
Abstract
Inositol-requiring enzyme 1 alpha (IRE1α) is a transmembrane sensor protein with both kinase and ribonuclease activity, which plays a crucial role in the unfolded protein response (UPR). Protein misfolding in the endoplasmic reticulum (ER) lumen triggers dimerization and subsequent trans-autophosphorylation of IRE1α. This leads to the activation of its endoribonuclease (RNase) domain and splicing of the mRNA of the transcriptional activator XBP1, ultimately generating an active XBP1 (XBP1s) implicated in multiple myeloma survival. Previously, we have identified human IRE1α as a target for the development of kinase inhibitors that could modulate the UPR in human cells, which has particular relevance for multiple myeloma and other secretory malignancies. Here we describe the development and validation of a 384-well high-throughput screening assay using DELFIA technology that is specific for IRE1α autophosphorylation. Using this format, a focused library of 2312 potential kinase inhibitors was screened, and several novel IRE1α kinase inhibitor scaffolds were identified that could potentially be developed toward new therapies to treat multiple myeloma.
Introduction
The endoplasmic reticulum (ER) is involved in protein synthesis, folding, assembly, and posttranslational modification. In eukaryotic cells, proteins enter the ER as unfolded polypeptide chains via translocation and are subsequently folded and assembled correctly in the ER lumen. 1 There is a balance between the load of unfolded proteins entering the ER and the capacity of the cellular machinery that handles the correct folding and modification. Disturbance of this balance causes misfolded protein to accumulate in the ER lumen, which is known as ER stress. The ER mediates a specific set of intracellular signaling pathways in response to this stress, known as the unfolded protein response (UPR). 2 The UPR triggers an increase in the amount of the ER membrane and its components, including chaperones and protein-modifying enzymes needed to fold proteins, as well as a decrease in translation and loading of proteins into the ER. In addition, unfolded proteins are sent for degradation. However, if the balance of protein production to the capacity of the ER cannot be reestablished, the UPR switches from pro-survival mode to triggering apoptosis.2,3
In mammalian cells, the UPR signaling cascades are initiated by three ER-localized protein sensors: PKR-like ER kinase (PERK), activating transcription factor 6 (ATF), and inositol-requiring enzyme 1 alpha (IRE1α). 1 IRE1α is located in the ER membrane and is composed of an N-terminal luminal domain, a transmembrane spanning region, and cytosolic serine/threonine kinase and ribonuclease domains. IRE1α senses the buildup of misfolded proteins in the ER lumen and facilitates the response signal from the ER to the cytoplasm and ultimately the nucleus. The requirement of IRE1α for this pathway was established by the discovery of the ire1/ERN1 gene in yeast and its role in the UPR. Later, human cDNA was isolated encoding a homolog to yeast in which the cytoplasmic domain is highly conserved, but the luminal domain is highly diverged from the yeast gene product. 1 In yeast, it has been shown that the IRE1α luminal domain forms a groove that, when dimerized, could bind unfolded proteins directly to aid dimerization. 1 The crystal structure of the luminal domain of human IRE1α, however, displays a groove too narrow for peptide binding. 4 Existing evidence suggests that the human ER luminal domain of IRE1α is maintained in its inactive monomeric state by binding to the ER-specific HSP70 chaperone BiP (GRP78). 1 Accumulation of misfolded proteins in the ER is thought to cause the release of BiP followed by dimerization of the IRE1α luminal domain. 4 Dimerization leads to trans-autophosphorylation of the kinase domain, which in turn leads to the activation of the RNase domain. 5 This results in activation of a nonclassical endonucleolytic cleavage and ligation of XBP1u mRNA. 5 The product of the splicing (XBP1s mRNA) is translated to an active transcription factor, XBP1s, that activates the promoter of various target genes of the UPR pathway. These include genes encoding proteins involved in ER protein folding and modification (Bip/GRP78 and GRP94) and proteins involved in phospholipid biosythesis (INO1). 2 Upregulating these UPR target genes also closes a feedback loop that adjusts the ER protein-folding response capacity according to need. 5
Multiple myeloma is an incurable disease affecting 3500 new patients each year in the United Kingdom and over 86 000 worldwide, and new treatments are urgently required. 6 It has been shown that XBP1 is crucial in myeloma cell biology.3,7–9 A partial UPR is maintained in secretory cells with high constitutive protein translation, including myeloma cells. XBP1 promotes the transcription of genes encoding chaperones and cytokines, mediating myeloma growth and survival via upregulation of the cytokine interleukin-6 and the adaptive stress response to eliminate misfolded immuglobulins.7–9 The expression of XBP1 and the ratio of XBP1s/u mRNA are higher in myeloma cells compared with B cells. In addition, high expression of XBP1s is an independent prognosis factor for poor overall survival in myeloma patients. 9 Myeloma cells with functionally deficient XBP1 undergo increased apoptosis in response to ER stress. Proteasome inhibitors used to treat myeloma induce ER stress and activate elements of the UPR while suppressing the activity of IRE1α. 10
We have previously shown that the kinase inhibitor sunitinib inhibits human IRE1α kinase in vitro and also inhibits the ER stress-induced RNase function in myeloma cell lines, leading to reduced splicing of XBP1 mRNA. 11 We hypothesize that inhibition of the activation of IRE1α, and hence of its RNase activity, to reduce expression of the active XBP1 transcription factor will have antiproliferative effects in multiple myeloma. We therefore set out to develop a robust high-throughput screening (HTS) assay to identify small-molecule inhibitors of the activation of IRE1α. Assays using a generic AlphaScreen format to measure dimerization/oligomerization or the overall phosphorylation state of IRE1α have been reported. 12 However, we sought to develop an assay based on the proposed mechanism of IRE1α activation, 11 measuring IRE1α autophosphorylation. We have adapted a low-throughput IRE1α autophosphorylation DELFIA assay using an antibody specific for IRE1α phosphorylated at serine 724. This residue is located in the activation loop and therefore involved in the activation process. 11 We have used the new high-throughput DELFIA assay to screen a kinase-focused compound library for inhibition of IRE1α autophosphorylation at three different concentrations. Here we describe the outcome of the screen and the chemical properties of several of the identified hits. The DELFIA assay format has previously been described as an HTS assay measuring serine threonine kinase substrate phosphorylation activity. 13 Our study shows that this high-throughput assay format can also be used to measure the inhibition of the autophosphorylation of dual-functionality enzymes such as IRE1α.
