency than that of the unlabeled parent IgG in a competition format.
Kinetics Assays Using Biolayer Interferometry
Kinetics assays were performed on the Octet RED384 (ForteBio) at 25 °C in assay buffer containing PBS, 0.1% v/v BSA, 0.01% v/v Tween-20 (Sigma, P9416) (pH 7.4) using flat-bottom (Greiner, Monroe, NC; 781209) or tilted-bottom (ForteBio, 18-5076) black 384-well plates. Assays were set up using streptavidin biosensors (ForteBio, 18-5020) according to the manufacturer’s instructions. Biotinylated human CGRPα (125 nM) was loaded onto biosensors for 5 min for a wavelength shift of approximately 1.5 nm. Association was measured by incubating loaded biosensors with the unpurified or purified scFv. Dissociation was measured following transfer into assay buffer. Data were analyzed using the Octet data analysis software version 7.0. The data were reference subtracted using either a buffer or negative control and aligned to the baseline prior to association. Global data analysis was used to analyze scFv titrations, while partial local data analysis was used to evaluate single concentrations of scFv. Both data analysis models assume a simple monovalent interaction.
Results
HTRF Binding Assays Identified CGRP-Specific, Species Cross-Reactive scFv
To isolate anti-human CGRP antibodies, phage display selections were performed on naïve scFv antibody libraries using biotinylated human CGRPα peptide in solution. Individual scFv antibodies were then prepared as unpurified E. coli periplasmic extracts and screened for binding to human CGRPα and for cross-reactivity to rodent CGRPα in a simple HTRF binding assay format. In this case, rodent CGRPα refers to mouse and rat CGRPα, as they are identical in sequence. 