
Abstract
Endoplasmic reticulum (ER) stress activates three distinct signal transducers on the ER membrane. Inositol-requiring protein 1 (IRE1), the most conserved signal transducer, plays a key role in ER stress-mediated signaling. During ER stress, IRE1 initiates two discrete signaling cascades: the “adaptive” signaling cascade mediated by the XBP1 pathway and the “alarm” signaling cascade mediated by stress-activated protein kinase pathways. Fine-tuning of the balance between the adaptive and alarm signals contributes significantly to cellular fate under ER stress. Thus, we propose that the design of high-throughput screening (HTS) assays to selectively monitor IRE1 mediated-signaling would be desirable for drug discovery. To this end, we report the generation of stable human neural cell lines and development of cell-based HTS luciferase (Luc) reporter gene assays for the identification of pathway-specific chemical modulators of IRE1. We implemented a cell-based Luc assay using a chimeric CHOP-Gal4 transcription factor in 384-well format for monitoring IRE1 kinase-mediated p38MAPK activation and an unfolded response pathway element (URPE)–Luc cell-based assay in 1536-well format for monitoring IRE1’s RNase-mediated activation of XBP1. Chemical library screening was successfully conducted with both the CHOP/Gal4-Luc cells and UPRE-Luc engineered cells. The studies demonstrate the feasibility of using these HTS assays for discovery of pathway-selective modulators of IRE1.
Introduction
The endoplasmic reticulum (ER) is a dynamic intracellular organelle essential for the regulation of protein homeostasis. Accumulation of unfolded/misfolded proteins inside the ER lumen causes ER stress and activates signal transducers located on the ER membranes, which initiate an integrated intracellular signal transduction pathway collectively known as the unfolded protein response (UPR). 1 The UPR initially increases ER folding capacity and decreases incoming protein load to restore protein homeostasis in the ER. However, if homeostasis cannot be restored, UPR signaling eventually promotes apoptosis. Three distinct ER signal transducers have been identified, including inositol-requiring protein 1 (IRE1), protein kinase RNA (PKR)–like ER kinase (PERK), and activating transcription factor 6 (ATF6). Among them, IRE1 represents the most ancient and conserved transducer of the UPR capable of regulating cell fate. 2
IRE1 is a type 1 transmembrane ER-resident protein that contains an N-terminal luminal domain, a single-pass transmembrane domain, and C-terminal kinase and endoribonuclease (RNase) effector domains that lie outside of the ER. The luminal domain senses protein folding status in the ER and initiates signal transduction from the ER lumen to the cytosol and nucleus via the effector domains. The only known substrate of IRE1 kinase is IRE1 itself. During ER stress, oligomerization of the luminal domain of IRE1 triggered by misfolded proteins juxtaposes the kinase domains of IRE1 within the oligomer, allowing trans-autophosphorylation, which provides positive feedback for oligomer assembly and, in turn, activates the RNase activity of IRE1. 3 Studies have shown that the conformational change triggered by phosphorylation, rather than phosphorylation per se, is crucial because RNase activity can be selectively activated bypassing kinase activity. 4 Activated IRE1 causes nonconventional cleavage of a 26-nucleotide (nt) intron from x-box binding protein 1 (XBP1) messenger RNA (mRNA), which shifts the open reading frame and encodes a potent transcription factor. 5 XBP1 enhances ER biogenesis and upregulates transcription of genes encoding ER-resident chaperones and components of ER-associated degradation (ERAD), thereby augmenting ER folding capacity and reducing misfolded protein load. 2
In addition to splicing XBP1 mRNA, IRE1 also initiates a signaling cascade independent from its RNase activity to promote apoptosis following sustained or severe ER stress. ER stress-induced activation of IRE1 kinase activity is essential for recruiting the adaptor protein TNFR-associated factor 2 (TRAF2). Association of IRE1 with TRAF2 triggers phosphorylation of apoptosis signal-regulating kinase 1 (ASK1) and activates the downstream stress-activated protein kinases c-Jun N-terminal kinase (JNK) and p38 mitogen-activated protein kinase (p38MAPK). 6 Activation of the ASK1 signaling cascade has been implicated as a key element in ER stress-induced cell death. 7 Thus, IRE1 has a dual function and initiates two discrete signaling cascades in ER stress: the “adaptive” signaling cascade mediated by the IRE1 RNase/XBP1 pathway and the “alarm” signaling cascade mediated by the IRE1 kinase/ASK1/JNK and p38MAPK pathways. Fine-tuning of the delicate balance between the adaptive and alarm signals determines cellular fate.
UPR activation has been observed in many common diseases, including cancer, neurodegenerative diseases, inflammation, diabetes, viral infections, atherosclerosis, and myocardial infarction. Emerging evidence supports a pathogenic role of IRE1-mediated signaling cascades in diverse diseases. For example, activation of the IRE1 RNase/XBP1 pathway is a common finding in solid tumors as well as multiple myeloma and is important for tumor cell survival under stressful conditions. XBP1 upregulates the transcription of proangiogenic factors, including vascular endothelial growth factor, and is crucial for angiogenesis in xenograft tumor models. Deletion of XBP1 increases tumor cell susceptibility to hypoxia-induced cell death; likewise, inhibition of IRE1-mediated XBP1 splicing strongly suppresses tumor cell growth. 8 The IRE1 RNase/XBP1 pathway also plays an important role in the pathogenesis of inflammatory bowel diseases. 9 XBP1 deletion results in spontaneous enteritis and increased susceptibility to induced colitis. Hypomorphic XBP1 variants have been associated with both Crohn disease and ulcerative colitis. 10 Both arms of the IRE1-mediated signaling cascades in ER stress have been linked to diabetes. 11 XBP1 is required for insulin production and glucose-stimulated insulin secretion. Mice deficient in XBP1 display glucose intolerance and modest hyperglycemia. On the other hand, hyperactivation of JNK during obesity-associated ER stress phosphorylates insulin receptor substrate 1 (IRS-1) and suppresses insulin receptor signaling.
Increasing evidence indicates that ASK1 activation plays a key role in the pathogenesis of neurodegenerative diseases such as Huntington disease, Parkinson disease, and Alzheimer disease. Expanded polyglutamine (polyQ), a hallmark of a number of neurodegenerative disorders, including Huntington disease, triggers ER stress through proteasomal dysfunction. 7 Deletion of ASK1 or inhibition of ASK1 activation protects neurons from ER stress-induced cell death. 7 Amyloid beta (Aβ), the main component of senile plaques in Alzheimer disease, activates ASK1, and deletion of ASK1 protects against Aβ-induced neurotoxicity. 12 ASK1 activity has been observed in the brains of transgenic mice that overexpress α-synuclein, a key pathogenic protein in Parkinson disease. Deletion of ASK1 in these animals attenuates neuronal damage and improves motor performance. 13 JNK activation during ER stress has also been demonstrated to contribute to macrophage apoptosis, leading to vulnerable plaque formation in atherosclerosis. 14 Importantly, ASK1 is involved in left ventricular remodeling, and deletion of ASK1 reduces myocardial cell apoptosis and protects from heart failure in mouse models. 15
Given the evidence for a context-dependent pathogenic role of IRE1-mediated signaling cascades in diverse disease processes, it has become attractive to selectively modulate IRE signaling cascades relevant to specific diseases. For example, inhibition of the IRE1 RNase/XBP1 pathway may inhibit angiogenesis and promote cancer cell apoptosis. On the other hand, inhibition of the IRE1 kinase/ASK1 pathway may prevent neuronal apoptosis in neurodegenerative disorders. Therefore, the design of high-throughput assays to selectively monitor stress kinase stimulation resulting from IRE1 activation versus IRE1’s RNase mediated-signaling would be desirable for drug discovery. In this regard, compounds that inhibit IRE1 RNase activity and promote its ability to stimulate stress kinases may be useful for eradicating some types of unwanted cells, such as for cancer therapeutics discovery and possibly for eliminating autoimmune lymphocytes. Conversely, compounds that inhibit IRE1-mediated stress kinase activation while enhancing IRE1’s RNase activity may have cytoprotective activity, which could be useful for various diseases, including neurodegenerative disorders, islet cell preservation (diabetes), and heart failure (reviewed in Sano and Reed 16 ). To this end, we report the generation of stable cell lines and the development of luciferase reporter gene-based high-throughput assays for the identification of pathway-specific chemical modulators of IRE1 ( Fig. 1 ). We performed a pilot screen with a goal to identify IRE1 dual modulators that inhibit IRE1-mediated stress kinase activation and enhance IRE1’s RNase activity. These assays may also be used for small interfering RNA (siRNA)–based functional genomic screening for identifying cellular modulators of IRE1 signaling.