Materials and Methods
Production and Purification of Dephosphorylated IRE1α
The coding sequence for residues G547 to L977 of human IRE1α was PCR amplified from the IMAGE clone 40146436 (accession #BC130405) and inserted into a modified version of the pFastBac1 vector that encodes an N-terminal 6xHis-tag followed by a human rhinovirus 3C protease cleavage site. Recombinant baculovirus was generated according to Bac-to-Bac protocols (Invitrogen, Paisley, UK). Sf9 insect cells were grown in sf-900 II media (Invitrogen) to a cell density around 2 × 106 cells per mL, infected with 30 to 100 µL of virus per 107 cells and harvested 3 days later. Cell pellets were resuspended in 3 volumes of Talon Buffer A (50 mM HEPES [pH 7.0], 100 mM NaCl, 1 mM CaCl2, 1 mM MgCl2, 10% [v/v] glycerol) containing 1× Complete EDTA-free protease inhibitors and 10 U/mL recombinant RNase-free DNase I (Roche, Basel, Switzerland) and lysed by sonication. Following centrifugation, supernatant was purified using 10 mL of Talon resin (Clontech, Mountain View, CA) and then dephosphorylated overnight with lambda phosphatase at 0.04 U/pmol (NEB, Ipswich, MA). Dephosphorylated protein was concentrated and loaded onto a Superdex 200 HR 26/60 column (GE Healthcare, Waukesha, WI), equilibrated with 50 mM HEPES (pH 7.4), 200 mM NaCl, 2 mM dithiothreitol (DTT), 1 mM EDTA, and 10% (v/v) glycerol. Fractions containing monomeric IRE1α were further purified using a 6-mL Resource Q column (GE Healthcare) equilibrated in 50 mM HEPES (pH 7.5), 20 mM NaCl, 1 mM EDTA, 1 mM DTT, and 10% (v/v) glycerol and eluted using a NaCl gradient. To obtain the phosphorylated form of IRE1α, phosphatase inhibitors (10 mM NaF, 1 mM Na3VO4, 25 mM β-glycerophosphate) were added to the lysis buffer, and treatment with lambda phosphatase was omitted.
High-Resolution LC/MS Analysis of Intact IRE1α Proteins
Liquid chromatography/mass spectrometry (LC/MS) CHROMASOLV solvents, formic acid, or alternative eluent modifiers were purchased from Sigma-Aldrich (Poole, UK) unless otherwise stated. Of the samples, IRE1α (2.4 mg/mL) and IRE1α + lambda phosphatase (1.3 mg/mL), 10 µL standard injections (with needle wash) were made onto a Purospher STAR RP-18 endcapped column (3 µm, 30 × 4 mm encased in LiChroCART assembly; Merck KGaA, Darmstadt, Germany). Chromatographic separation at 30 °C was carried out using a 1200 Series HPLC (Agilent, Santa Clara, CA) over a 15-min gradient elution from 95:5 to 0:100 water and acetonitrile (both modified with 0.1% formic acid) at a flow rate of 1 and 0.5 mL/min. UV-Vis spectra were acquired at 280 and 210 nm on a 1200 Series diode array detector (Agilent). The postcolumn eluent flow from the diode array detector was infused into a 6520 Series qToF mass spectrometer fitted with a dual electrospray ionization (ESI) source (Agilent). A 3-min divert to waste was used to aid desalting the LC eluent, and nebulizing gas was introduced into the grounded nebulizer with spray direction orthogonal to the capillary axis. The aerosol was dried by heated gas (10 L/min of nitrogen at 350 °C, 50 psi), producing ions by ESI. Ions entered the transfer capillary, along which a potential difference of 4 kV was applied. The fragmentor voltage was set at 190 V and skimmer at 65 V. Signal was optimized by internal AutoTune methodology. Profile mass spectrometry data were acquired in positive ionization mode over a scan range of m/z 100 to 3200 (scan rate 1.0) with reference mass correction at m/z 195.09 (caffeine) and 922.01 (hexakis(1H,1H,3H-perfluoropropoxy)phosphazene). Raw data were processed using Agilent MassHunter Qualitative Analysis B.04.00 and MagTran. 14
Antibody Specificity DELFIA
An 96-well Immulon 2B plate (Thermo Fisher Scientific, Leicestershire, UK) was coated with the nonphosphorylated (CLAVGRHSFSRRSG) and phosphorylated peptides (CLAVGRHS(P)FSRRSG) at 1, 10, and 100 µg/mL in phosphate-buffered saline (PBS tablets; BR0014G; Oxoid, Hampshire, UK) (50 µL/well) and left overnight to coat at 4 °C. The plate was subsequently washed in 0.1% water Tween-20 (P1379; Sigma-Aldrich) four times using the Wellwash 4 mk 2 (Thermo Fisher Scientific), then blocked with 5% Marvel milk formula (Marvel, Lincolnshire, UK; 50 µL/well), and the plate was washed once again using the same method. The α-P724 antibody (rabbit anti-IRE1α phosphoserine 724 primary antibody), raised to a KLH conjugate of the peptide CLAVGRHS(P)FSRRSG and affinity purified using a nonphosphorylated peptide (Open Biosystems, Huntsville, AL), was diluted from 12 µg/mL to 12 pg/mL, coated onto the plate at 25 µL/well, and incubated for 2 h at 37 °C. The plate was washed again as before. The secondary antibody (Europium-labeled anti-rabbit IgG, AD0105; PerkinElmer Life Sciences, Waltham, MA) was added to the wells at 0.25 µg/mL, diluted in DELFIA assay buffer (4002-0010; PerkinElmer Life Sciences) at 25 µL/well, and incubated for 1 h at 37 °C. After another wash step, 25 µL/well of DELFIA Enhancement solution (4001-0010; Perkin Elmer Life Sciences) was added to each well and the plates were shaken for 10 min. All assay plates were read on an EnVision 2103 multilabel microplate reader (Perkin Elmer Life Sciences).
Western Blot Analysis
The dephosphorylated (lambda phosphatase treated) IRE1α or phosphorylated IRE1α were incubated with adenosine triphosphate (ATP) and MgCl2 for 60 min and analyzed on a 10% Tris-glycine sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) gel containing 0.5% 2,2,2 -trichloroethanol. Total protein was visualized using a UV transilluminator prior to transfer to a 0.45-µM nitrocellulose membrane (Bio-Rad, Hercules, CA). Following transfer, the membrane was blocked with 5% low-fat milk in Tris-buffered saline with 0.1% Tween-20 (TBST) and subsequently incubated with α-P724 antibody diluted 1 in 1000 in block solution. The blot was washed three times with TBST prior to addition of a goat antirabbit horseradish peroxidase (HRP) conjugate (DAKO, Glostrup, Denmark; cat. PO217) diluted 1 in 2000 in block solution. After incubation and further washing, the blot was developed using ECL Western Blotting Substrate (Pierce/Thermo Scientific, Rockford, IL) and exposed to X-ray film.