24 When scFv bind to CGRP, a fluorescence resonance energy transfer (FRET) complex is formed in which biotinylated CGRP is detected with streptavidin–europium cryptate fluorescent donor and the scFv is detected with an anti-His tag antibody labeled with the acceptor fluorophore, XL665. Binding of the scFv to CGRP brings the donor cryptate molecule and the XL665 acceptor fluorophore into close proximity such that excitation of the donor results in FRET to the acceptor, which then emits a fluorescent signal. To ensure only the strongest CGRP binding scFv were identified, CGRP scFv binders were defined as giving a signal of greater than 200% Delta F. From the 3512 unpurified scFv tested, 173 (4.9%) showed binding to both human CGRPα and rodent CGRPα, 259 (7.4%) to human CGRPα alone, and 85 (2.4%) to rodent CGRPα alone. The 173 human/rodent cross-reactive antibodies were sequenced, and a panel of 68 scFv that had unique and diverse amino acid sequences across all complementarity-determining regions (CDRs) were purified for further characterization ( Fig. 1A ).
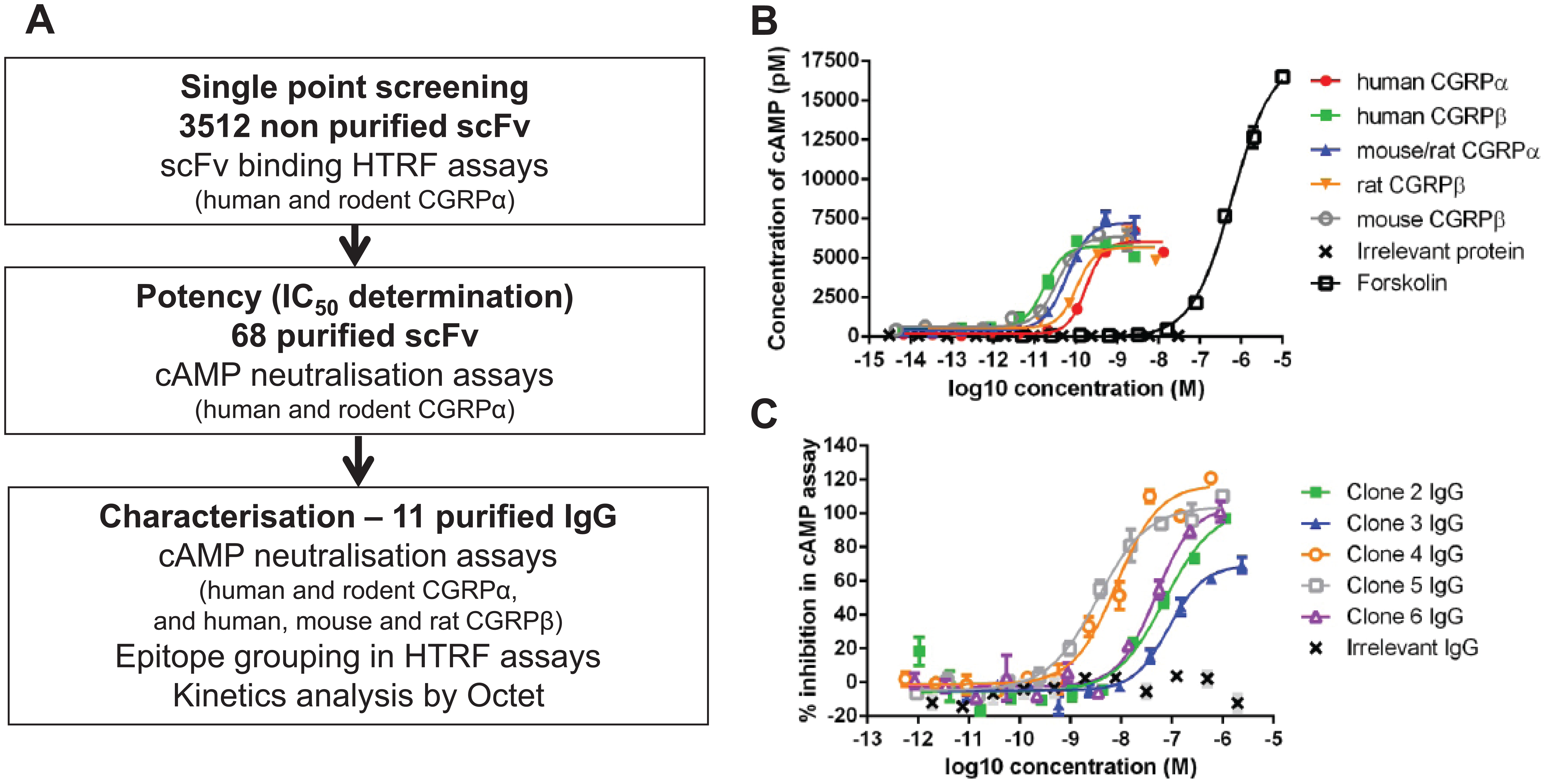
Isolation of anti-CGRP neutralizing antibodies. (
Diverse Panel of CGRP Neutralizing scFv Were Identified in a Functional Assay
The ability of scFv and IgG to inhibit human and rodent CGRP-mediated functional responses was assessed in SK-N-MC cells. This human neuronal epithelial cell line endogenously expresses CALRLR and its associated receptor proteins. cAMP production was stimulated using EC50–80 concentrations of human, mouse, and rat CGRPα and CGRPβ in order to produce equivalent levels of cAMP for each assay ( Fig. 1B ). The assays were used to rank antibodies according to their activities across the CGRP panel rather than for determination of absolute selectivity values.
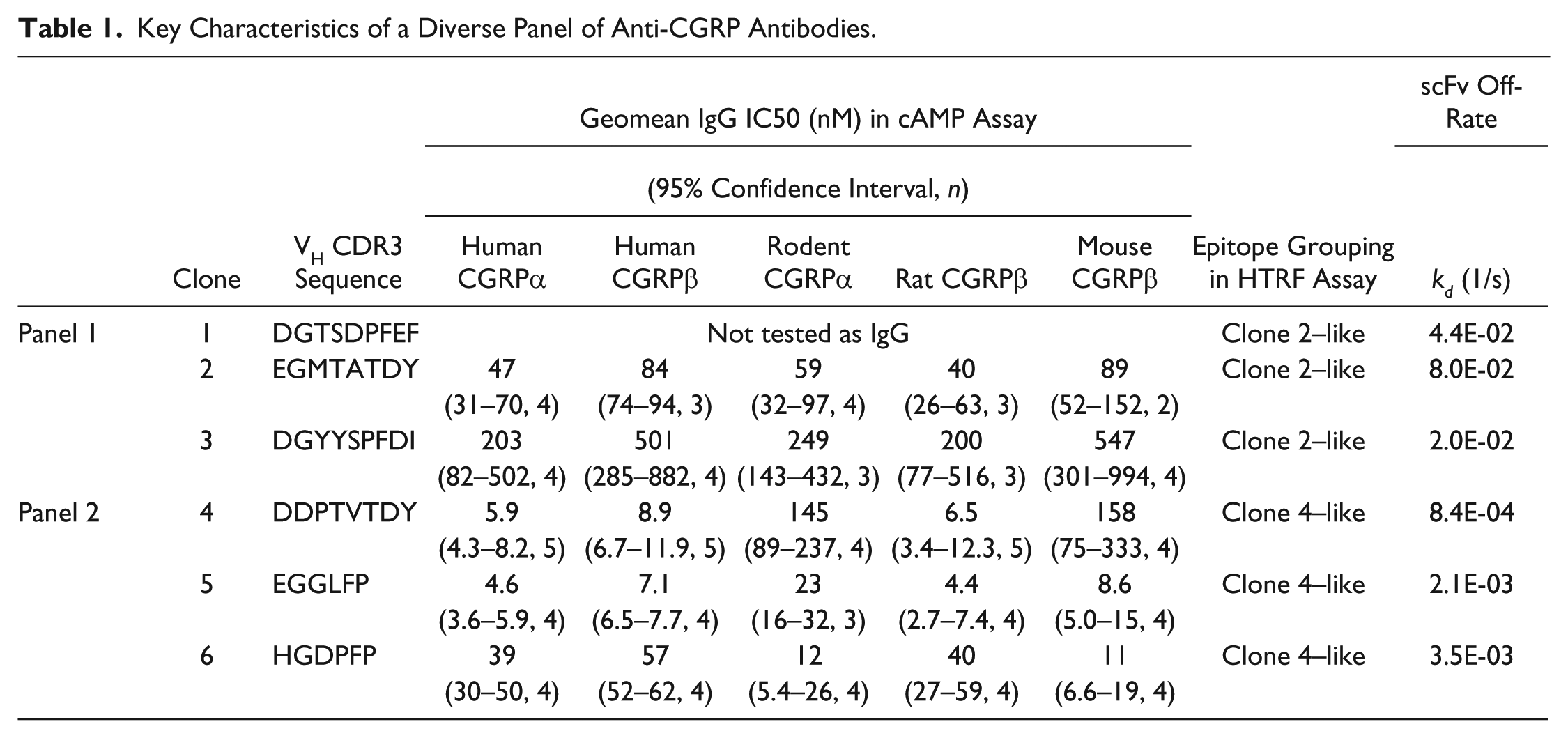
