
Abstract
Broad sequencing enterprises such as the FANTOM or ENCODE projects have substantially extended our knowledge of the human transcriptome. They have revealed that a large portion of genomic DNA is actively transcribed and have identified a plethora of novel transcripts. Many newly identified transcripts belong to the class of long noncoding RNAs (lncRNAs), which range from a few hundred bases to multiple kilobases in length and harbor no protein-coding potential. Although the biological activity of some lncRNAs is understood, the functions of most lncRNAs remain elusive. Tools that allow rapid and cost-effective access to functional data of lncRNAs are therefore essential. Here, we describe the construction and validation of an endoribonuclease-prepared siRNA (esiRNA) library designed to target 1779 individual human lncRNAs by RNA interference. We present a compendium of lncRNA expression data for 11 human cancer cell lines. Furthermore, we show that the resource is suitable for combined knockdown and localization analysis. We discuss challenges in sequence annotation of lncRNAs with respect to their often low and cell type–specific expression and specify esiRNAs that are suitable for targeting lncRNAs in commonly used human cell lines.
Introduction
The full sequence of the human genome has been available for more than a decade, 1 and we have since learned a great deal about the genetic information stored in almost every cell of the body. In contrast, our understanding of the human transcriptome is much less complete. Recent advances in sequencing technologies have paved the way for a more comprehensive catalog of transcribed genomic loci. Major contributions came from large sequencing projects such as FANTOM, 2 ENCODE, 3 or GENCODE,4,5 which revealed that about 75% of genomic DNA is actively transcribed 6 but less than 2% of the RNAs contain exons with protein coding potential. 3 The most frequently encoded transcripts with a total count of 113,513 7 belong to the class of long noncoding RNAs (lncRNAs), which range from a few hundred bases up to multiple kilobases in length.3,5 Thus far, only a few lncRNAs have been studied in great detail, but their functional relevance in the regulation of diverse biological processes including chromatin modification, 8 imprinting, 9 gene expression, 10 and splicing 11 has been demonstrated. For example, the lncRNA Xist mediates global changes in the chromatin structure of the X-chromosome, leading to its inactivation for dosage compensation in females. 8 In genomic imprinting, maternally and paternally inherited alleles are differentially regulated, a process that is partially mediated by the lncRNA AIR. 9 AIR recruits H3 lysine 9 methylase G9a in cis, leading to transcriptional silencing of the respective genomic loci in the paternal allele. 9 Long noncoding transcripts have also been found to directly regulate gene transcription. The lncRNA PANDA was shown to associate with the transcription factor NF-YA, displacing it from its promoter sites and leading to transcriptional shut down of the respective genes. 10 In addition, IncRNAs have been implicated in the regulation of alternative splicing. MALAT1 interacts with serine/arginine splicing factors and influences their distribution in nuclear speckles. 11 Furthermore, lncRNAs have been shown to play an important role in the onset of diverse human diseases such as cancer12,13 and Alzheimer’s disease. 14 These and other features make lncRNAs a relevant and interesting subject for future functional studies. Biological characterization of lncRNAs, however, comes with substantial technical challenges. In contrast to coding transcripts, noncoding RNAs are more difficult to annotate because of their often reduced and tissue-specific expression5,15,16 and low phylogenetic conservation. 5 These challenges influence the methods applied for their functional dissection. Therefore, the available technologies for the systematic functional characterization of lncRNAs are limited.
Endoribonuclease-prepared siRNA (esiRNA) technology has been widely used as an efficient tool for mediating RNA interference (RNAi) for coding transcripts in mammalian cells, ranging from single transcript targeting to genome-wide loss-of-function (LOF) screening.17–26 Furthermore, esiRNAs have been shown to be efficient in various cell types, including diverse human cancer cell lines,17–24 mouse embryonic stem cells, 25 and cells in the developing mouse brain. 26 esiRNAs are pools of several hundreds of individual siRNAs generated by enzymatic digestion of a typically 300- to 600-base-pairs-long double-stranded RNA (dsRNA) derived from a single target transcript. 27 Pooling of siRNAs has been shown to increase on-target specificity by decreasing off-target effects,27,28 because the siRNAs that make up the pool exist in comparable amounts and have the same on-target competence. Therefore, the silencing capacity for the intended target is additive, whereas off-target effects are diluted out. Genome-scale esiRNA libraries for coding transcripts of Mus musculus and Homo sapiens origins have been used intensively for LOF screening17–22,24,25 and are commercially available. Furthermore, an esiRNA library for targeting mouse lncRNAs has been successfully used in an LOF screen identifying genes implicated in the maintenance of pluripotency in embryonal stem cells. 29 We reasoned that the same advantages of the esiRNA technology should apply for silencing human lncRNAs. The generation of a library for the systematic screening of human lncRNAs by RNAi and localization profiling may therefore be a suitable method to perform a small-, medium-, or large-scale investigation of lncRNA function. Here, we present a first-generation human esiRNA library targeting 1779 lncRNAs. We demonstrate that esiRNAs designed against specific lncRNAs effectively deplete their targets, and fluorescence in situ hybridization (FISH) probes generated from the same source can detect the subcellular localization of long noncoding transcripts in fixed human cells. For screening, we recommend selecting target lncRNAs that are expressed in the chosen cell line to lower costs and labor. To facilitate screening, we provide the expression pattern of the targeted lncRNAs in 11 commonly used cell lines.