DELFIA Autophosphorylation Assay
Dephosphorylated IRE1α (700 nM) was incubated for 25 min with 100 µM ATP (A7699) in 15 µL assay buffer consisting of 40 mM Tris (pH 7.5) (T2663), 20 mM MgCl2 (M1028), and 1 mM DTT (DL-Dithiothreitol, D0632). All reagents were obtained from Sigma-Aldrich. The assay was stopped by the addition of 15 µL of 40 mM EDTA (100935V; BDH, Dorset, UK). Samples were transferred to 384-well high-binding plates (781061; Greiner Bio-One, Frickenhausen, Germany) and incubated overnight at 4 °C. Plates were washed three times in 0.1% water Tween-20 as described above, followed by a blocking step using 5% Marvel in PBS (50 µL/well), and incubated for 30 min at 37 °C. The plates were washed three times in 0.1% water Tween-20 again before 25 µL of the α-P724 phospho- specific IRE1α primary antibody (160 pg/mL) in PBS was added to each well and incubated at 37 °C for 1.5 h. After incubation, plates were washed before the secondary antibody, Europium-labeled antirabbit IgG diluted in DELFIA assay buffer, was added. The plates were subsequently incubated for 60 min at 37 °C. Another wash step was followed by the addition of 25 µL/well of DELFIA enhancement solution, and the plates were shaken for 10 min. All these additions were carried out using a Multidrop Combi (Thermo Scientific). All assay plates were read on an EnVision 2103 multilabel microplate reader (PerkinElmer Life Sciences).
IRE1α Kinase-Focused Screen
The assay was carried out in 384-well polypropylene plates (781280; Greiner Bio-One). Compounds were added to the assay plates using an Echo 550 acoustic dispenser (Labcyte, Sunnyvale, CA). Compound volumes of 5 nL, 30 nL, or 150 nL from 10-mM stock solution in 100% DMSO were replicated directly into individual assay plates, and the wells were backfilled with 100% DMSO to make the total volume 150 nL (final compound concentrations 1.7 µM, 10 µM, and 50 µM in 1% DMSO). Control wells containing just 150 nL 100% DMSO were also dispensed using the Echo 550 acoustic dispenser. Subsequently, the His-tagged dephosphorylated IRE1α enzyme was added at 7.5 µL/well to give a final concentration of 700 nM, followed by 7.5 µL of 100 µM ATP in assay buffer or assay buffer alone. These additions were made using a Matrix electronic pipette (Thermo Scientific). In addition to the test wells, each plate contained total enzyme activity control wells and wells with enzyme but no ATP for data normalization. Phosphophorylated IRE1α enzyme was added to two wells per plate to act as an extra positive control for the DELFIA reagents. Each plate was sealed with an adhesive seal (ABSM-PSEAL; Anachem, Bedfordshire, UK). Plates were spun at 1000 rpm for 1 min in a Harrier 18/80 centrifuge (MSE, Buckinghamshire, UK) and then incubated for 25 min at 37 °C. The assay was stopped by the addition of 15 µL of 40 mM EDTA using a Matrix electronic pipette. Plates were spun at 1000 rpm for 1 min, and 25 µL of the contents of each well was transferred to a high-binding 384 Greiner plate (781061). The assay was left to coat for at least 48 h at 4 °C. The DELFIA was then carried out as in the detection methodology described above. Primary screening data were processed in Studies (Dotmatics Ltd, Herts, UK). Percentage inhibition was calculated as follows: [100 – (S – B)/(T – B)] × 100, where S represents the counts from each compound well, B the counts in the wells containing no ATP, and T the counts in the total activity wells. The Z′ factor was calculated as described previously. 15
IC50 Generation and Hit Confirmation
Hits were prioritized for confirmation by analysis of the 3-point screening data. The list of hits was further refined by eliminating known actives (e.g., analogues of staurosporine or sunitinib), structures containing functional groups known to be problematic or unproductively promiscuous, 16 or compounds likely to be poorly soluble at high concentrations. 17 Hits were confirmed and validated by IC50 determination from 8-point concentration-response curves with a final DMSO concentration of 1%. Independent serial dilutions were obtained by dispensing the compounds from 10-mM and 0.1-mM stock solutions in 100% DMSO directly into assay plates using the Echo 550 acoustic dispenser followed by backfilling with 100% DMSO. Confirmed hits were defined as those with Hill slopes in the range of 0.6 to 1.6.
XBP1 Splicing Assay Method
H929 multiple myeloma cells were resuspended at 5 × 105 cells/mL in RPMI 1640 medium (61870-044; Life Technologies, Carlsbad, CA) supplemented with 10% fetal bovine serum (FBS) (F7524; Sigma-Aldrich) and seeded in a 6-well plate (353046; BD, Oxford, UK) at a total density of 2 × 106 cells/well. Following a 2-h incubation at 37 °C, the cells were treated with either a combination of 10 µg/mL tunicamycin (T7765; Sigma-Aldrich) and 1 µM nilotinib or 13.5 µM sunitinib, or compound alone. The concentration of compound was predetermined based on the GI50 value obtained from a WST-1 cell proliferation assay (11644807001; Roche). The plate was incubated at 37 °C for a further 4 h and resuspended in buffer RLT Plus (74134; Qiagen, Manchester, UK) containing 40 mM DTT (D9779; Sigma-Aldrich). Samples were frozen at −80 °C. RNA was extracted using an RNeasy Plus Mini Kit (74134; Qiagen), eluted in nuclease-free water, and the concentration was measured using a Nanodrop ND-1000 spectrophotometer (Thermo Scientific). cDNA was synthesized using the iScript Select cDNA synthesis kit (170-8897; Bio-Rad) and amplified with the Platinum Taq DNA polymerase kit (10966-034; Life Technologies). The PCR reaction was performed as described previously, 1 using the following primers: 5′-CCTTGTAGTTGAGAACCAGG-3′ (forward) and 5′-GGGGCTTGGTATATATGTGG-3′ (reverse) (Life Technologies). Conditions for the PCR reaction consisted of an initial hold step at 94 °C for 2 min, followed by 35 cycles of 94 °C for 15 s, 60 °C for 1 min, and 72 °C for 30 s. Then, 20 µL of each sample was run on a 2% agarose gel containing GelRed Nucleic Acid Stain (BT41003; Cambridge Bioscience, UK) at 90 V until the spliced and unspliced XBP1 bands could be resolved.
Results
Assay Development
The pSer724 IRE1α antibody specificity was tested in the DELFIA assay format using a nonphosphorylated and a phosphorylated IRE1α peptide. It was shown that the pSer724 IRE1α antibody was able to detect the phosphorylated IRE1α peptide but not the nonphosphorylated IRE1α peptide ( Fig. 1A ). In addition, it was demonstrated that the pSer724 IRE1α antibody does not detect the purified dephosphorylated IRE1α by Western blotting unless it has been incubated in the presence of ATP and magnesium chloride ( Fig. 1B ).