Of the 68 purified scFv profiled for inhibition of human and rodent CGRPα-stimulated cAMP accumulation, 19 were shown to inhibit human and rodent CGRPα, 5 were human CGRPα–only inhibitors, 3 were rodent CGRPα–only inhibitors, and 41 did not inhibit. IC50 values could only be determined for 14 out of the 24 human CGRPα inhibitors and 9 out of the 22 rodent CGRPα inhibitors, as full dose responses were not obtained with the remaining scFv. A panel of 11 of the most potent human and rodent cross-reactive scFv in the cAMP assays were converted to IgG format for activity testing against the human CGRPβ isoform and species cross-reactivity against mouse and rat CGRPβ (the 5 most potent IgG are shown in
Table 1
and
Fig. 1C
;
Key Characteristics of a Diverse Panel of Anti-CGRP Antibodies.
Competition and Off-Rate Ranking Assays Identified Two Distinct Groups of Antibodies
HTRF epitope competition assays were used to characterize the epitope on CGRP recognized by the neutralizing antibodies. In these assays, a FRET complex is formed between biotinylated CGRP, streptavidin–europium cryptate donor, and IgG directly labeled with the acceptor fluorophore. The epitopes bound by unlabeled competing antibodies can be broadly categorized using this assay. Antibodies that bind an overlapping epitope to that bound by the labeled antibody can be detected by a reduction in assay signal. If the antibody binds a discrete nonoverlapping epitope, then simultaneous binding of the labeled and unlabeled antibodies can occur and no competition will be observed. Two epitope competition assays were established using clone 2 IgG and clone 4 IgG, as they showed different species cross-reactivity profiles in the cAMP assays, and therefore were likely to bind to different epitopes. The six most potent purified scFv in the human CGRPα cAMP assay were profiled in the two epitope competition assays as either scFv (clone 1 due to lack of expression as an IgG) or IgG (clones 2–6). It was clear that the antibodies fell into two distinct categories, with nonoverlapping epitopes ( Fig. 2A,B and Table 1 ).