Materials and Methods
esiRNA Synthesis
esiRNAs were synthesized as described previously.
30
Briefly, we amplified the lncRNA transcript region of interest in a two-step PCR using transcript-specific primers with sequence tags added to their termini (left tag: 5′-TGACACTATAGAAGTG-3′, right tag: 5′-CTCACTA-TAGGGAGA-3′). The sequences of all primers are given in
Transcriptome Analysis
Analysis of gene expression was carried out using RNA-Seq data generated by the Cold Spring Harbor Laboratory as a part of the ENCODE project. 6 . Raw sequencing reads obtained from polyA+ cytosolic fractions of the 11 analyzed cell lines were downloaded from the UCSC repository and aligned to the human reference genome (hg19 assembly, February 2009) using STAR version 2.4.0e. 31 The read alignment process was guided by a splice junction database constructed from a set of protein-coding transcripts obtained from the Ensembl database (release 74) combined with a set of noncoding transcripts from LNCipedia (www.lncipedia.org, release 3.0). Gene expression levels were estimated using an in-house developed application, which calculates fragments per kilobase of expressed exons per million mapped reads (FPKM values) in a manner similar to NEUMA. 32 In our approach, to calculate an effective length of genes, instead of using simulated data, we used a pooled set of aligned RNA-Seq reads for assessing genome mapability. Further analyses of gene expression results and generation of plots were performed in R (version 3.1.2), with the aid of “plyr” and “ggpot2” packages. Two-dimensional kernel density estimations were calculated using the “kde2d” function from the “MASS” package, with 500 grid points in each direction.
Quantification of lncRNAs
For quantitative PCR assays, HeLa cells were transfected in 12-well cell culture plates with 300 ng esiRNA using 4.2 µL oligofectamine (Life Technologies) per well. After 24 h incubation, the cells were harvested and total RNA was extracted using the RNeasy Kit (Qiagen, Venlo, the Netherlands) including an on-column DNase I digest (Qiagen) as given in the manufacturer’s protocol. Total RNA was reverse transcribed using SuperScript III reverse transcriptase (Life Technologies) and Oligo-(dT)16-20 or random hexamer primers. Quantification of the targeted RNA was conducted using the ABsolute QPCR SYBR Green Kit (Thermo Scientific, Waltham, MA) according to the manufacture’s protocol using a CFX96 Touch real-time PCR machine (Bio-Rad, Hercules, CA). The expression levels of either GAPDH or TBP transcripts were used as endogenous controls. All primer sequences are provided in
Combined Immunofluorescence and RNA FISH
HeLa cells were grown on Labtek chamber slides (Thermo Scientific Nunc) until they reached confluency. The chambers were washed with 1× phosphate buffer saline (PBS) and fixed with 4% paraformaldehyde for 10 min at room temperature. The cells were then washed two times with 1× PBS and permeabilized with 0.5% (v/v) triton X-100 in PBS and 2 mM vanadium ribonucleoside for 5 min. Following permeabilization, the cells were washed three times with 1× PBS and blocked with 1% bovine serum albumin (BSA) in PBS for 15 min at room temperature. Primary antibodies were diluted in 1% BSA, and cells were incubated in a humidity chamber with the antibody solution for 20 to 30 min. Subsequently, the chambers were washed three times for 5 min each with PBS and incubated with secondary antibodies (diluted in 1% BSA) in a dark humidity chamber for 1 h. Thereafter, the cells were postfixed with freshly made 4% paraformaldehyde for 10 min at room temperature. The cells were then washed twice in 2× saline-sodium citrate (SSC) buffer for 5 min, and RNA FISH was performed as described previously. 