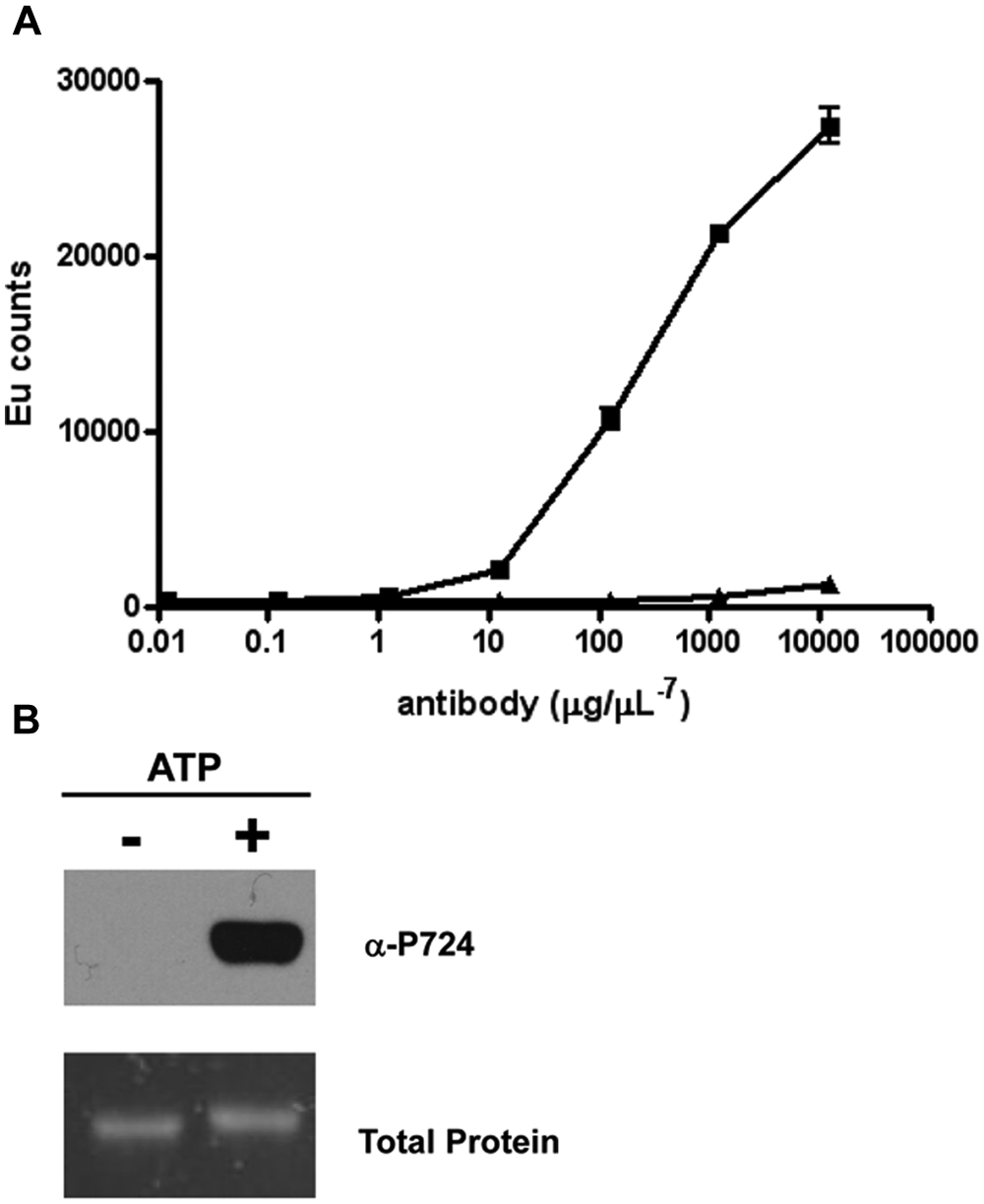
Specificity for the anti–inositol-requiring enzyme 1 alpha (IRE1α) phosphorylated Ser 724 antibody. (
The untreated (phosphorylated) and lambda phosphatase–treated (dephosphorylated) IRE1α proteins were analyzed by high-resolution LC/MS to confirm the phosphorylation state of the proteins used in the DELFIA assay. In the lambda phosphatase–treated IRE1α sample, a single peak was observed, whereas in the untreated IRE1α sample, 2 peaks were observed with higher molecular weights of +80 Da and +160 Da, respectively, which is indicative of protein phosphorylation (see
Both the deconvoluted spectra of the untreated IRE1α and of IRE1α + lambda phosphatase indicated a protein of molecular weight 51380 Da, which is +43 Da higher than the predicted molecular weight of 51337 Da for the expected protein sequence. No sequencing errors were found when the PCR product was DNA sequenced, and the deconvoluted molecular weight for IRE1α with the N-terminal His-tag removed by proteolytic cleavage with Precision Protease (GE Healthcare, Buckinghamshire, UK) was consistent with that predicted for the protein sequence. This suggests that N-terminal acetylation gave rise to the +43 Da molecular weight observed in the IRE1α His-tagged samples.
In general, kinase screening assays are run at ATP concentrations below the Km to provide bias toward ATP-competitive compounds. For IRE1α, we hypothesized that ATP-competitive compounds should give a desired mechanism of action, and therefore we titrated the magnesium chloride and ATP concentrations to determine the optimum screening conditions to achieve this bias in our assay. The maximum signal in the DELFIA assay was obtained with an excess of magnesium chloride. The IRE1α Km for ATP was 135 µM, which was determined by running the assay at seven different ATP concentrations from 0 to 800 µM for 30 min and fitting the initial rates to a Michaelis-Menten equation ( Fig. 2A , B ). An ATP concentration of 100 µM, below our calculated ATP Km, was chosen for the screening, resulting in a signal-to-background (S/B) ratio of 24, which is acceptable for a DELFIA assay.