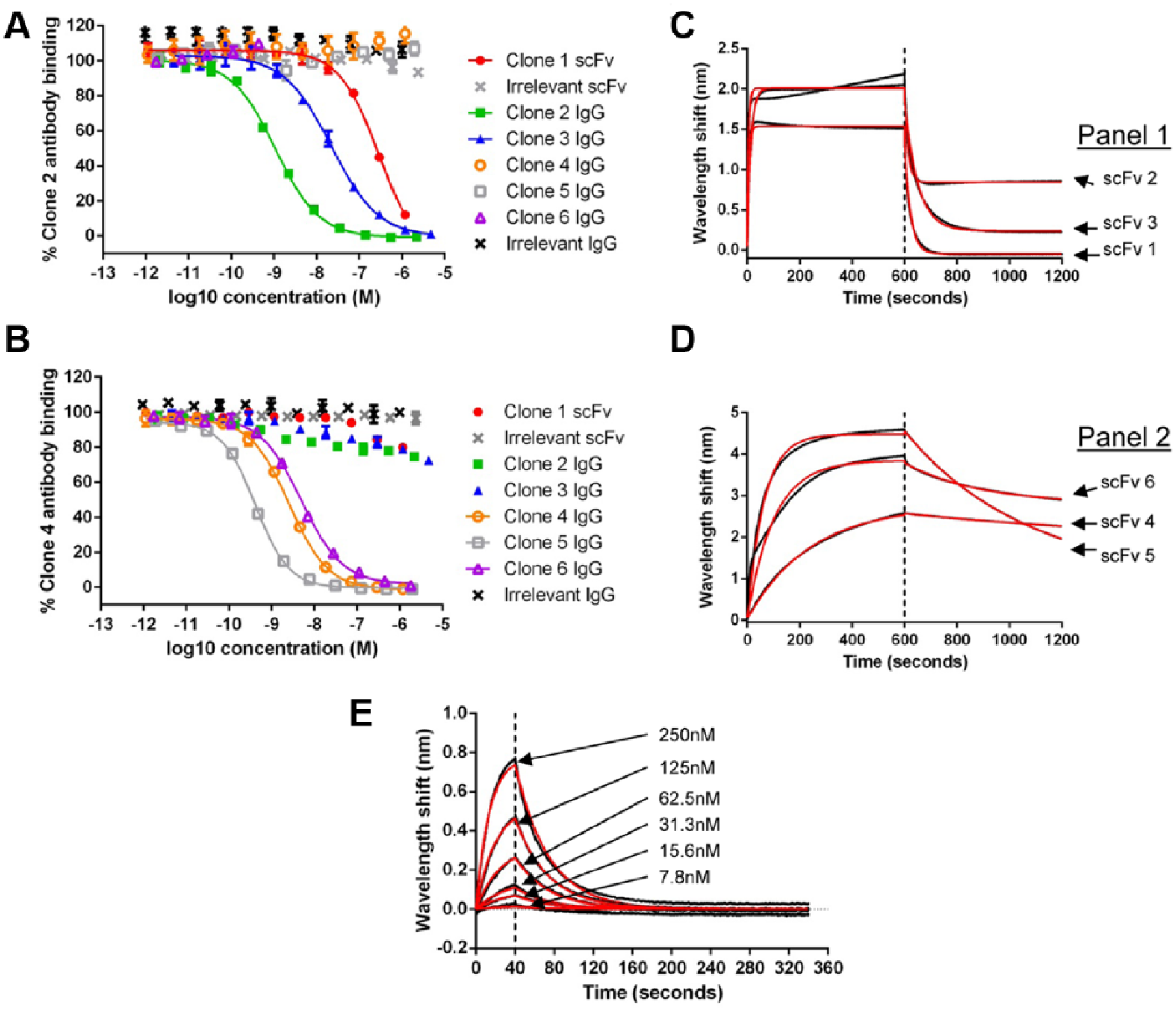
Anti-CGRP scFv recognize distinct epitopes. Representative potency data for six scFv or IgG for inhibition in the clone 2 (
The kinetic profiles of the six scFv were also characterized using BLI (Octet RED384) in a human CGRPα off-rate ranking assay using 1 µM purified scFv. Two distinct off-rate profiles were observed, which was consistent with the groupings in the epitope competition assays. ScFv that competed with clone 2 had fast off-rates, and scFv that competed with clone 4 had approximately 10-fold slower off-rates ( Fig. 2C,D and Table 1 ), providing further support for differences in these two panels of antibodies.