29 To prepare probes for RNA FISH using the c-KLAN approach, 29 the first round PCR products were amplified with forward primer containing full SP6 sequence (5′-GAATTTAGGTGACACTA-TAGAAGTG-3′) and reverse primer containing full T7 sequence (5′-GCTAATACGACTCACTATAGGGAGA-3′). The amplicons were transcribed in vitro using Chromatide Alexa Fluor-546–tagged UTPs (Invitrogen, Carlsbad, CA) with either T7 polymerase for antisense riboprobe or SP6 polymerase for sense riboprobe (according to the manufacturer’s instructions). The RNA probes were purified using the RNeasy mini kit (Qiagen) according to the manufacturer’s instructions and diluted in 2× hybridization buffer (20% 20× SSC, 50% dextran sulfate, 20% BSA, and 10% vanadyl ribonucleoside). For hybridization, 15 to 20 ng probe, 10 µg salmon sperm DNA (Invitrogen), 10 µg human Cot-1 DNA (Invitrogen), and 10 µg yeast tRNA (Sigma Aldrich, St. Louis, MO) were mixed with two volumes of 100% ethanol and dried. The dried mixture was then resuspended in 5 µL of 100% formamide at 37 °C for 10 min followed by denaturation at 74 °C for 7 min and then resuspended in 5 mL hybridization buffer. Twenty microliters of probe was then added to the cells and placed in a humidifying chamber overnight with gentle rocking. Slides were washed the next day with 4× SSC followed by washes (three times each) with 2× SSC, 50% formamide, at 39 °C for 5 min and 2× SSC at 39 °C for 5 min and washed with 1× SSC at room temperature for 10 min followed by staining with DAPI. FISH images were acquired using Delta Vision Core Widefield deconvolution fluorescence microscope (Applied Precision Inc., Mississauga, Ontario, Canada) using an Olympus UPlanSApo 100×/1.4 oil immersion lens. The refractive index of immersion oil (Applied Precision Inc.) used was 1.518. For RNA FISH, the cells were covered with 4× SSC buffer, and images were taken at a resolution of 15.528 pixels per micron and voxel size of 0.06 × 0.06 × 0.20 micron. Other parameters of image acquisition for RNA FISH are as follows: bits per pixel = 16, dimension order = XYCZT, camera type = CoolSNAPHQ2/HQ2-ICX285, illumination = SSI-lumencor transmitted light (LED), dichromatic mirrors = DAPI/FITC/TRITC/Cy5, excitation bands = 381-399 (DAPI), 529-556 (Alexa 546), 650-670 (Cy5). Image processing for all RNA FISH images was done as follows: images were maximally projected using the Z Project tool of Fiji. The maximum and minimum displayed values were then manually adjusted to make the background invisible using the brightness and contrast settings tool. The same value ranges were applied to all figures wherever comparisons were made. The images were cropped to zoom into specific regions in the figure followed by a conversion to an eight-bit image. Merged images were generated using LUTs green for Alexa 546, red for Cy5, and blue for DAPI.
Results and Discussion
esiRNA Library Design
Systematic investigations of lncRNAs require resources to functionally test the roles of these molecules in cells. Indeed, some companies have developed reagents to study lncRNAs at larger scale. General Electric offers a Lincode collection of predesigned siRNAs against 2231 characterized human lncRNAs, and Exiqon applies chemically derived single-stranded LNA-based antisense oligomers complementary to the target lncRNA. To build a first-generation esiRNA library targeting human lncRNAs, we investigated the current annotation of long noncoding transcripts. We used LNCipedia,
7
an integrated database, which compiles the content of several other resources. Currently, the LNCipedia database reports a total of 113,513 long noncoding transcripts expressed from 63,038 genes of the human genome.