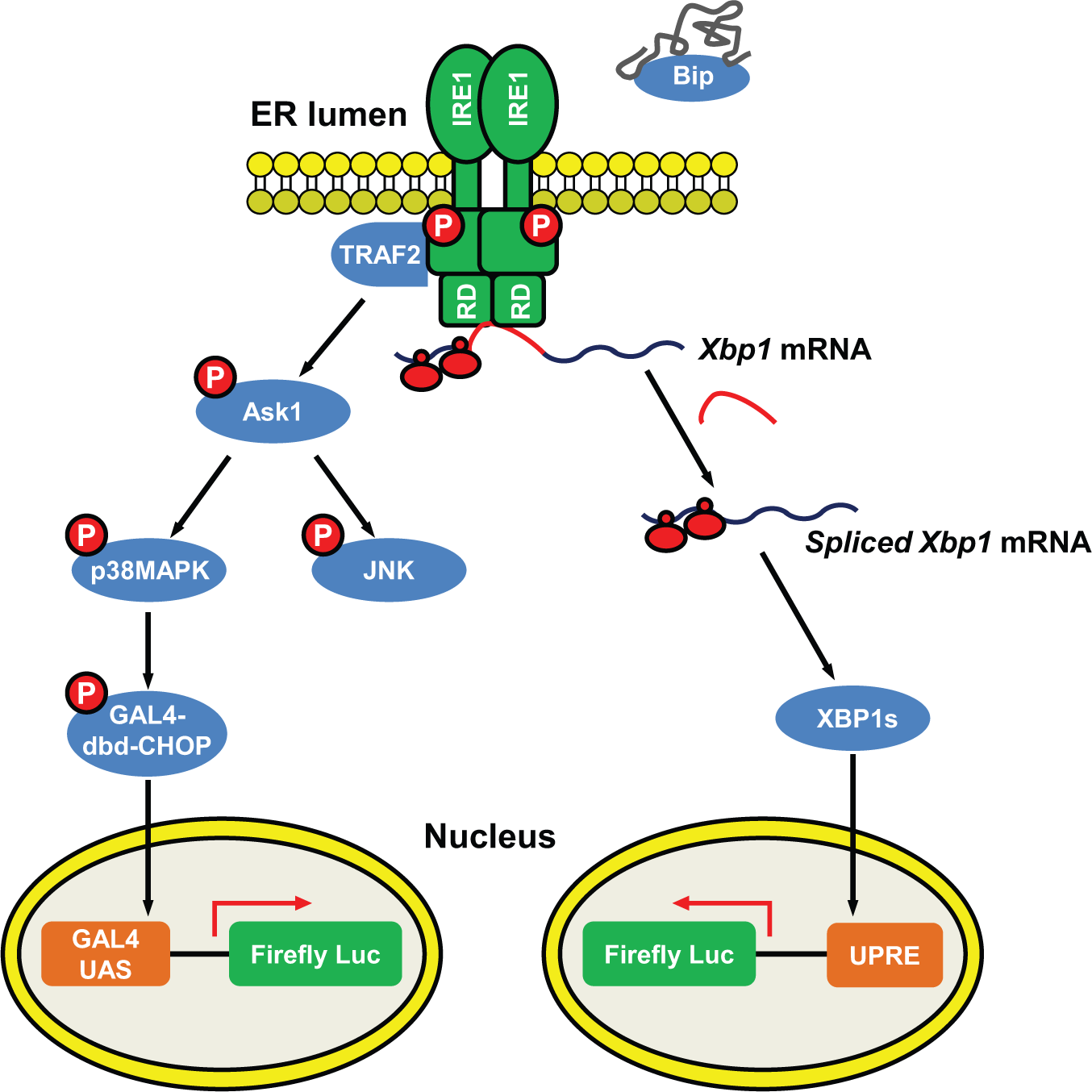
Strategy for selectively monitoring inositol-requiring protein 1 (IRE1)–mediated signaling in cells. IRE1 is an endoplasmic reticulum (ER)–resident protein containing an N-terminal ER luminal domain, a transmembrane domain, and C-terminal cytosolic kinase and RNase effector domains. During ER stress, ER chaperone Bip binds to unfolded proteins and dissociates from the luminal domain of IRE1. IRE1’s RNase domain (RD) is allosterically activated by oligomerization and trans-autophosphorylation of the kinase domain. IRE1 RNase domain splices XBP1 messenger RNA (mRNA), which shifts the reading frame and encodes a potent transcriptional factor, XBP1s. The activated IRE1 kinase domain recruits TRAF2 and ASK1, which activates downstream stress kinases JNK and p38MAPK. To monitor IRE1-XBP1 activation, we generated cells stably expressing the firefly luciferase reporter under the control of UPRE. UPRE-mediated transcriptional induction is dependent on XBP1 activity. To monitor IRE1-stress kinase activity, we generated cells stably expressing the PathDetect CHOP trans-reporting system (Agilent Technologies, Santa Clara, CA), which consists of a chimeric transcription factor containing the p38MAPK-responsive transactivation domain of CHOP.
Materials and Methods
Cell Culture and Plasmid Transfection
Human neuroglioma H4 cells and HeLa cells were propagated and maintained in Dulbecco’s modified Eagle’s medium (DMEM; Corning Life Sciences, Corning, NY) supplemented with 10% fetal bovine serum (FBS; HyClone, Logan, UT), 100 IU/mL penicillin, and 100 µg/mL streptomycin with 5% CO2 at 37 °C. Lipofectamine 2000 (Life Technologies, Carlsbad, CA) was used for plasmid transfection. Briefly, cells were cultured overnight and then transfected with plasmid DNA and Lipofectamine 2000 mix in Opti-MEM I–reduced serum medium at a 1:1 (µg/µL) ratio according to the manufacturer’s protocol.
High-Throughput Screening Assays
H4-CHOP-Luc cells were dispensed into Greiner (Monroe, NC) 384-well TC-treated cell culture white plates at a cell density of 103 cells per well in 50 µL DMEM (without phenol red), 10% FBS, 100 IU/mL penicillin, and 100 µg/mL streptomycin using a Mutidrop Combi (Thermo Fisher Scientific Inc, Pittsburgh, PA), followed by brief low-speed (1000 rpm) centrifugation. After 24 h of incubation, the medium was removed by brief inverted low-speed (1000 rpm) centrifugation. First, 20 µL serum-free DMEM (without phenol red) with 100 IU/mL penicillin and 100 µg/mL streptomycin was added to the wells using a Mutidrop Combi. Next, 50 nL of 10-mM compounds in DMSO or control DMSO was dispensed into the wells using a Labcyte Echo 550 acoustic pipetter (Agilent Technologies, Santa Clara, CA). Last, 25 µL serum-free DMEM (without phenol red) with 100 IU/mL penicillin and 100 µg/mL streptomycin was added using a Mutidrop Combi. After a 1-h incubation at 37 °C with 5% CO2, 5 µL of 100 nM thapsigargin in 1% DMSO or control DMSO was added to the wells using a Biomek FX liquid handler (Beckman Coulter, Brea, CA). The final concentration of thapsigargin was 10 nM, at approximately EC80, for the assay. The final concentration of compounds was 10 µM. The final concentration of DMSO was 0.2% (v/v). The plates were incubated with 5% CO2 at 37 °C for an additional 18 h. After a 30-min room temperature equilibration, 25 µL Bright-Glo reagents (Promega, Madison, WI) was added to each well using a Mutidrop Combi, and the plates were imaged and analyzed with a ViewLux uHTS Plate Imager (PerkinElmer, Waltham, MA). We screened the ChemBridge DIVERSet Library (57,600 compounds) in a 384-well format in the Conrad Prebys Center for Chemical Genomics at the Sanford-Burnham Medical Research Institute (La Jolla, CA). All plates were analyzed for Z factor with a minimum acceptable value of 0.5 for thapsigargin control. Signal to noise was also measured to confirm assay quality. The data were normalized to the positive control (10 nM thapsigargin) as 0% inhibition and cells alone without thapsigargin as 100% inhibition. Compounds that caused 50% inhibition were selected as primary hits and retested in triplets.