Competition and Off-Rate Ranking Assays Identified Antibodies with Increased Potency
The combination of the HTRF binding assay, cAMP functional assay, HTRF epitope competition assay, and Octet off-rate ranking enabled identification of a panel of six CGRP neutralizing antibodies (
Table 1
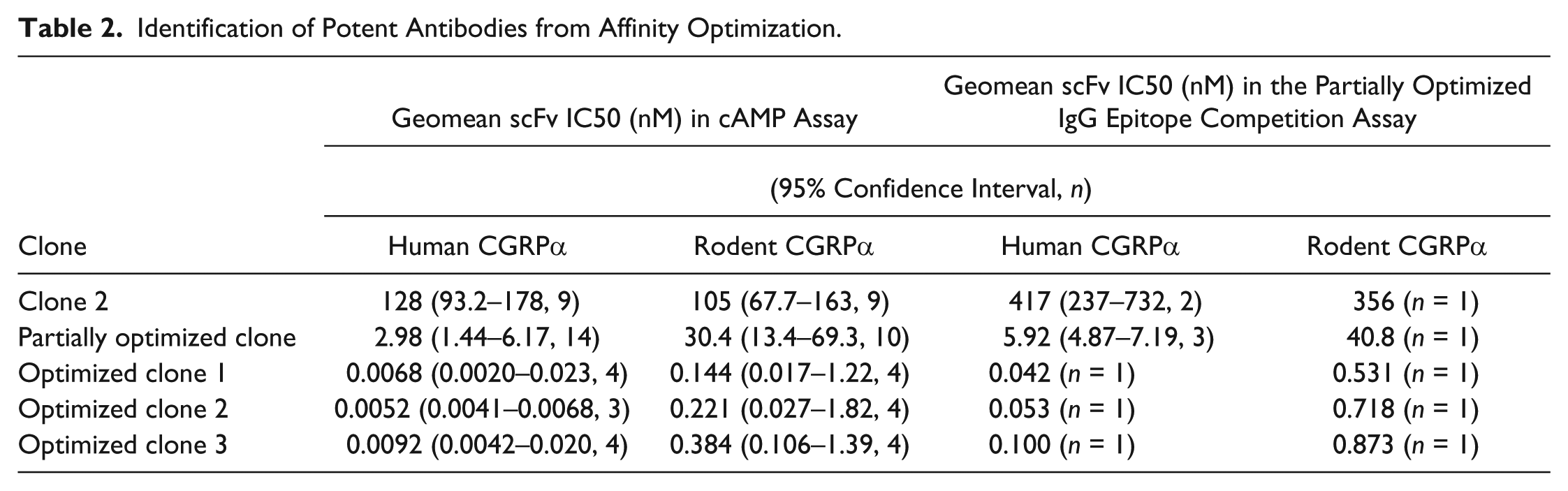
) that had the desired species cross-reactivity and selectivity. From this panel of antibodies, clone 2 exhibited the most favorable species cross-reactivity profile and was estimated to have an affinity of 415 nM using BLI (
Fig. 2E
; additional affinity data are shown in
Affinity optimization of clone 2 was performed by introducing random mutations across the variable regions of clone 2 and its derivatives by error-prone mutagenesis in ribosome display format. Improved variants were enriched by selection on decreasing concentrations of human CGRPα. Affinity-optimized variants derived from clone 2 were identified from this approach by screening unpurified scFv variants in an HTRF epitope competition assay in which the binding of fluorescently labeled clone 2 to biotinylated human CGRPα was measured. ScFv variants that showed greater inhibition than clone 2 parental scFv in this assay were purified and ranked by potency in the same assay to identify those with the greatest increases in affinity. A total of 5632 unpurified scFv were tested in the clone 2 (parent) epitope competition assay ( Fig. 3A ), identifying 426 scFv with increased potency relative to clone 2.
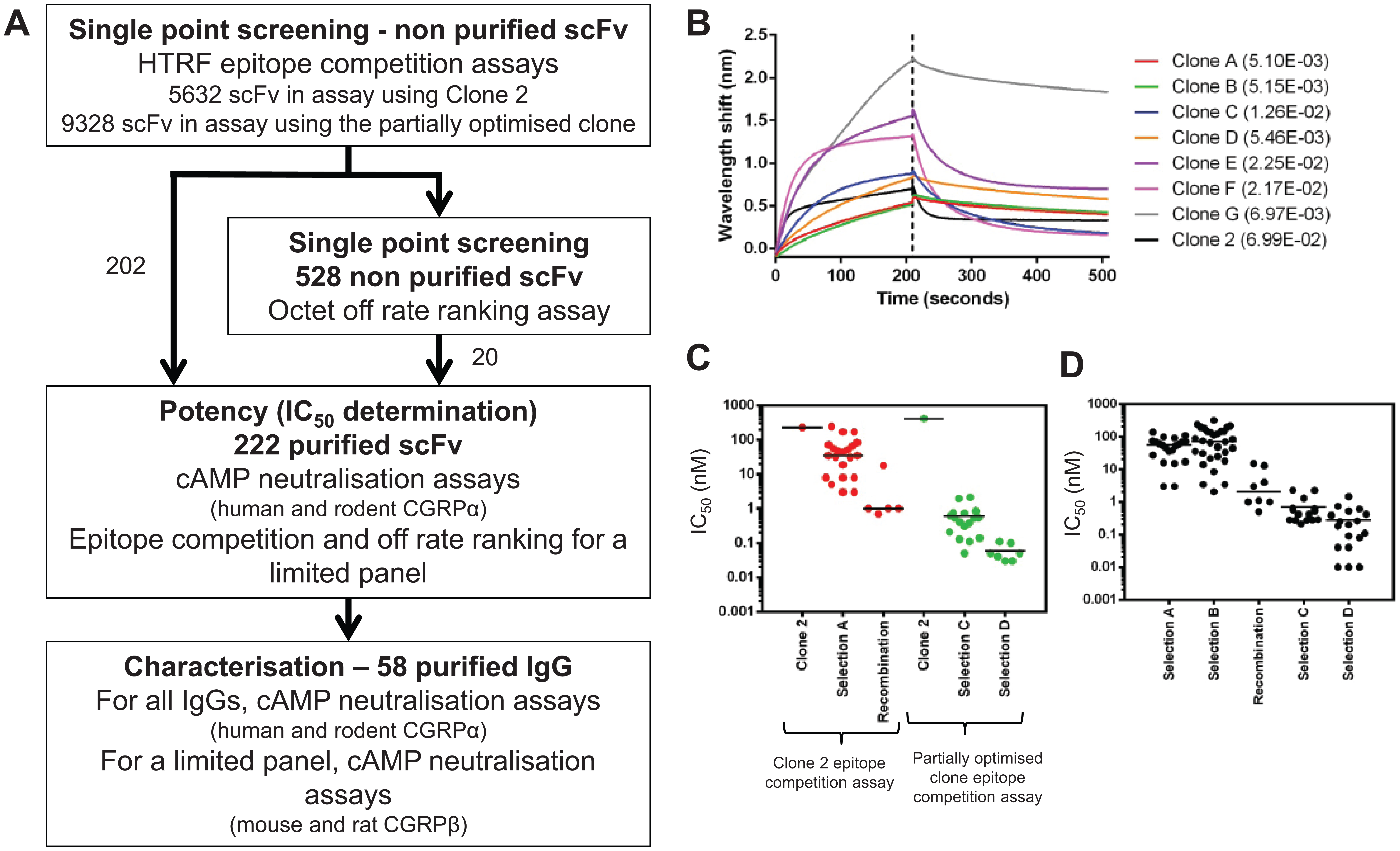
Affinity optimization of clone 2 identified antibodies with improved potency and off-rates. (
As the affinity of antibodies identified in the optimization process increased, a point was reached within the epitope competition assay at which the concentration of CGRP exceeded the affinity of the antibodies being tested. At this stage, the assay could no longer resolve the potency differences of the antibodies, and a more sensitive assay using a higher-affinity antibody was required to enable continued discrimination of the affinity-optimized variants. For this reason, a second, more sensitive epitope competition assay was developed