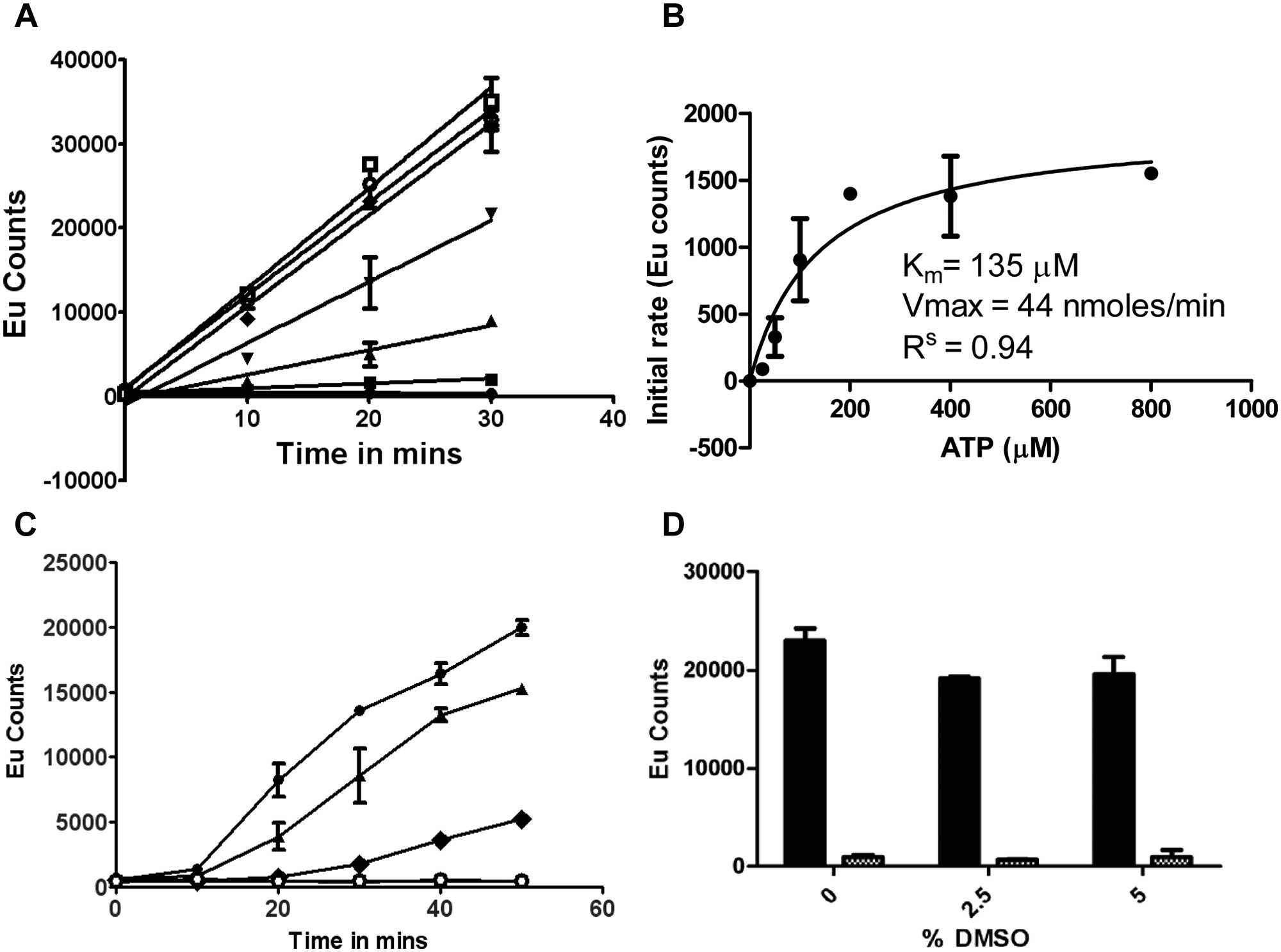
Optimization of conditions for the inositol-requiring enzyme 1 alpha (IRE1α) autophosphorylation DELFIA assay. Conditions for the DELFIA were established according to Materials and Methods. (
To optimize the enzyme concentration in the autophosphorylation assay, we carried out a titration with dephosphorylated IRE1α. A good window (S/B ratio = 47) could be obtained with 700 nM enzyme. Lower concentrations of enzyme resulted in a lower S/B ratio not suitable for a reproducible and robust assay. A time course demonstrated that with an ATP concentration of 100 µM and enzyme concentration of 700 nM, IRE1α showed linear kinetics for 25 min at 37 °C ( Fig. 2C ). Use of 700 nM enzyme gave product concentrations that fell within the linear detection range of the DELFIA readout, and reading from a standard curve generated using phosphorylated IRE1α showed that typically 30% to 50% of the protein was phosphorylated under assay conditions (data not shown).
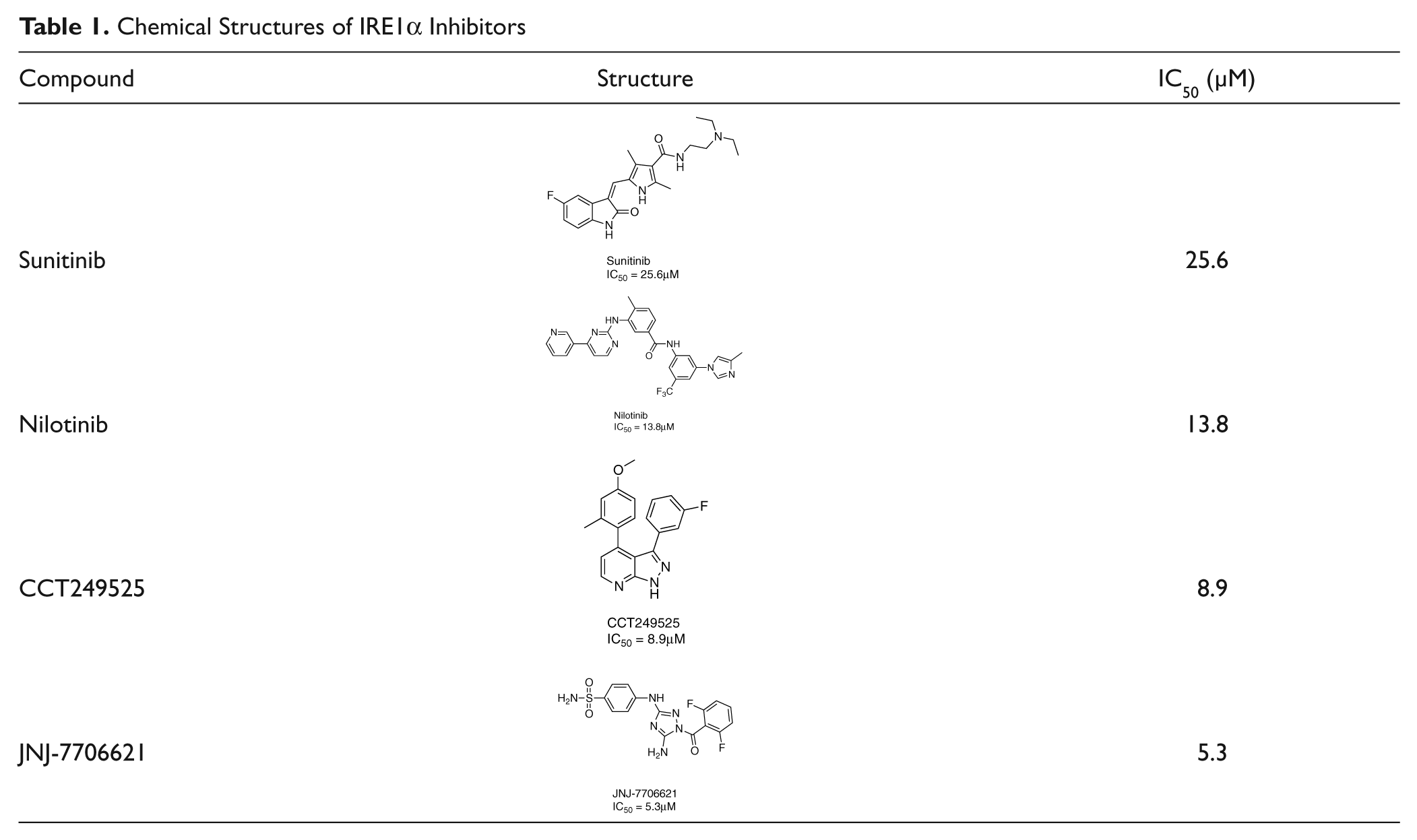
The assay was tested for tolerance to DMSO, and no significant effect was observed up to 5% ( Fig. 2D ). IRE1α without the addition of ATP was used as the 100% inhibition control for the screen, because although we had previously identified sunitinib as an IRE1α inhibitor, 11 it is only a weak inhibitor ( Table 1 ), and a high concentration would have been required to give 100% inhibition.