H4-UPRE-Luc cells were dispensed into Corning (Corning, NY) 1536-well TC-treated cell culture white plates at a cell density of 103 cells per well in 5 µL DMEM (without phenol red) with 10% FBS, 100 IU/mL penicillin, and 100 µg/mL streptomycin using a Mutidrop Combi, followed with brief low-speed (1000 rpm) centrifugation. After overnight (16–18 h) incubation, 5 nL of 10-mM compounds in DMSO or control DMSO was dispensed into the wells using a Labcyte Echo 550 acoustic pipetter. Thapsigargin was used as a qualitative assay control. The final concentration of compounds was 10 µM. The final concentration of DMSO was 0.1%. The plates were covered with Kalypsys (San Diego, CA) lids and incubated with 5% CO2 at 37 °C for an additional 24 h. After a 30-min room temperature equilibration, 3 µL Bright-Glo reagents was added to each well using a Mutidrop Combi, and the plates were imaged and analyzed with a ViewLux uHTS Plate Imager (PerkinElmer). We screened the Sanford-Burnham Medical Research Institute’s high-throughput screening (HTS) library (320,000 compounds) in a 1536-well format in the Conrad Prebys Center for Chemical Genomics at the Sanford-Burnham Medical Research Institute. All plates were analyzed for Z factor with a minimum acceptable value of 0.5 for thapsigargin control. Signal to background was also measured to confirm assay quality. The entire data set was used to calculate a Z score, and compounds that were six times the Z score were selected as primary hits and retested in triplets using a hit criterion of 3 times the Z score.
Reverse Transcriptase PCR Analysis of XBP1 Splicing
H4 cells were seeded at 1.5 × 105 cells in 96-well plates and cultured in medium containing 10% FBS. The next day, the cells were treated with the ER stress inducer tunicamycin or compounds in serum-free medium for 7 h. Total RNA was extracted using the RNeasy plus mini kit (Qiagen, Valencia, CA). Complementary DNA (cDNA) was synthesized using the SuperScript III first-strand synthesis kit (Invitrogen, Carlsbad, CA). Reverse transcriptase (RT)–PCR was performed as previously described. 17 Primers used for XBP1 were 5′-TTACGAGAGAAAACTCATGGC-3′ and 5′-GGGTCCA AGTTGTCCAGAATGC-3′. The following RT-PCR conditions were used: 50 °C for 30 min; 94 °C for 2 min; 35 cycles of 94 °C for 30 s, 58 °C for 30 s, and 72 °C for 30 s; and 72 °C for 10 min. The PCR products were resolved on a 3% Metaphor agarose gel.
Statistical Analysis
Statistical significance (p < 0.05) was assessed by using the Student t test and one-way analysis of variance (ANOVA). Calculations were performed by using Prism 5 software (GraphPad Software, La Jolla, CA). Four-parameter nonlinear regression dose-response curve fits and calculations of Hill slope, EC80, EC50, and IC50 values were performed by using Prism 5 software (GraphPad Software).
Results
Strategy for Selectively Monitoring IRE1-Mediated Signaling
IRE1 initiates two distinct signaling cascades in ER stress: (1) the kinase-dependent recruitment of TRAF2, leading to activation of ASK and downstream JNK and p38MAPK stress kinases, and (2) the RNase-dependent splicing of XBP1, leading to generation of a potent transcriptional factor XBP1. We proposed to establish stable cells and selectively monitor each of these signaling cascades using luciferase-based reporter gene assays ( Fig. 1 ). To monitor p38MAPK activation, we used the PathDetect CHOP trans-Reporting System (Agilent Technologies). In this system, activation of endogenous p38MAPK phosphorylates the CHOP transactivation domain fused downstream of the GAL4 DNA binding domain (GAL4 DBD), which binds the GAL4 upstream activation sequence (GAL4 UAS) and activates transcription of the luciferase reporter. Therefore, luciferase induction reflects the p38MAPK activation status. Thus, this assay differs from a prior report 18 that described methods for measuring CHOP gene promoter activity, which is regulated by all three ER stress transducers: IRE1, PERK, and ATF6. 16 To monitor XBP1 activity, we used a luciferase reporter fused downstream of a cis-acting mammalian UPR element (UPRE). UPRE-mediated transcriptional induction was reported to be solely dependent on XBP1 activity. 19 We also generated a luciferase reporter fused immediately downstream of the XBP1 splicing site to directly monitor XBP1 splicing. Removal of the 26-nt intron from XBP1 mRNA introduces a frame shift and allows transcription of luciferase.