using a fluorescently labeled partially optimized IgG, which was identified by recombination of individual beneficial mutations. This antibody showed the greatest potency improvement in the cAMP assay compared to all the other antibodies that had been profiled at this stage of affinity optimization. A further 9328 unpurified scFv were tested in the more sensitive second epitope competition assay ( Fig. 3A ), identifying 556 scFv with increased potency relative to clone 2.
In addition to potency ranking in the epitope competition assays, a subset of 528 unpurified scFv were evaluated for off-rate improvements early in the course of optimizing clone 2. These clones were chosen based on activity in the epitope competition assay as unpurified scFv. Off-rate was investigated, as typically antibody off-rate is improved during affinity optimization.
26
Using BLI technology, a scFv off-rate ranking Octet assay was developed and validated for use as a single-point screen with unpurified scFv (
Identification of Potent Antibodies from Affinity Optimization.
To confirm that we had identified the most relevant scFv, a comparison of the IC50 values obtained in the assays was performed ( Fig. 4 ). The correlation between off-rate ranking in the Octet assay and potency improvements in the cAMP assay was high, with a Pearson r value of 0.72 (62 data points, 95% confidence interval 0.57–0.82). The correlation between potency improvements in cAMP and the clone 2 epitope competition assay was very high, with a Pearson r value of 0.92 (26 data points, 95% confidence interval 0.82–0.96), but was lower, Pearson r value of 0.55, for the more sensitive epitope competition assay (24 data points, 95% confidence interval 0.19–0.78). The Pearson r value was lower for the second epitope competition assay, as it is likely that it had reached its limit of sensitivity and could not discriminate the most potent antibodies. These results demonstrate that the data obtained from the epitope competition and Octet off-rate assays correlated well with functional potency in the cAMP assay, highlighting that both assays were suitable for use as surrogate screening assays for functional activity.