7
However, not every cell or tissue expresses the full set of lncRNAs. To investigate the expression pattern of lncRNAs in different human cells, we analyzed RNA-sequencing data generated by the ENCODE consortium
3
for 11 commonly used cell lines derived from different tissues and cell types (GM12878, K562, A549, HelaS3, HepG2, HUVEC, IMR90, MCF-7, SK-N-SH, NHEK, H1hESC;
Table 1
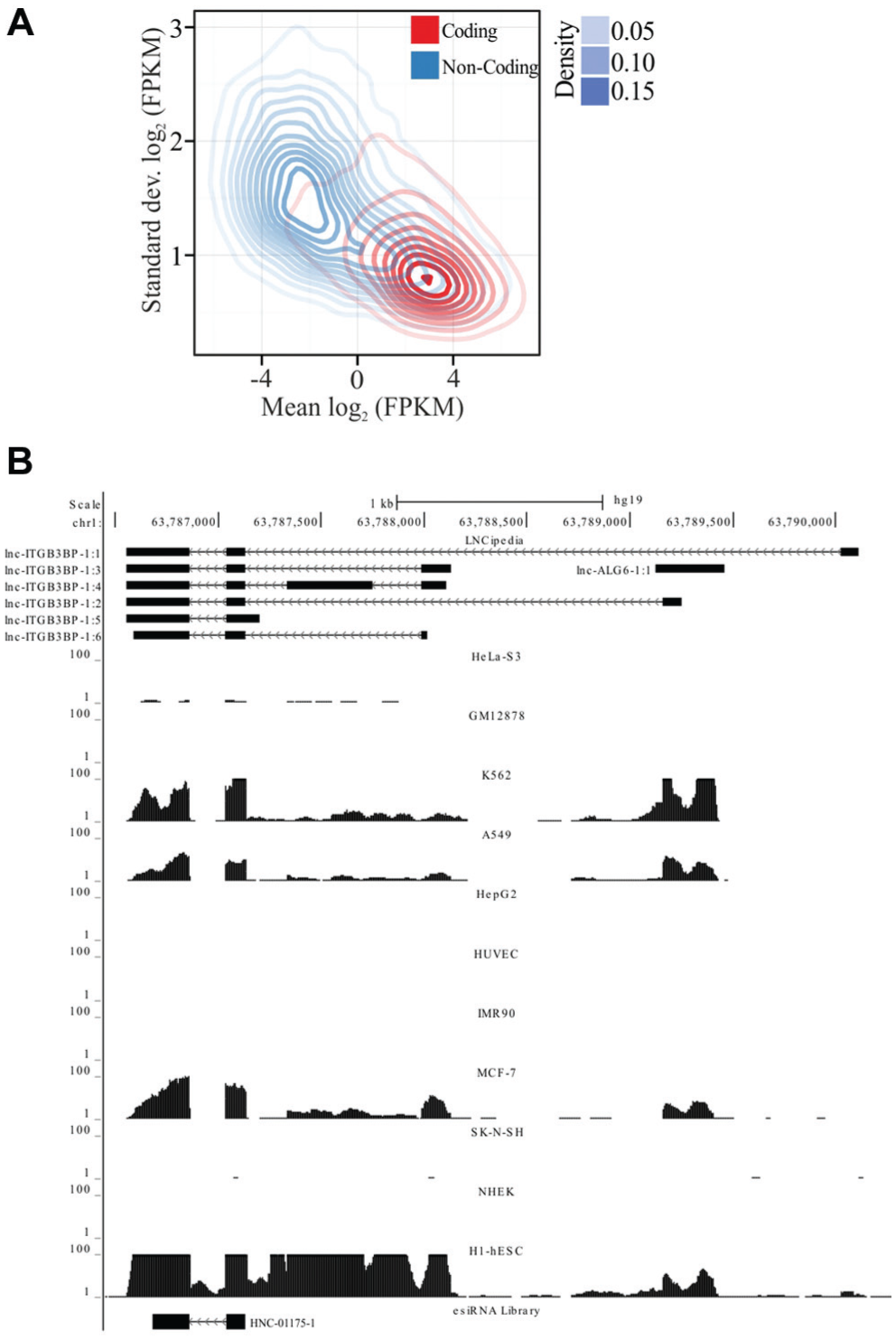
) and compared their transcriptome composition. We found that noncoding transcripts were on average lower in expression than protein-coding transcripts in all cell lines tested (
Fig. 1A
). We also observed a significant number of lncRNAs being expressed at comparable levels to highly expressed coding RNAs. However, a larger number of lncRNAs are expressed at a much lower level than most coding RNAs (