Establishment of Stable H4-CHOP-Luc Cells to Monitor IRE1-Mediated Stress Kinase (p38MAPK) Activation
We generated human neuroglioma H4 cell lines stably expressing a luciferase reporter gene downstream of the GAL4 UAS by cotransfection of pFR-Luc and pPUR, followed with puromycin selection. Of the 11 puromycin-resistant clones, we selected one clone with the highest luciferase induction when transiently transfected with plasmids encoding the c-Jun transactivation domain fused downstream of GAL4DBD and the upstream MAP kinase kinase kinase (MEKK) (
To determine the optimal incubation duration, luciferase activity was measured after H4-CHOP-Luc cells were treated with thapsigargin for 6 h, 16 h, or 21 h. We determined that 16 h treatment was sufficient to induce luciferase production (
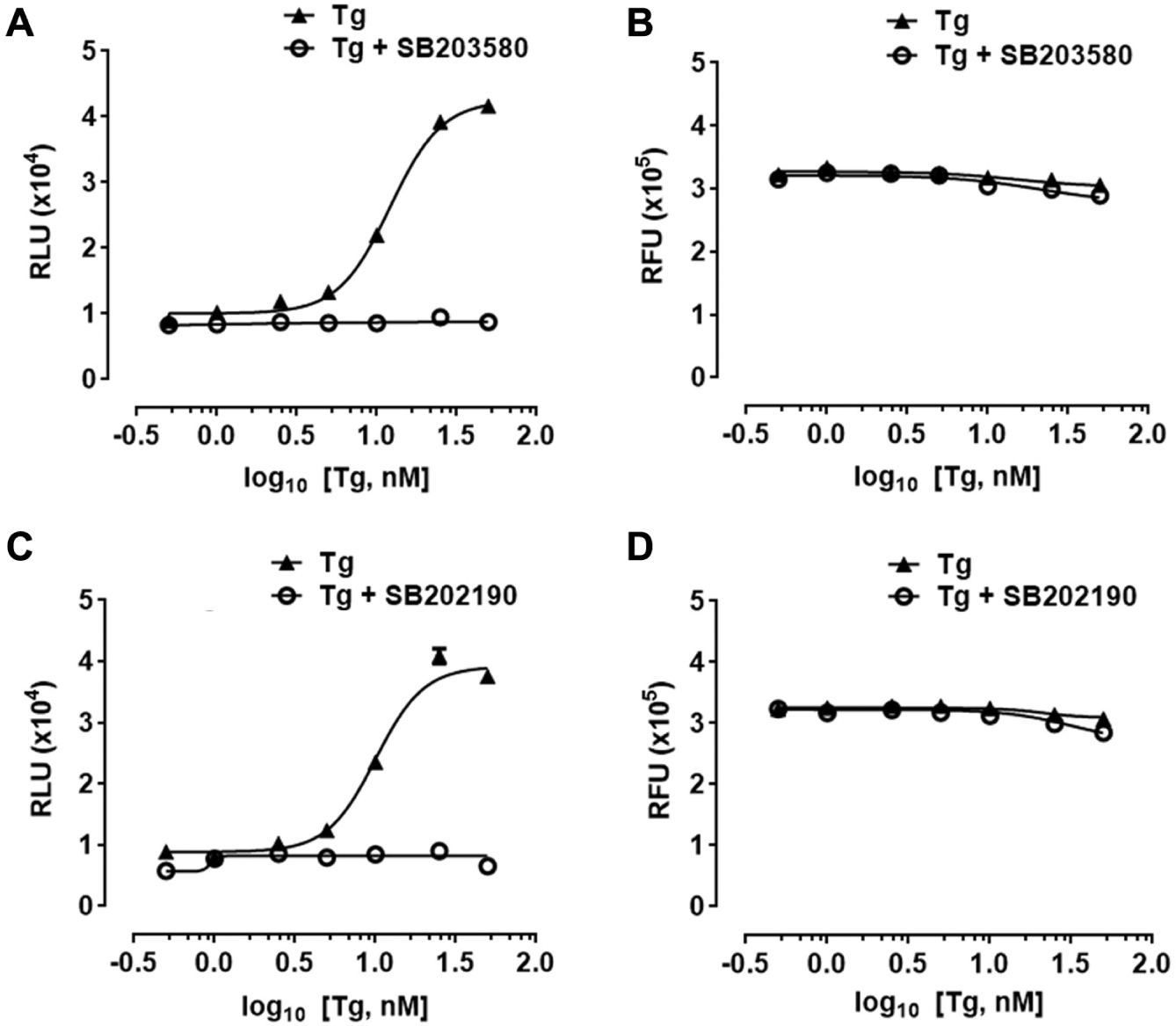
Thapsigargin-induced expression of luciferase in H4-CHOP-Luc cells is p38 MAPK-dependent. (
Thapsigargin-Induced Expression of Luciferase in H4-CHOP-Luc Cells Is p38MAPK and IRE1 Dependent
We compared thapsigargin-induced luciferase production in H4-CHOP-Luc cells with or without p38MAPK inhibitor treatment ( Fig. 2A,C ). Thapsigargin caused a dose-dependent increase in luciferase production in H4-CHOP-Luc cells. However, thapsigargin-induced luciferase production was completely abolished at all concentrations tested in the presence of 10 µM p38MAPK inhibitors SB203580 or SB202190. The inhibitory effect of p38MAPK inhibitors on luciferase induction was not due to cellular toxicity because cellular viability was not significantly affected by the inhibitors ( Fig. 2B,D ). The estimated IC50 of p38MAPK inhibitor SB202190 is 0.58 µM in H4-CHOP-Luc cells, which is higher than the reported IC50 of approximately 50 to 100 nM for p38MAPKα/β obtained by using in vitro biochemical assays employing purified proteins.
We further tested whether thapsigargin-induced luciferase production was dependent on intact IRE1. Three IRE1 siRNAs caused over 80% reduction of IRE1 mRNA levels in H4-CHOP-Luc cells ( Fig. 3A ). Significantly, thapsigargin-induced luciferase production was greatly reduced in the presence of IRE1 siRNAs ( Fig. 3B ). The remaining luciferase level was correlated with residual IRE1 mRNA levels ( Fig. 3B ). Compared with control siRNA-transfected cells, no significant alteration of cellular viability of IRE1 siRNA-transfected cells was observed when treated with 10 nM thapsigargin ( Fig. 3C ).
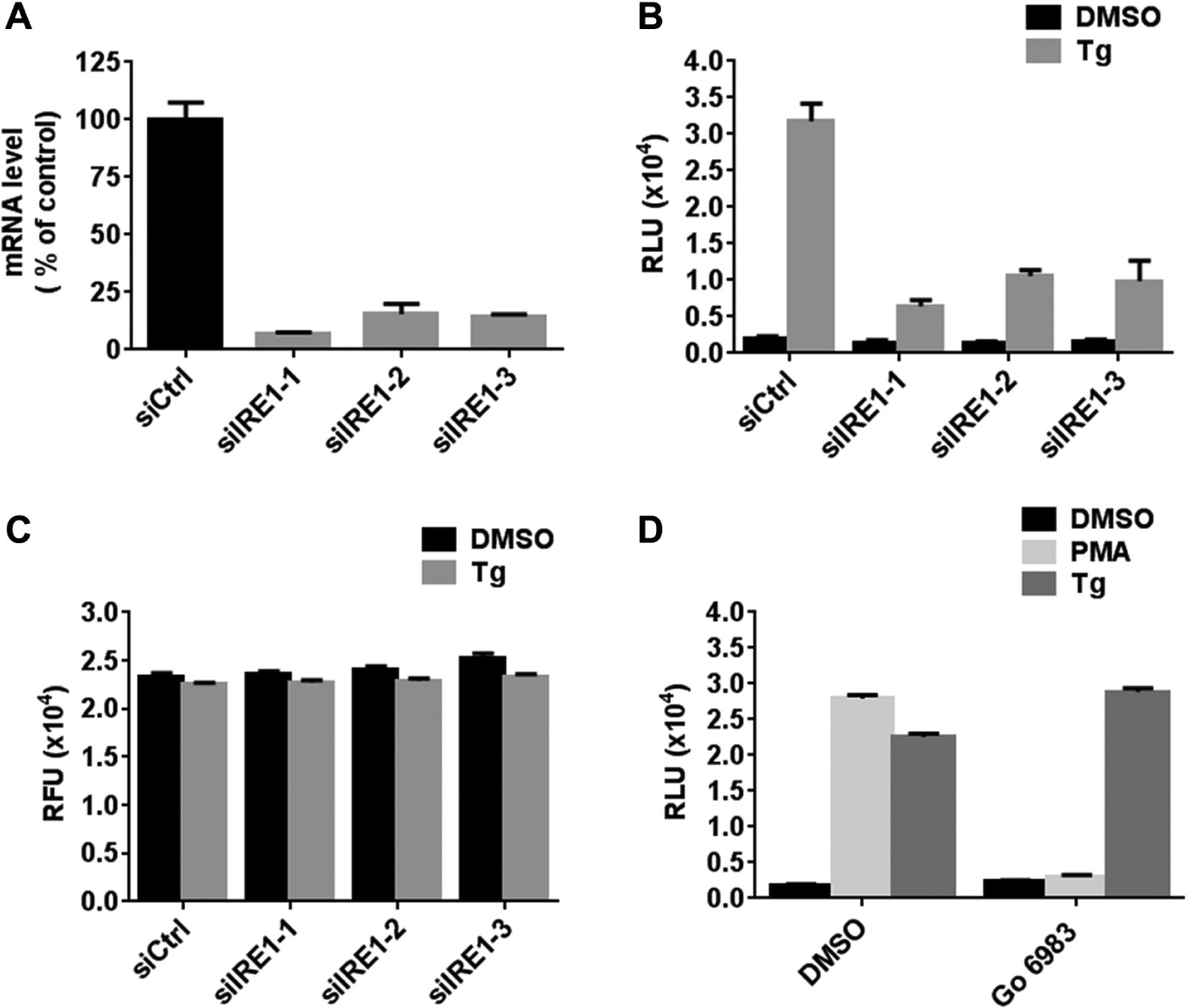
Thapsigargin-induced expression of luciferase in H4-CHOP-Luc cells is inositol-requiring protein 1 (IRE1) dependent. (
Because thapsigargin elevates cytoplasmic Ca2+, we considered that this event might stimulate activation of Ca2+-dependent protein kinases (PKCs) that are capable of initiating kinase cascades leading to p38-MAPK activation independent of ER stress signaling pathways. For example, it was reported that stimulation of PKC by phorbol 12-myristate 13-acetate (PMA) leads to the activation of p38 MAPK and mediates PKC-induced apoptosis in cancer cells. 20 We therefore tested whether thapsigargin activates p38MAPK and induces luciferase production through activation of PKC in H4-CHOP-Luc cells. At a 10-nM concentration, thapsigargin-induced luciferase production was not due to activation of PKC, as illustrated by experiments using PKC inhibitor Gö 6983. In contrast, Gö 6983 completely abolished PMA-induced luciferase production ( Fig. 3D ).