Chemical Structures of IRE1α Inhibitors
Screening Results
A total of 2312 compounds from small in-house and commercial-focused libraries enriched with kinase scaffolds and structures likely to be ATP-competitive kinase inhibitors were screened at final concentrations of 1.7 µM, 10 µM, and 50 µM in the high-throughput DELFIA.
18
The three concentrations were chosen to maximize the possibility of finding hits and at the same time give an indication of a concentration-dependent dose response. The reproducibility of the screen is illustrated in
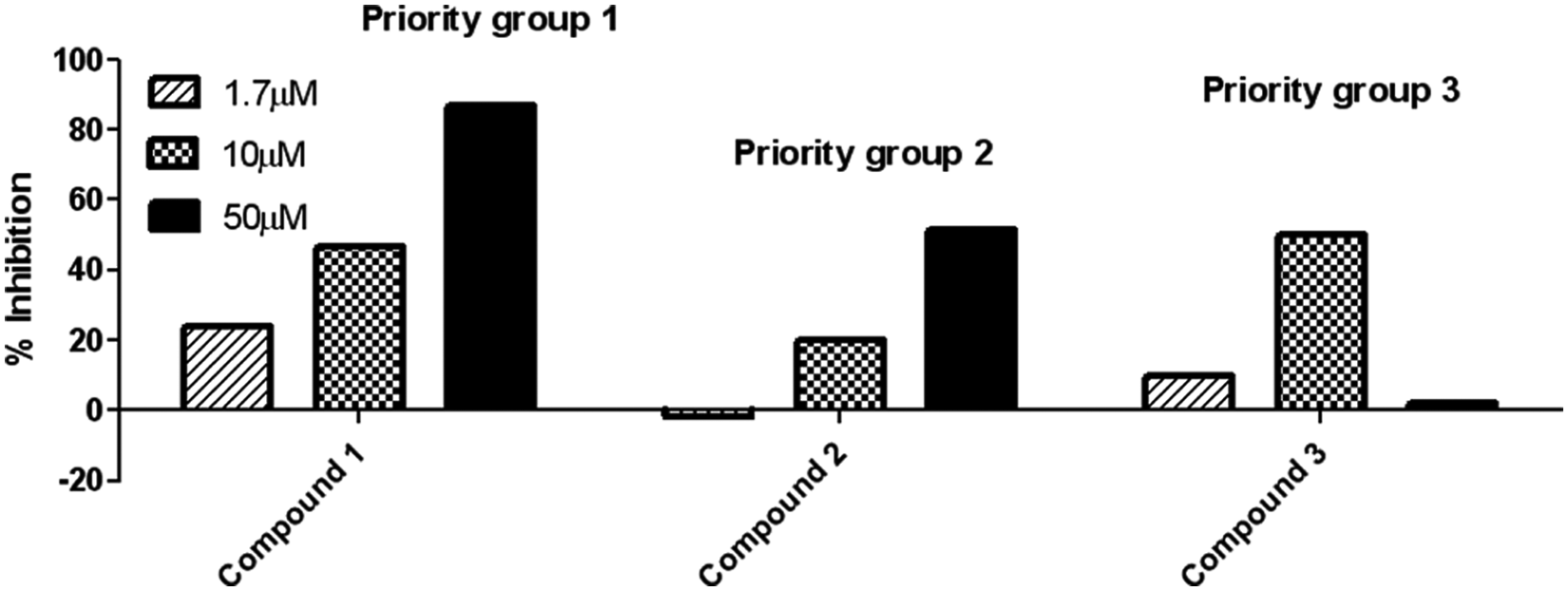
Examples of single-point (n = 1) compound data to illustrate the three priority groups used to triage the hits from the high-throughput screen as described in screening results.
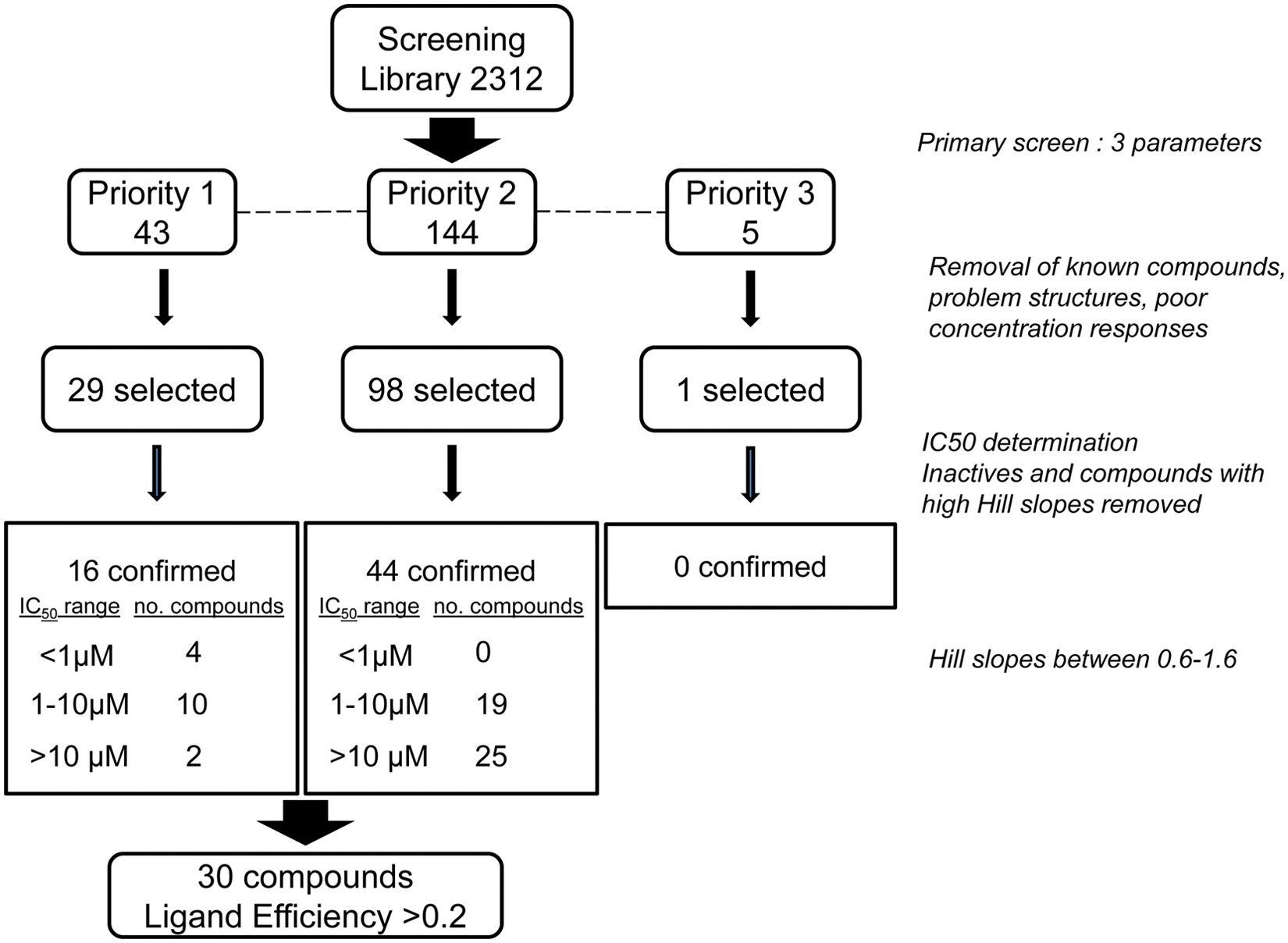
Summary of compound screening cascade and hit validation showing number of primary hits from the high-throughput screen and how these were prioritized and filtered.