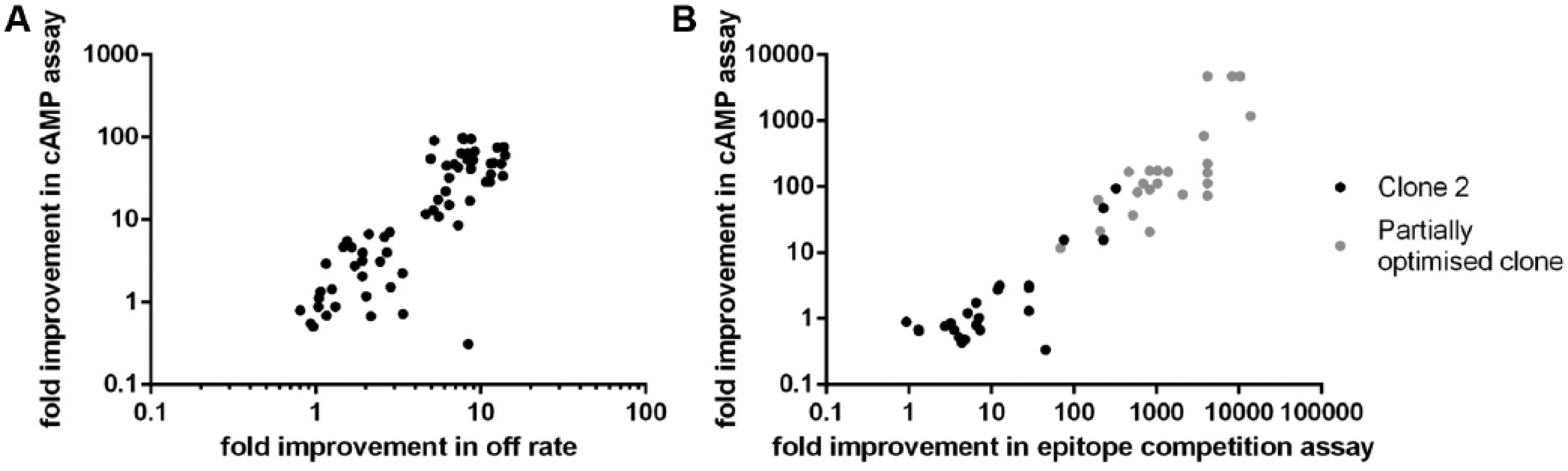
Good correlation was observed between Octet, epitope competition, and cAMP assays. (
Discussion
Four simple assays were used to identify antibodies to the therapeutic target CGRP and to profile these antibodies for desirable characteristics, such as species cross-reactivity. When using phage and ribosome display technologies for antibody isolation or optimization, the screening cascade needs to consider sample tolerance to unpurified periplasmic extracts. The benefits of biochemical assays are that they offer improved tolerance over cell-based assay formats and they can be run in homogenous formats. For the identification of CGRP antibodies, the biochemical assays used were simple mix and read HTRF assays, and thus were preferable to heterogeneous plate-based assays such as ELISA or dissociation-enhanced lanthanide fluorescent immunoassay (DELFIA). Furthermore, the time-resolved measurement of fluorescent emission in the HTRF assays minimized potential optical interference from the unpurified sample preparation. 27 HTRF assays can be optimized quickly using off-the-shelf tool box detection reagents and run in high-density plate formats, thus reducing reagent and sample volume requirements. The limitation of biochemical assays, such as HTRF, is that they may not be predictive of functional activity. In particular, for receptor targets, the purified conformation may not always reflect the native conformation of the target of interest, resulting in an attrition of hits between the biochemical and downstream functional cell assays.
Here we describe HTRF assays for the isolation of antibodies that bind CGRP. In order to reduce high numbers of binders, unpurified scFv were initially screened for binding to human and rodent CGRPα, which share 89% amino acid identity. 28 Cross-reactivity to rodent CGRP was considered a desirable property to enable in vivo pharmacology studies. Even at this high-percentage species identity, rodent cross-reactivity may not necessarily be expected, as just one single amino acid change in an epitope can disrupt the antibody–antigen interaction. 29 However, by screening for human and rodent binding, we successfully identified large numbers of species cross-reactive scFv. As the concentration of scFv in the periplasmic extract was not measured or normalized in the primary screen, the assay did not discriminate between scFv with the strongest binding interaction and those with the highest expression levels. Quantitation of high numbers of unpurified scFv would increase workload and cost; therefore, assays are optimized to detect scFv in the range of 0.1–10 µg/mL, based on in-house 96-well expression data, with the understanding that low-expressing scFv may not be detected in the assay. In addition, the scFv binding assay did not provide an indication of the ability of the antibody to inhibit the binding of CGRP to its receptor. A receptor/ligand competition assay would have been a preferred screen; however, the CGRP receptor is an integral membrane protein, thus endogenous expression levels in cells are typically low and therefore unlikely to give a suitable assay signal. Furthermore, to develop such a homogenous high-throughput assay would potentially require engineering of cell lines for higher expression of the receptor complex.