Optimization of H4-CHOP-Luc Cells for High-Throughput Small-Molecule Screen
We set out to optimize the H4-CHOP-Luc assay in a 384-well format at a cell density of 1000 cells per well with time of thapsigargin stimulation of 18 h. Because DMSO is the most common solvent for dissolving chemical compounds, H4-CHOP-Luc cells were tested with varying concentrations of DMSO in the presence or absence of 5 nM thapsigargin to assess DMSO tolerance (
Development of Stable H4-UPRE-Luc and H4-XBP1-Luc Cells
We generated stable H4 cell lines expressing luciferase downstream of UPREs by cotransfection of p5xUPRE-GL3 and pPUR, then followed by puromycin selection. A total of 40 puromycin-resistant clones were further tested by treatment with 10 nM thapsigargin. We selected four clones that showed significant luciferase induction after 8 h of treatment (
H4-UPRE-Luc cells were treated with other ER stress inducers, including tunicamycin (Tm) and brefeldin A (BFA). Both induced significant luciferase production, although less potently than thapsigargin at the concentrations tested (
We also generated stable H4 cell lines expressing luciferase fused immediately downstream of a XBP1 mRNA splicing site by cotransfection of pCAX-F-XBP1delDBD-Luc2 and pPUR, followed by puromycin selection. Removal of the 26-nt intron from XBP1 mRNA introduces a frame shift and allows transcription of luciferase. Unlike H4-UPRE-Luc cells, which monitor XBP1 transcription factor activity, these cells directly monitor XBP1 mRNA splicing. Of 15 puromycin-resistant clones, two showed luciferase induction after thapsigargin treatment (
ER Stress-Induced Expression of Luciferase in H4-UPRE-Luc Cells Is XBP1 and IRE1 Dependent
We tested whether ER stress-induced expression of luciferase in H4-UPRE-Luc cells was dependent on intact XBP1 and IRE1 by using RNA interference (RNAi). The effectiveness of XBP1 siRNA or IRE1 siRNA in gene knockdown is shown in Figure 4A and Figure 3A . All siRNAs tested caused at least 75% reduction in the mRNA level of the target mRNAs. Importantly, compared with control double-strand RNA, XBP1 siRNA pretreatment completely abolished tunicamycin-induced luciferase production in all four H4-UPRE-Luc cell lines ( Fig. 4B ). Furthermore, thapsigargin-induced luciferase production was greatly reduced in H4-UPRE-Luc cells pretreated with each of the three verified IRE1 siRNAs ( Fig. 4C ). Compared with control siRNA-transfected cells, no significant alteration of cellular viability of IRE1 siRNA-transfected cells was observed when treated with 10 nM thapsigargin ( Fig. 4D ). Thus, XBP1 and IRE1 were indispensable for ER stress-induced transcription of luciferase in H4-UPRE-Luc cells.
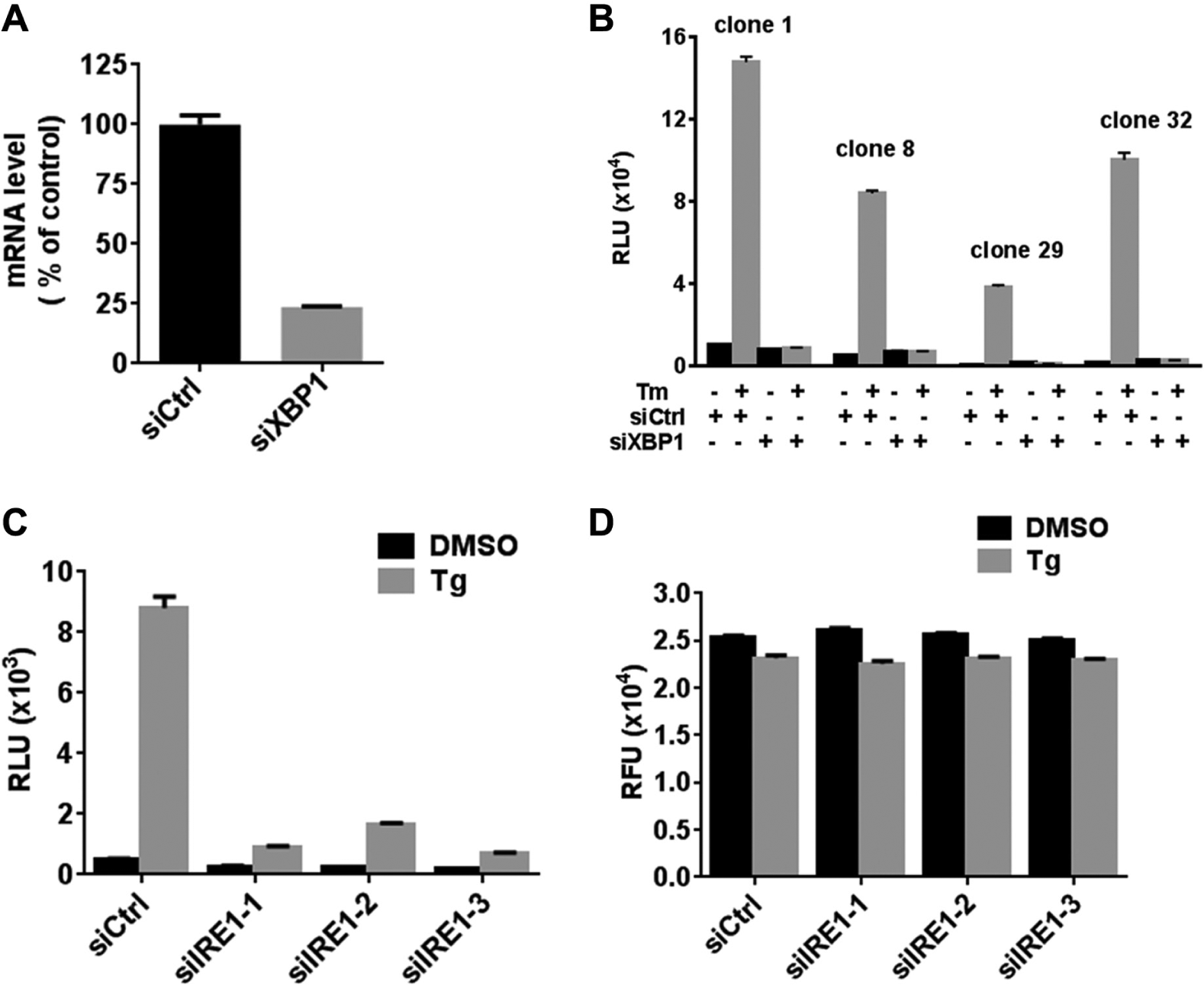
Thapsigargin-induced expression of luciferase in H4-UPRE-Luc cells is XBP1 and inositol-requiring protein 1 (IRE1) dependent. (
Optimization of H4-UPRE-Luc Cells for High-Throughput Chemical Screen
We set out to optimize the UPRE-Luc assay in a 1536-well format with time of thapsigargin stimulation of 24 h. We determined the optimal cell density in a 1536-well format for HTS and calculated the EC50 of thapsigargin for luciferase induction in H4-UPRE-Luc cells. H4-UPRE-Luc cells were seeded at varying cell densities in 1536-well plates, and thapsigargin dose-response experiments were conducted at a final DMSO concentration of 0.1% (v/v). EC50 values and Hill slopes were calculated (
Validation of H4-UPRE-Luc and H4-CHOP-Luc Cell Assays with IRE1 or ER Stress Inhibitors
We validated H4-UPRE-Luc and H4-CHOP-Luc cell assays using a pathway-specific inhibitor of IRE1. Chemical compound 4-methyl umbelliferone 8-carbaldehyde (4µ8C) was previously identified as the most potent inhibitor of IRE1 RNase activity from a chemical screen using an in vitro fluorescence resonance energy transfer (FRET)–depression assay. 21 We tested 4µ8C using the H4-UPRE-Luc and H4-CHOP-Luc cell-based assays. 4µ8C was a potent inhibitor of thapsigargin-induced XBP1 activation and caused dose-dependent inhibition of luciferase production following thapsigargin treatment in H4-UPRE-Luc cells (IC50 = 3.1 µM; Fig. 5A ). Significantly, 4µ8C had no effect on thapsigargin-induced luciferase production in H4-CHOP-Luc cells, which monitor the IRE1 kinase-initiated stress kinase signaling pathway ( Fig. 5B ).