Fig. 1A
), suggesting that noncoding RNAs are generally more versatile in their expression than translated RNAs. The broad range of expression was observed between different cell types and also between lncRNA and mRNA levels in the same cell type (
Fig. 1A
;
Long noncoding RNA (lncRNA) expression analysis. a
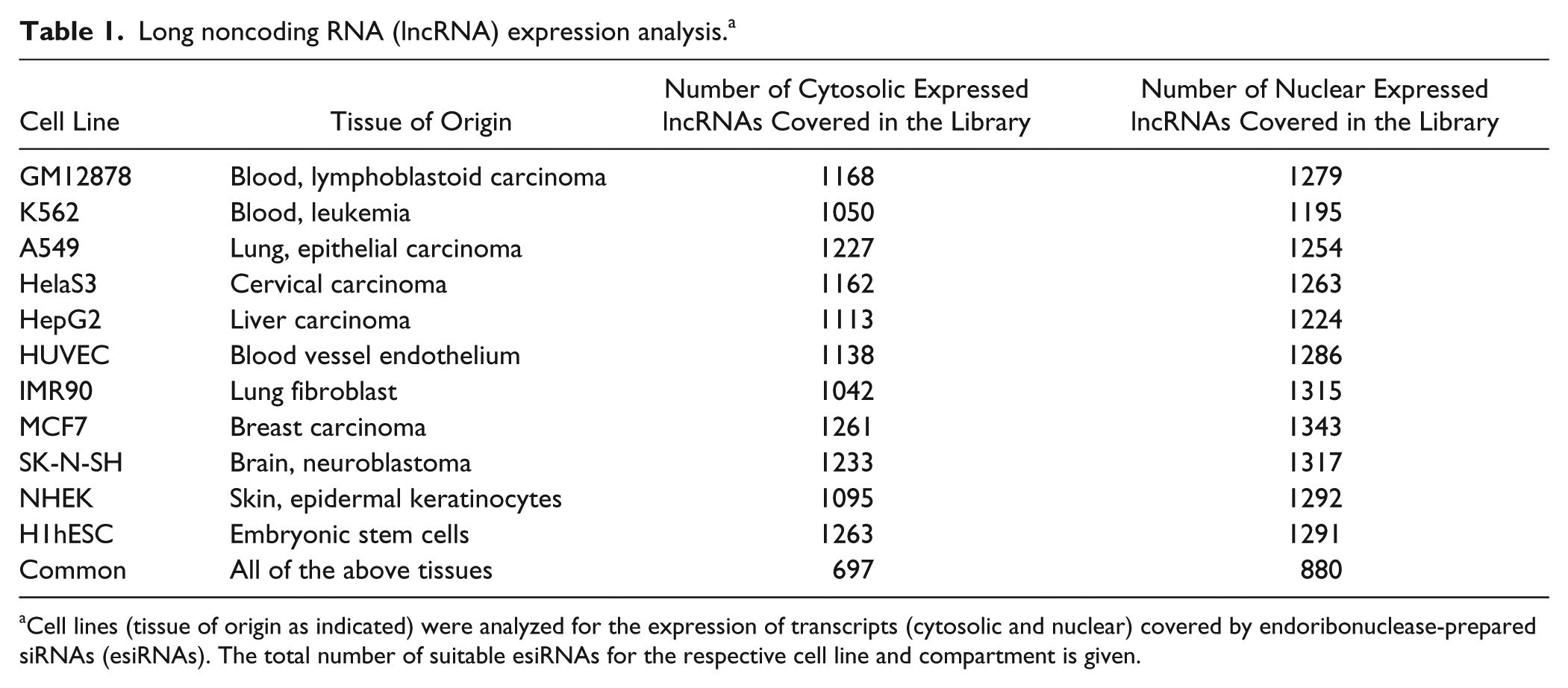
Cell lines (tissue of origin as indicated) were analyzed for the expression of transcripts (cytosolic and nuclear) covered by endoribonuclease-prepared siRNAs (esiRNAs). The total number of suitable esiRNAs for the respective cell line and compartment is given.