Despite the drawbacks of using a binding assay as the primary screen, the parallel screening for binding to rodent CGRPα successfully triaged the samples to a manageable number for purification and profiling in a functional assay. To confirm the antibody inhibited the interaction of CGRP with its receptor, we used a 384-well functional assay measuring cAMP to profile the purified scFv and IgG. A fluorometric imaging plate reader (FLIPR) assay to measure intracellular Ca2+ release and an HTRF IPone assay with the SK-N-MC cells were evaluated; however, the assay signals were not sufficient for robust potency testing. A number of commercially available functional assays could have been evaluated, such as β-arrestin recruitment (DiscoveRx) or reporter gene assays (GeneBLAzer, Life Technologies); however, these use cells that overexpress CALRLR and RAMP1. It is preferable to develop assays that use cells endogenously expressing receptor and measure an intrinsic response to best mimic an in vivo setting. CGRP can signal through adenylate cyclase to increase cAMP levels, 5 and we chose to use a well-established CGRP cAMP assay with SK-N-MC cells, 30 which endogenously express CALRLR and RAMP1, for functional profiling of purified scFv and IgG. This provided a robust high-throughput (384-well) format for profiling large numbers of samples in a cell-based assay. The cAMP assay would have ideally been used as the primary screen to confirm functional inhibition and thus reduce attrition rates, but it was not tolerant to unpurified scFv. However, it did provide a single-assay format for comparison of the activity of purified samples for functional inhibition of CGRPα and CGRPβ isoforms (human CGRPα and CGRPβ differ only by three amino acids 28 ) and species orthologs. The sequence identity for human CGRPβ compared to rat CGRPβ is 92%, 28 and although the rat and mouse CGRPα have the same sequence, the CGRPβ isoforms differ by three amino acids. 24 Despite the relatively high sequence homology between the CGRP isoforms for each species, preferential binding was observed for some of the IgG. Therefore, the cAMP assay enabled selection of antibodies that were active across the CGRP isoforms and species orthologs.
Species cross-reactivity was monitored throughout affinity optimization by profiling purified scFv in the human and rodent CGRPα cAMP assays. At the early stages of affinity optimization, the potency improvements against rodent CGRPα were less than those observed against human CGRPα (
Epitope competition assays and Octet off-rate measurements complemented each other to provide information that guided the selection of a single antibody for affinity optimization. Epitope competition assays enabled grouping of the antibodies into two clear categories according to the epitope recognized on CGRP. The Octet off-rate assay supported this epitope grouping and provided information regarding the kinetic profile of the IgG, facilitating selection of an antibody for optimization based on its relatively fast off-rate.
For antibody optimization, we typically use epitope competition assays as surrogate assays for affinity improvements, as these are high-throughput cost-effective assays that use a low sample volume. However, the disadvantage of using these as single-point assays is that they are highly dependent on scFv expression. Therefore, an alternative higher-throughput primary screening assay that is concentration independent and directly measures kinetic properties was evaluated as part of the screening campaign. We used the HTRF epitope competition assay to rapidly triage samples, and ranked by off-rate a select subset of scFv inhibitors to remove any potential bias in scFv selection due to sample concentration. This strategy was adopted because it would be less cost-effective to run the BLI off-rate assay as a primary screen, as the biosensors were not regenerated. For the BLI assay, maximum wavelength shifts did vary between samples, and this was linked to concentration, but it had no effect on the ability to measure off-rate, except for when the wavelength shift was insufficient for measurement. While affinity ranking would be beneficial, this is not possible with unpurified scFv, as on-rates are not always fitted well (as seen in
Fig. 2C
); however, off-rates can be successfully ranked using unpurified sample types.
17
The BLI assay was useful for the early stages of affinity optimization where the off-rate data fit well (as seen in
Fig. 3B
and
In conclusion, we have shown that there are advantages and disadvantages to using each assay format; however, by using a combination of diverse assay technologies at different stages of the screening cascade, we were able to isolate a panel of functionally diverse antibodies to CGRP. Furthermore, we demonstrate that the use of a combination of HTRF epitope competition and BLI assays can serve as a suitable substitute for a cell-based screen for identification of functional antibodies. This is of significance, as it is the first time off-rate ranking on CGRP has been shown to be predictive of function for the majority of clones; however, there are limitations to this assay, as discussed earlier. Off-rate BLI is potentially applicable to other monomeric soluble targets, but it would require empirical determination. Finally, we have demonstrated how a novel combination of a simple set of assays can be used for the isolation and characterization of antibodies to CGRP. The true value of any anti-CGRP antibody as therapeutics would have to be determined in more disease-relevant cell-based assays and in vivo models. This combination of assay technologies described could also be applied for isolation of antibodies to other soluble monomeric therapeutic targets.
Footnotes
Acknowledgements
We would like to thank all of the support functions at MedImmune who have contributed to the success of this project. In particular, we would like to thank the Biologics Expression and Tissue Culture teams, Bioinformatics and Laboratory Support Services.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.