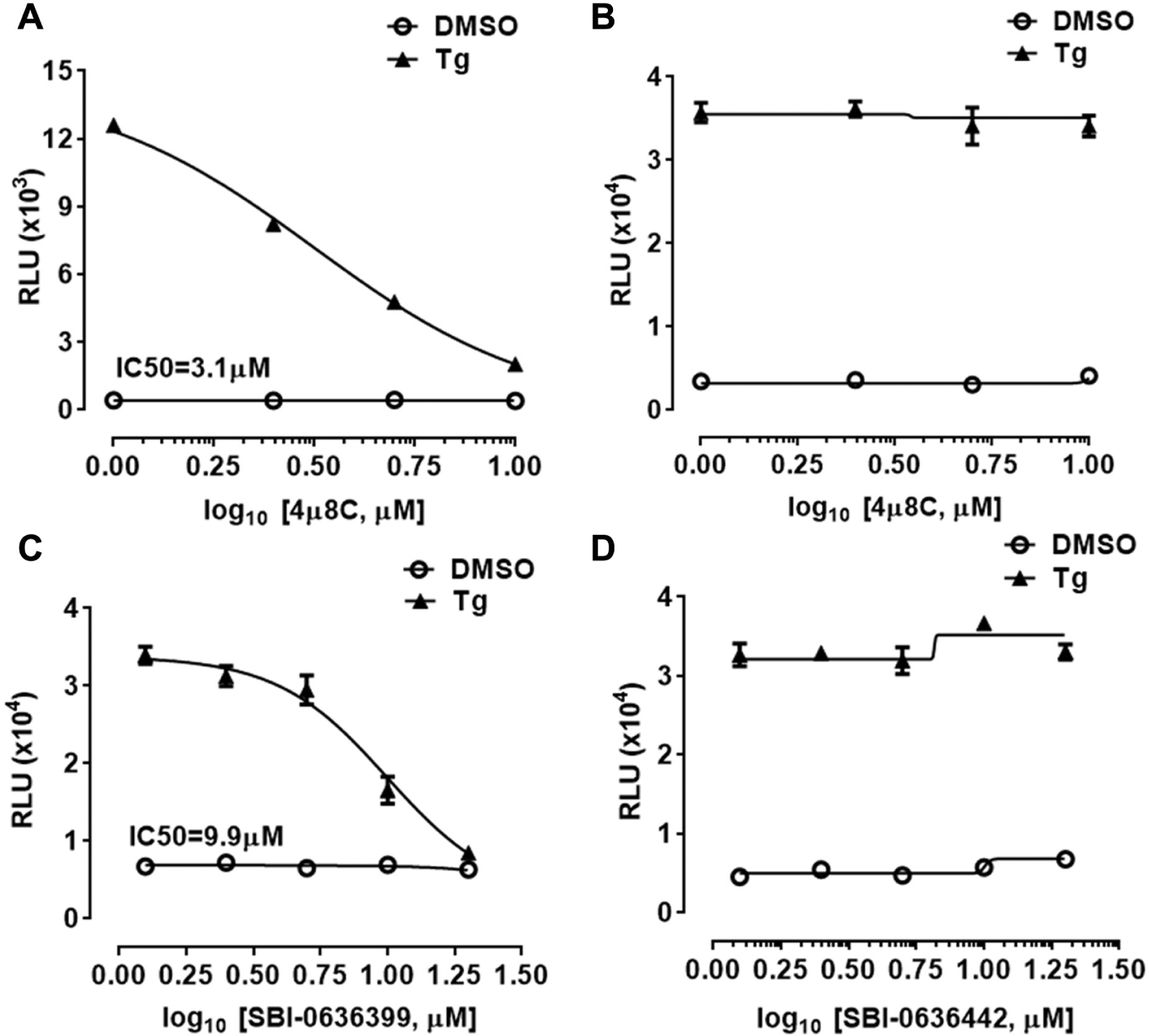
Validation of H4-UPRE-Luc and H4-CHOP-Luc cell assays using inositol-requiring protein 1 (IRE1) or endoplasmic reticulum (ER) stress inhibitors. (
We also tested novel ER stress inhibitors using the H4-CHOP-Luc cell-based assay. We previously identified benzodiazepinone derivative hits from a phenotypic cell-based high-throughput screen that inhibit ER stress-mediated neuronal cell death. 22 The benzodiazepinone derivatives protect against ER stress-mediated cell death via inhibiting ASK1 signaling, downstream of IRE1 activation and upstream of p38MAPK activation. Structure-activity relationship (SAR) studies of the screening hits led to the discovery of novel ER stress inhibitors suppressing ASK1 signaling. 23 We tested a novel ER stress inhibitor of the benzodiazepinone derivative SBI-0636399 (11-[4-(diethylamino)phenyl]-8-chloro-3,3-dimethyl-2,3,4-trihydro-5H,10H,11H-benzo[1,2-f]benzo[b]1,4-diazepin-1-one) and an inactive analogue SBI-0636442 (7-benzoyl-11-(4-hydroxyphenyl)-3,3-dimethyl-2,3,4,5,10,11-hexahydro-1H-dibenzo[b,e][1,4]diazepin-1-one) using the H4-CHOP-Luc cell-based assay. SBI-0636399 showed dose-dependent inhibition of thapsigargin-induced luciferase production (IC50 = 9.9 µM; Fig. 5C ). In contrast, the inactive analogue SBI-0636442 had no effect on thapsigargin-induced luciferase production ( Fig. 5D ). Both compounds showed no significant cytotoxicity at the concentrations tested (data not shown).
HTS of Small-Molecule Libraries
Because the CHOP-Luc assay performed robustly in a 384-well format, while the UPRE-Luc assay was readily formatted for 1536-well plates, we conducted pilot screens using a smaller chemical library for CHOP-Luc and a larger library for UPRE-Luc. First, we performed screening of the ChemBridge DIVERSet small-molecule library (57,600 compounds) using H4-CHOP-Luc cells to identify inhibitors of IRE1-initiated stress kinase pathway activity, monitoring p38MAPK inhibition via the CHOP transactivation domain-dependent reporter gene as described above. Compounds were screened in a 384-well plate format at a final concentration of 10 µM and a final concentration of DMSO 0.2% (v/v). H4-CHOP-Luc cells were pretreated with the compounds for 1 h in serum-free medium followed by incubation with 10 nM thapsigargin in the presence of the compounds for an additional 18 h. The plates were imaged and analyzed with a ViewLux uHTS Plate Imager (PerkinElmer). We used a hit criterion cutoff of 50% inhibition and obtained 1544 hits. The primary hits were cherry-picked and retested in triplets, with 58% of the primary hits confirming inhibitory activity. The Z′ factor was 0.55, with a signal-to-background ratio of 8.49. The hit rate was 1.56% after confirmation.