Hit Validation
Of the 128 primary hits selected for IC50 determination in the same assay format, an IC50 value could be obtained for 108 compounds. A further reduction of the number of hits was carried out by removing hits with IC50 curves with high Hill slopes. We allowed Hill slopes between 0.6 and 1.6, 19 leaving 60 compounds that were classed as confirmed hits (2.6%) ( Fig. 4 ). The IC50 values of the confirmed hits ranged from 0.3 to 64 µM. Binning the hits into three potency ranges (<1 µM, 1–10 µM, 10–100 µM) revealed that the priority 1 compounds generally tracked with the more potent compounds ( Fig. 4 ). Of these compounds, 30 had ligand efficiency greater than 0.2. The primary hit rate was high due to the bias in the compound library toward kinase inhibitors. The confirmed hit rate is higher than previously reported for other screens performed using the DELFIA assay format, 13 which could be due to the high proportion of kinase scaffolds in the library and to screening each compound at three concentrations. Screening at multiple concentrations allowed us to focus on the most potent compounds that demonstrated a concentration-dependent response. The false positives that did not meet our Hill slope criteria at the IC50 stage were likely due to the promiscuous properties of these compounds.
Five clusters and several singletons of drug-like compounds were identified as IRE1α autophosphorylation inhibitors, with a cluster of hits comprising three or more close structural analogues. One hit cluster included a group of 3,4-diaryl substituted pyrazolo[3,4-b]pyridines, represented by the compound CCT249525 ( Table 1 , 8.9 µM). Although the pyrazolo[3,4-b]pyridine bicycle is a known kinase inhibitor scaffold,20–22 to our knowledge, the simplest 3,4-diphenylpyrazolo[3,4-b]pyridines such as CCT249525 have not been reported as kinase inhibitors to date. More complex pyrazolo[3,4-b]pyridines, including 3,4-di(hetero)aryl substitution, recently have been proposed as inhibitors of ALK. 23
Among the singleton hits identified in the screen was the known kinase inhibitor, 4-((5-amino-1-(2,6-difluorobenzoyl)-1H-1,2,4-triazol-3-yl)amino)benzenesulfonamide, also known as JNJ-7706621 (
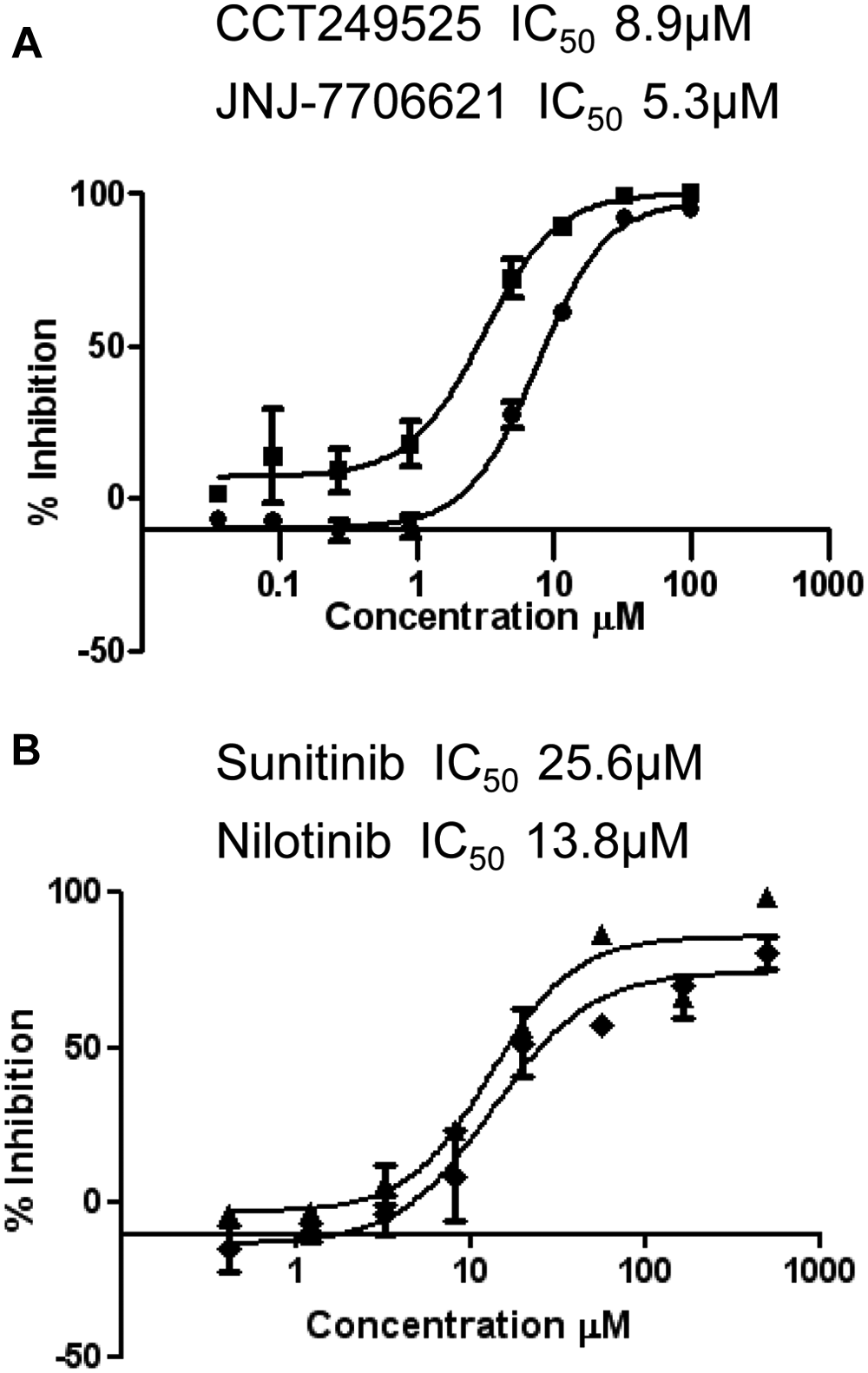
(
The screening library contained a small subset of known clinically used kinase inhibitors, and several compounds in this group were identified as IRE1α autophosphorylation inhibitors. One of these is sunitinib ( Table 1 and Fig. 5B ), which has been identified previously as an inhibitor of IRE1α autophosphorylation in both human and yeast. 11 Sunitinib is a well-characterized ATP competitive type I kinase inhibitor, typically binding to target proteins in their kinase-active, DFG-in conformation. Intriguingly, the screen also identified nilotinib as an inhibitor of IRE1α autophosphorylation ( Table 1 and Fig. 5B ). In contrast to sunitinib, nilotinib is an ATP competitive type II inhibitor that characteristically binds to, not only the ATP-binding site, but also an adjacent hydrophobic binding site accessible only when a kinase is in the DFG-out conformation. 27
XBP1 Splicing Assay Results
There is clear evidence that the ribonuclease activity of IRE1α is dependent on the kinase activity,
11
and therefore inhibiting the autophosphorylation of IRE1α should ultimately lead to inhibition of XBP1s. An XBP1 splicing assay was performed to show that the compounds not only demonstrated inhibition in the biochemical autophosphorylation DELFIA assay but also reduced or inhibited the splicing of XBP1 mRNA in cells. This assay demonstrated that sunitinib and nilotinib were able to effectively reduce tunicamycin-induced XBP1 mRNA splicing in H929 multiple myeloma cells (
Discussion
IRE1α inhibitors could play an important role in the treatment of cancer, particularly multiple myeloma. Kinases in general have proved to be highly druggable targets, and HTS has played a key role in the discovery of several kinase inhibitors. 13 However, many of the HTS assay formats used are based on either ATP turnover or substrate phosphorylation. Because trans-autophosphorylation is a crucial step in the activation of human IRE1α, we have developed an easy to automate, cost-effective, high-throughput, and specific enzymatic assay for identification of IRE1α small-molecule inhibitors.