(
To design esiRNAs for the silencing of the 2944 lncRNAs, we analyzed the transcript sequences in more detail. We found that 1963 (67%) genes have more than one splicing variant and 129 genes have close paralogs in the human genome (
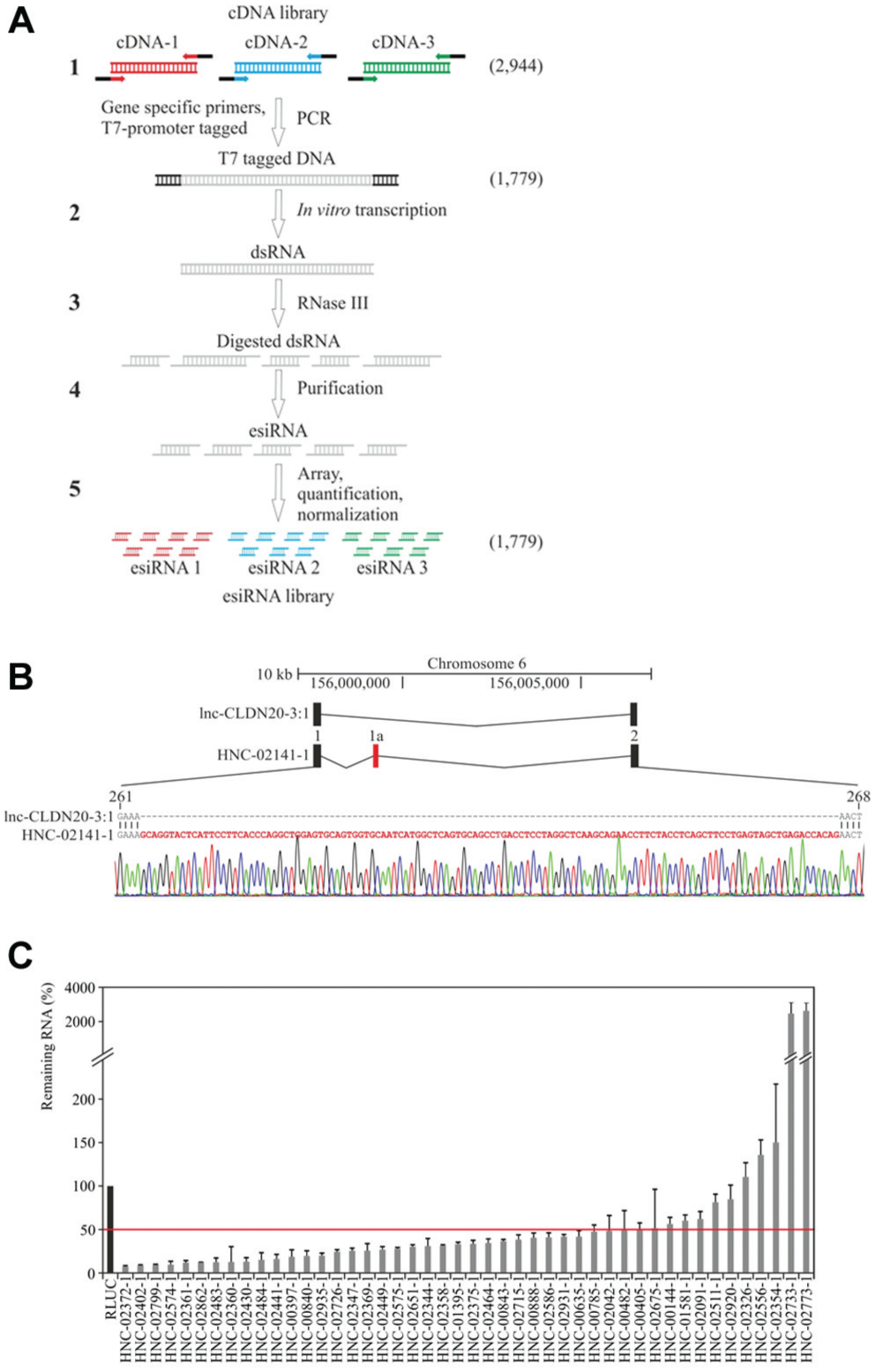
Endoribonuclease-prepared siRNA (esiRNA) synthesis and validation. (
esiRNA Library Validation
To validate the quality of our library and to quantify its RNAi capability, we nominated 46 esiRNAs that target lncRNAs expressed in HeLa cells for quantitative real-time PCR (qRT-PCR) analysis (
Fig. 2C
). To ensure that the 46 chosen target genes are representative of the library, we selected transcripts that covered the full range of lncRNA expression levels (
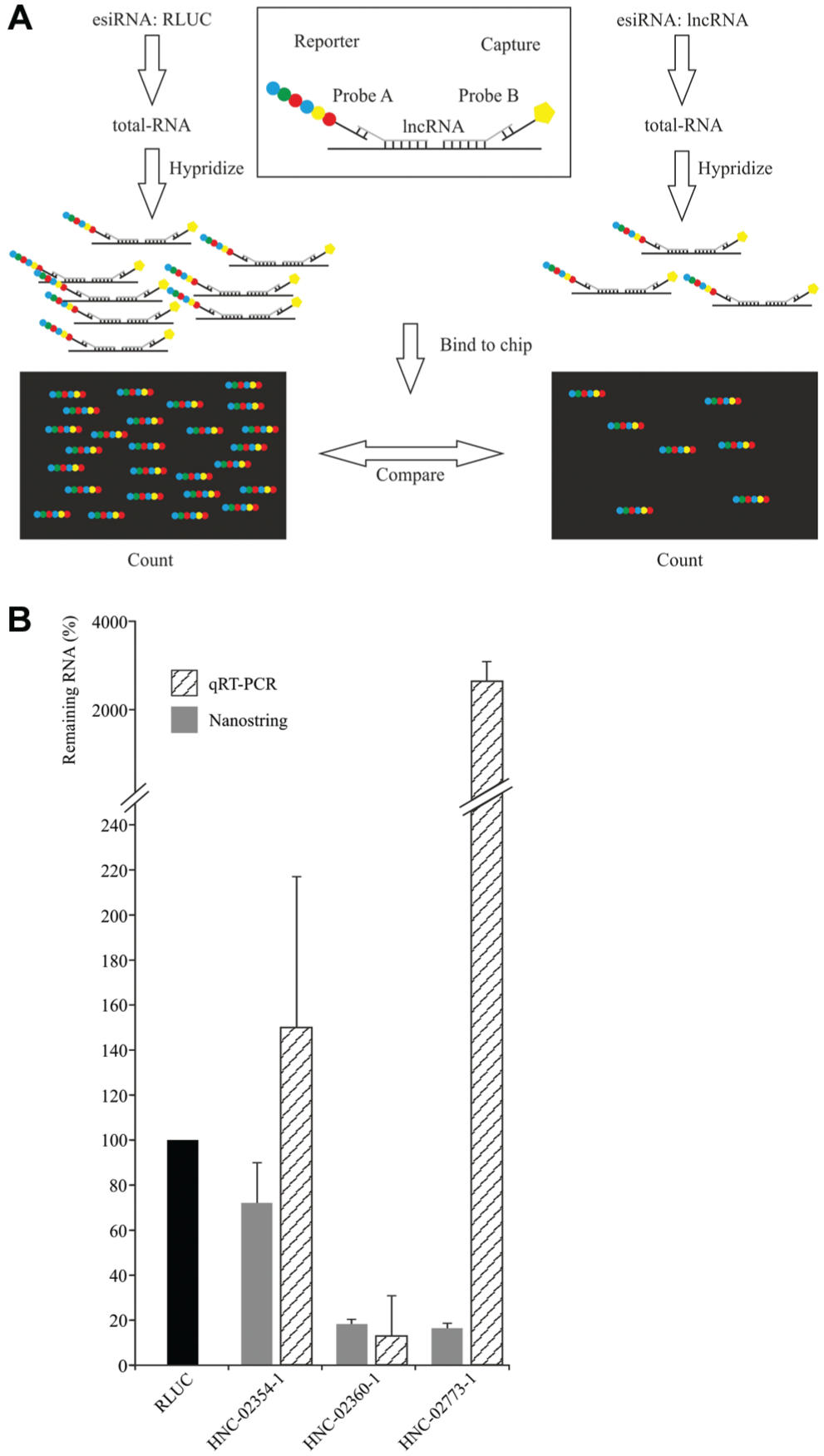
Transcript quantification using Nanostring technology. (
We conclude that our esiRNA library is capable of silencing the majority of human IncRNA transcripts. Hence, the presented esiRNA resource represents a valuable screening tool that should expedite the molecular characterization of human IncRNAs.
c-KLAN
The generation of LOF phenotypes by RNAi provides a valuable approach to gaining insights into the biological functions of lncRNAs. Additional understanding may come from determining the subcellular localization of lncRNAs.18,29,36 To combine knockdown and localization experiments, we previously established a method
29
that allows the generation of sense- and antisense-labeled riboprobes for RNA FISH and dsRNAs for esiRNA synthesis from a single source. c-KLAN
29
was successfully applied to the functional dissection of mouse long noncoding transcripts.