Second, we screened the Sanford-Burnham Medical Research Institute’s HTS library (320,000 compounds) using H4-UPRE-Luc cells in search of inducers of IRE1 RNase activity (XBP1 splicing and activation). Compounds were screened in a 1536-well plate format at a final concentration of 10 µM and final DMSO concentration of 0.1% (v/v). H4-UPRE-Luc cells were treated with the compounds in 10% FBS medium for 24 h followed by quantitative intensity analysis using a ViewLux uHTS Plate Imager (PerkinElmer). Thapsigargin was used as a qualitative assay control. We used a hit criterion of 6 times the Z score of the mean luciferase activity of the compounds. This resulted in 2909 primary hits. The hit rate was 0.9%. The primary hits were cherry-picked and retested in triplets in the primary assay. The signal-to-background ratio was 12.30. Using a hit criterion of 3 times the Z score, this produced 103 confirmed hits. The high false-positive hit rate may reflect the biology of the IRE1/XBP1 pathway, which is sensitive to general inducers of cell stress.
To interrogate the hits from this primary screen, we devised a testing funnel with the goal to identify IRE1 dual modulators. For cytoprotection, we hypothesized that the ideal hits would inhibit IRE1-initiated stress kinase pathway activation while promoting IRE1 RNase activity. Analyzing the hit compounds from both high-throughput screens, we identified three compounds that inhibited thapsigargin-induced luciferase production in H4-CHOP-Luc cells while spontaneously promoting UPRE-dependent luciferase production in H4-UPRE-Luc cells in dose-dependent manners: SBI-0091194 (N-(4-methoxyphenyl)-N′-[4-(2-thienyl)-6-(trifluoromethyl)-2-pyrimidinyl]guanidine), SBI-0138527 (N-(4-{[6-(4-morpholinyl)-3pyridazinyl]amino}phenyl)benzamide), and SBI-0117439 (4-[2-(3-methoxybenzyl)-4-morpholinyl]-6-methyl-2-pyrimidinamine) ( Fig. 6 A–C ). In particular, compound SBI-0091194 was of interest due to its potency at inhibiting the H4-CHOP-Luc assay and activating the H4-UPRE-Luc assay ( Fig. 6A ).
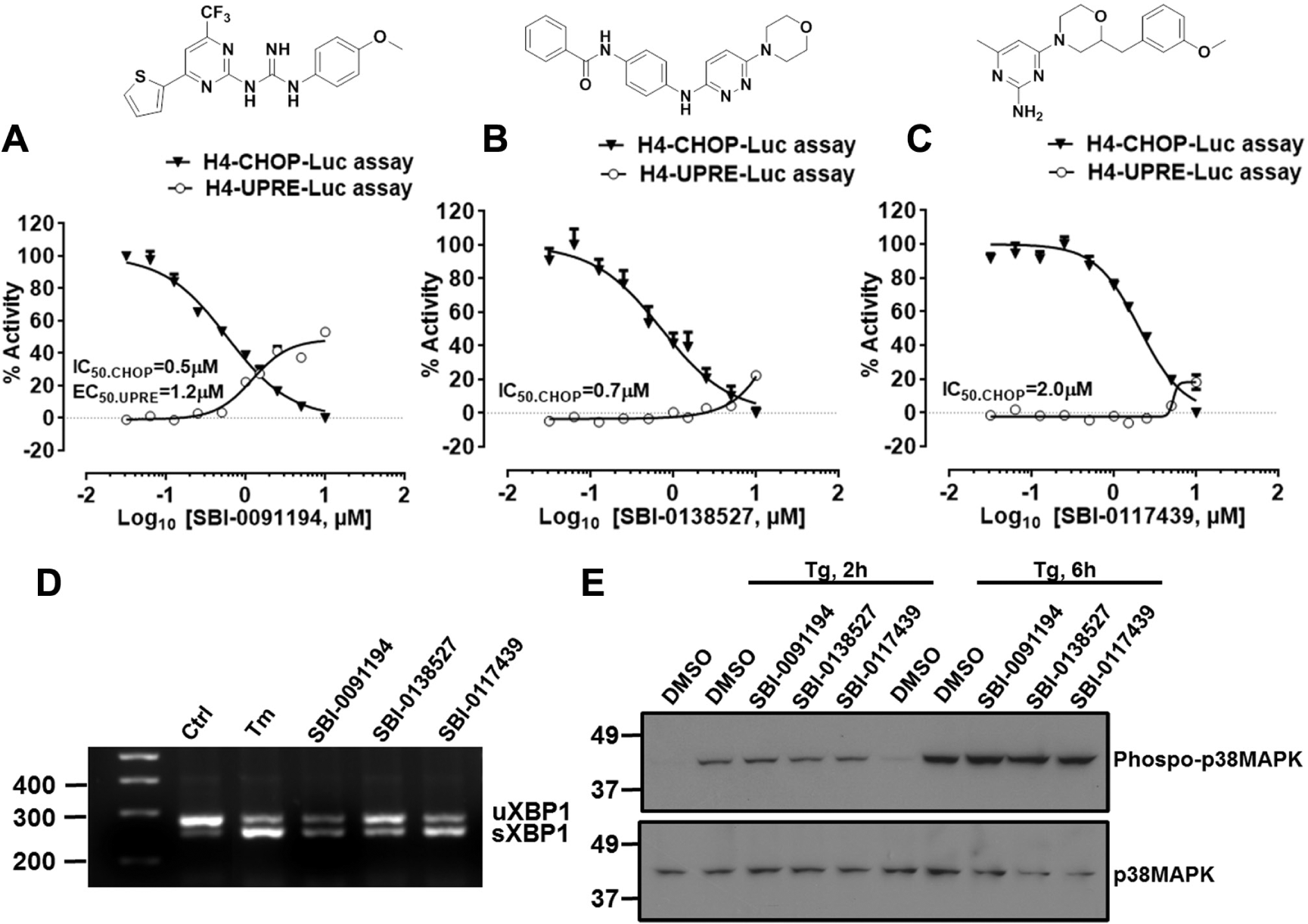
Dose-response studies of the screening hits and counterassays. (
We followed with counterscreens that were independent of luciferase-based assays, including RT-PCR for XBP1 mRNA splicing and immunoblot analysis for p38MAPK phosphorylation. Consistent with the H4-UPRE-Luc assay results, all three compounds induced XBP1 splicing at a concentration of 10 µM ( Fig. 6D ). However, none of these compounds inhibited thapsigargin-induced phosphorylation of p38MAPK ( Fig. 6E ), suggesting the possibility that the compounds act downstream of p38MAPK phosphorylation to inhibit its activity of the chimeric transcription factor containing the p38MAPK-responsive CHOP transactivation domain. Taken together, these results illustrate the performance of these cell-based assays for monitoring specific steps involved in ER stress signaling and show their applicability for HTS-based interrogation of chemical compound collections.
Discussion
The UPR is triggered by accumulation of misfolded proteins in the ER and has been observed in a variety of diseases. Although UPR signaling initially plays an adaptive role to restore cellular homeostasis, prolonged or excessive UPR signaling is detrimental and triggers apoptosis. Both the adaptive and the proapoptotic mechanisms of UPR signaling have been implicated in the pathogenesis of various diseases in a context-dependent manner. Therefore, the development of screening strategies to selectively monitor each of the adaptive and the proapoptotic signaling branches in the UPR is of high interest. It may be useful to identify small-molecule compounds that modulate discrete UPR signaling branches for achieving a desired therapeutic benefit. Luciferase-based assays are well suited for high-throughput applications due to sensitivity, broad linearity, and robustness.
Here we focus on IRE1, the most evolutionarily conserved UPR signal transducer, and describe how we developed cell-based luciferase reporter gene screening strategies to monitor IRE1-initiated signaling cascades. Of note, cell-based assays for HTS aimed at identifying compounds that selectively activate the CHOP gene promoter but not the XBP1 pathway have been described that use Chinese hamster ovary (CHO-K1) cells together with engineered luciferase reporter genes. 18 The assays are complementary to our assays described here as they monitor distinctly different signaling events associated with ER stress.