Compared with other kinase assays, a high concentration of IRE1α was required, which could potentially limit the sensitivity of the assay. However, for the homogeneous time-resolved fluorescence (HTRF) assay we reported previously, 11 enzyme concentrations in the range of 200 to 800 nM were required to observe the phosphorylation of substrate peptide, present in 200 nM concentration. In addition, in a published AlphaScreen assay, measuring dimerization/oligomerization or the overall phosphorylation state of IRE1α, use of an enzyme concentration of 470 nM was reported to give the best S/B ratio. 12 Therefore, the concentration used in our autophosphorylation assay is in keeping with those used in IRE1α assays using other formats. One of the reasons for the need for a high IRE1α concentration in the autophosphorylation assay could be the use of a truncated IRE1α construct without the luminal domain, which may have resulted in impaired dimerization and therefore catalytic activity. In addition, in the autophosphorylation assay, IRE1α acted as its own intermolecular substrate in the reaction, which contributed to the need for a high enzyme concentration.
Despite the high enzyme concentration, the assay was successfully used in an HTS format to test 2312 compounds for the inhibition of IRE1α autophosphorylation. The compounds were screened at three concentrations, which was a valuable way of prioritizing hits for further confirmation and eliminating false positives. We identified 192 primary hits, of which 60 were confirmed as active by IC50 determination. A further triage based on ligand efficiency enabled us to prioritize 30 compounds for further investigation. Several drug-like series of compounds have been identified with a potency range of 0.3 to 65 µM against dephosphorylated IRE1α. The hits included the known kinase inhibitor JNJ-7706621 and the previously identified IRE1α inhibitor, sunitinib, demonstrating the validity of the screen. We confirmed our previous finding that sunitinib blocks XBP1 splicing in multiple myeloma cells. As well as identifying ATP-competitive type I inhibitors, we have shown inhibition of IRE1α autophosphorylation with the ATP-competitive type II inhibitor and DFG-out binder, nilotinib. 27 This is an interesting finding as it suggests that the activation loop of IRE1α may be able to adopt a DFG-out conformation. Since the sequence and structural homology between kinase ATP-binding sites can present a general challenge to the design of selective protein kinase inhibitors, targeting a DFG-out conformation of the kinase domain of IRE1α could be a way of introducing desired kinase selectivity, in a similar manner as demonstrated for the Bcr-Abl inhibitor imatinib. 28 In addition, different chemotypes unknown to be inhibitors of IRE1α have been identified as ligand-efficient inhibitors of IRE1α autophosphorylation as exemplified by CCT249525. The discovery of ATP-competitive compounds, with both DFG-in and DFG-out binding scaffolds represented, that not only show inhibition of the IRE1α autophosphorylation but also block XBP1 splicing in myeloma cells suggests that more potent and selective compounds could be developed toward new therapies to treat multiple myeloma.
Footnotes
Acknowledgements
We would like to dedicate this paper in loving memory of Anthea Hardcastle (November 1, 1947, to August 17, 2012).
We acknowledge NHS funding to the NIHR Biomedical Research Centre. The authors thank their colleagues and collaborators in the Division of Cancer Therapeutics for valuable discussions. We thank Dr. Wynne Aherne for her help and advice. We thank Dr. Maruf Ali and Professor Laurence Pearl for helpful discussions. We thank the Institute of Cancer Research for funding. We would also like to thank the Kay Kendall Leukaemia Foundation
Declaration of Conflicting Interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: The authors are present or past employers of The Institute of Cancer Research, which has a commercial interest in the IRE1α inhibitors. Please note that all authors who are, or have been, employed by The Institute of Cancer Research are subject to a “Rewards to Inventors Scheme,” which may reward contributors to a program that is subsequently licensed.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by Cancer Research UK (grant number C309/A8274). Faith E. Davies is a Cancer Research UK senior cancer fellow (grant number C20826/A12103) (http://www.cancerresearchuk.org/), (![]() ).
).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.