29
To extend c-KLAN to human lncRNAs, we selected two PCR products (HNC-02360-1, HNC-02586-1) designed to target the lncRNAs lnc-APC-6 and lnc-HSP90AA1-9 from our library and generated labeled FISH probes for hybridization in HeLa cells. We detected distinct signals for both probes using fluorescence microscopy and observed diverse localization patterns for the different lncRNAs. lnc-APC-6 mainly showed nucleolar localization, whereas lnc-HSP90AA1-9 was found dispersed as spots in the nucleus and cytoplasm (
Fig. 4
). In both cases, we did not observe a signal for the sense probes (
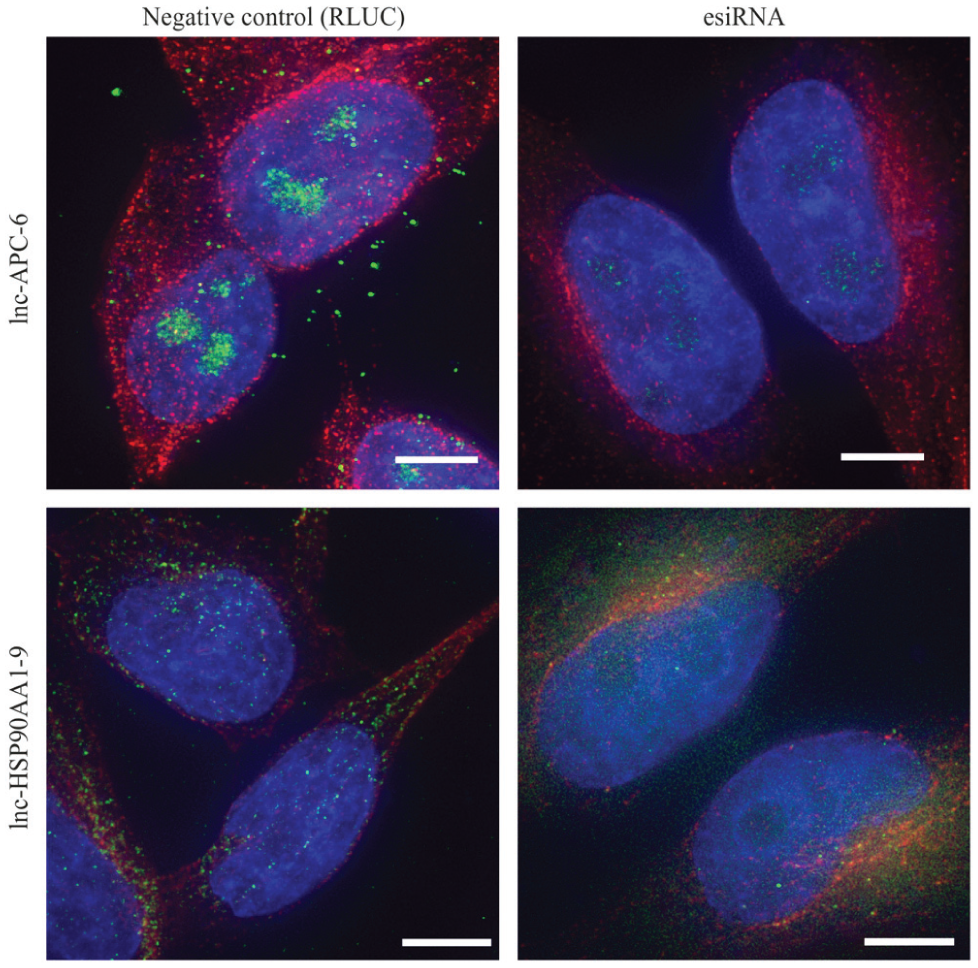
Localization of long noncoding RNAs (lncRNAs) by fluorescence in situ hybridization. HeLa cells stained by fluorescently labeled RNA probes (green), anti-tubulin antibody (red), and DAPI (blue) after transfection with a negative control endoribonuclease-prepared siRNA (esiRNA; Renilla Luciferase, left) and an esiRNA (right) designed against the target lncRNAs lnc-APC-6 (HNC-02360-1, top panel) and lnc-HSP90AA1-9 (HNC-02586-1, bottom panel). All images were processed using the same exposure settings and with identical threshold adjustments to represent the actual signal intensities. Scale bars: 5 µm.
In conclusion, we present in this study synthesis and utility of a renewable resource of esiRNA and FISH probes for the dissection of human lncRNA functions by RNAi and by localization profiling. This scalable resource allows the systematic characterization of lncRNAs and can be extended to a genome-wide collection covering the entire noncoding transcriptome, allowing for a comprehensive investigation of lncRNA function in human biology and disease.
Footnotes
Acknowledgements
The authors thank Sebastian Rose and Romy Heinze for technical assistance and Annett Erkes for computational support.
Declaration of Conflicting Interests
The authors M.T., M.P.-R., and I.W. declare a conflict of interest due to an affiliation with Eupheria Biotech GmbH. F.B. is an adviser for Eupheria and therefore declares a conflict of interest. D.C. declares no conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The work was funded by the Bundesministerium für Bildung und Forschung (BMBF) grant Go-Bio 2 (0315980).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.