IRE1 initiates two distinct signaling cascades that affect ER stress signaling through its C-terminal kinase and RNase effector domains: the adaptive signaling cascade mediated by IRE1 RNase/XBP1 pathway and the proapoptotic signaling cascade mediated by the IRE1 kinase/ASK/JNK-p38MAPK stress kinase pathway. We generated stable H4-CHOP-Luc cells to monitor IRE1-mediated p38MAPK activation. In optimizing the assay conditions, we found that cellular density and serum concentration dramatically affected the signal-to-background ratio in H4-CHOP-Luc cells. Therefore, the cells were typically seeded at a low density and incubated in serum-free medium for assay profiling and compound library screening. Using the H4-CHOP-Luc assay, we observed that a number of known ER stress inducers were able to stimulate luciferase production. The most potent among them was thapsigargin, an irreversible inhibitor of the SERCA. At concentrations as low as 10 nM, thapsigargin was sufficient to stimulate robust luciferase responses (~EC80). Small-molecule inhibitors of p38MAPK, SB203580, and SB202190 completely abolished the thapsigargin-induced luciferase response in H4-CHOP-Luc cells. In contrast, a small-molecule inhibitor of PKC did not interfere with thapsigargin-mediated luciferase induction in H4-CHOP-Luc cells, excluding a role for this Ca2+-dependent protein kinase that is capable of initiating kinase cascades, leading to p38-MAPK activation in some cellular contexts. Also, siRNA treatment of cells to reduce expression of IRE1 confirmed that thapsigargin-driven stimulation of luciferase activity in H4-CHOP-Luc cells is IRE1 dependent. Altogether, these studies validate usage of H4-CHOP-Luc cells for monitoring p38MAPK activation resulting from ER stress.
The H4-CHOP-Luc cell assay performed robustly in a 384-well plate format. However, optimization of the H4-CHOP-Luc assay in a 1536-well plate format was attempted but was not consistently successful due to decreased signal-to-noise ratio. To conduct a pilot screen with this cell-based assay, we screened a collection of 57,600 diverse compounds at 10 µM, obtaining a hit rate of 1.56% when using a hit criteria cutoff of 50% luciferase activity inhibition. Prior experience with cell-based HTS assays at the Sanford-Burnham has revealed hit rates ranging from 3.0% to 0.1% (mean = 1.0% based on 26 HTS campaigns). Thus, the hit rate is not substantially higher than many cell-based assays and indicates the need for a rigorous downstream testing funnel for elimination of false positives. We note that a chemical probe (ML291) has been reported that stimulates the CHOP gene promoter and that has cytotoxic activity against a variety of cell lines. 18 Thus, this chemical probe differs markedly from the noncytotoxic compounds identified here that stimulate UPRE-Luc. Even though XBP-1 contributes to CHOP gene expression, along with ATF4 and ATF6, we surmise that compounds that can be identified by our UPRE-Luc assay will in many cases differ markedly in their cellular phenotype from compounds identified by the previously described CHOP promoter reporter gene assay.
We also generated stable H4-UPRE-Luc cells to monitor IRE1-mediated production of XBP1. A number of ER stress inducers stimulated luciferase production. Consistent with the H4-CHOP-Luc cell assay, thapsigargin at 10 nM was sufficient to stimulate a robust luciferase response. The H4-UPRE-Luc cell assay was further validated with XBP1 and IRE1 siRNAs. Compared with the H4-CHOP-Luc cell assay, the H4-UPRE-Luc cell assay was more robust in that it achieved a higher signal-to-background ratio with less incubation time. In addition, the H4-UPRE-Luc cells had no restriction on serum and provided an excellent signal window in both 10% FBS medium and serum-free medium when stimulated with ER stress-inducing compounds. The H4-UPRE-Luc cell assay performed robustly in the 1536-well plate format, and as a result, we used a larger library for the H4-UPRE-Luc screen than for the H4-CHOP-Luc screen for the pilot screens. Using this cell-based assay in a 1536-well format, we obtained a hit rate of 0.9% when screening a diverse collection of 320,000 compounds using a hit criteria cutoff of 6 times the Z score of the mean luciferase activity of the compounds. Significantly, thapsigargin was among our hits and actually was the most potent luciferase inducer identified by the screen. We also generated H4-XBP1-Luc cells to directly monitor XBP1 splicing. We observed that the H4-XBP1-Luc assay was not as robust as the H4-UPRE-Luc assay and generally achieved a lower signal-to-background ratio. Nevertheless, the assay directly monitors XBP1 splicing rather than downstream XBP1-mediated gene transcription. Thus, it could serve as a confirmatory assay.
IRE1 RNase inhibitors have been identified from compound screening using in vitro biochemical screening assays or cell-based assays similar to our H4-XBP1-Luc cell assay described here.21,24–26 We tested the IRE1 inhibitor 4µ8C using our H4-CHOP-Luc and H4-UPRE-Luc cell assays. Consistent with a previous report, 4µ8C was a potent IRE1 RNase inhibitor as assessed by our H4-UPRE-Luc cell assay, without affecting IRE1 kinase activity as monitored using our H4-CHOP-Luc cell assay, thus further validating our cell-based assays for profiling small-molecule modulators of IRE1. Using the H4-CHOP-Luc cell-based assay, we also tested our novel benzodiazepinone derivatives that inhibit ER stress-mediated ASK1 signaling downstream of IRE1 kinase activation. The active ER stress inhibitor SBI-0636399, but not an inactive analogue SBI-0636442, specifically inhibited thapsigargin-induced luciferase production in a dose-dependent manner.
IRE1 has a dual function in ER stress, initiating both adaptive and alarm (cell stress) signaling cascades through the IRE1 RNase domain and kinase domain, respectively. Although autophosphorylation of the IRE1 kinase domain is required for activation of its RNase domain, it has been established that conformational changes triggered by occupancy of the active site with a ligand in the kinase domain, rather than phosphorylation per se, are crucial for IRE1 RNase activation. Thus, kinase inhibitors could act as potent activators of IRE1 RNase activity. 27 Furthermore, cytoplasmic ligands may act at sites distinct from the kinase domain to modulate IRE1 activity. 28 These concepts lay the foundation for a screen for compounds that selectively modulate IRE1 kinase or RNase activities or downstream signaling cascades relevant to specific diseases. Compounds that inhibit IRE1 RNase activity and promote its ability to stimulate stress kinases (e.g., activation of ASK1) may be of value as cancer therapeutics or potentially for eradicating autoimmune lymphocytes. Conversely, compounds that inhibit IRE1 kinase activity and promote IRE1 RNase activity may be of therapeutic benefit in neurodegenerative disorders, for islet cell protection, and for heart failure, among other applications. 16 Although a pilot screen using the ChemBridge DIVERSet library failed to identify IRE dual modulators that promote IRE1 RNase activity and inhibit its activation of stress kinases, the high-throughput assays described here could be used for screening large compound libraries to identify novel chemical scaffolds that may be starting points for cytoprotective drug discovery. Alternatively, the high-throughput assays could be used to screen for novel anticancer chemical scaffolds that activate stress kinases in an IRE1-dependent manner and that inhibit IRE1’s RNase activity as novel cytotoxics. The compounds identified in the course of validating the assays will be further evaluated for these properties. The assays described here may also be useful for siRNA/small hairpin RNA-based functional genomic screening in search of cellular modulators of IRE1 signaling.
Footnotes
Acknowledgements
We thank Melanie Hanaii for manuscript preparation, Drs. Ron Prywes and Masayuki Miura for providing plasmids, Drs. Guang Chen and Daniel DiSepio for helpful scientific discussion, Dr. Douglas Sheffler for review of the manuscript, and the Conrad Prebys Center for Chemical Genomics High-Throughput Screening Facility and Staff for technical assistance.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research and/or authorship of this article: This work was supported by funding from Janssen Research and Development LLC.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.