
Abstract
Our approach aims to optimize postscreening target validation strategies using viral vector–driven RNA interference (RNAi) cell models. The RNAiONE validation platform is an array of plasmid-based expression vectors that each drives tandem expression of the gene of interest (GOI) with one small hairpin RNA (shRNA) from a set of computed candidate sequences. The best-performing shRNA (>85% silencing efficiency) is then integrated in an inducible, all-in-one lentiviral vector to transduce pharmacologically relevant cell types that endogenously express the GOI. VariCHECK is used subsequently to combine the inducible knockdown with an equally inducible rescue of the GOI for ON-target phenotype verification. The complete RNAiONE–VariCHECK system relies on three key elements to ensure high predictability: (1) maximized silencing efficiencies by a focused shRNA validation process, (2) homogeneity of the RNAi cell pools by application of sophisticated viral vector technologies, and (3) exploiting the advantages of inducible expression systems. By using a reversible expression system, our strategy adds critical information to hot candidates from RNAi screens and avoids potential side effects that may be caused by other, irreversible genomic manipulation methods such as transcription activator-like effector nucleases (TALEN) or clustered regularly interspaced short palindromic repeats/Cas9 (CRISPR/Cas). This approach will add credibility to top-hit screening candidates and protect researchers from costly misinterpretations early in the preclinical drug development process.
Introduction
Since the discovery of RNA interference (RNAi) phenomena in plants 1 and nematodes in the 1990s,2,3 controlled manipulation of transcript levels to silence gene function quickly advanced to become a staple technique for the investigation of gene function and signaling pathways in early drug discovery. Comparably easy handling and flexible manipulation of short RNA sequences made this technology an ideal tool for large-scale screening applications. This rapid development of RNAi techniques has led to a collection of genome-wide libraries to address the hot topics of today’s medical research.4–10 Even in vivo applications of RNAi libraries in mice have been made possible recently when combined with the newest advancements in animal genetics. 11
As the number of RNAi screening applications grew, so did awareness of the challenges that lie hidden within this technology.12–18 Throughout the years, the output of reproducible hits has been low, and a distinctive lack of overlap has been reported from screen to screen. Analyzing data from 30 publications that consisted of various screening approaches, Bhinder and Djaballah 19 demonstrated this lack of agreement in the obtained data as well as a low degree of predictability. Apparent paper-to-paper differences were linked to the types of RNAi libraries used (arrayed versus pooled) and the types of RNAi [small hairpin RNA (shRNA) versus small interfering RNA (siRNA) duplexes] that were applied in the screen.
A proximate cause for these major inconsistencies may be found in the overall differences in experiment design as well as in postscreen analysis, demonstrating the necessity of standardization for screening procedures. The ultimate causes lie deeper, however, and have become a major topic in the field under the term off-target effects (OTEs). This concept describes observed side effects on the expression levels of a large number of genes that are not originally intended as targets of the shRNA or siRNA approach. It is thought that this effect is driven mainly by partial sequence complementarities with the unintentionally targeted genes, mimicking microRNA (miRNA) activity that leads to phenotypic overlaps with the silenced target gene. 20 Imprecise cleavage during Dicer activity can lead to further inaccuracies when using shRNA constructs. 21
Learning from past research, the movement within the field clearly aims toward improving the output of true positive hits from RNAi screening methods. A great number of ideas are being explored to help standardize approaches and reduce the number of off-target effects during screening. Recent refinement of the clustered regularly interspaced short palindromic repeats/Cas9 (CRISPR/Cas) gene-editing technology for eukaryotic cells 22 will likely add a further dimension to the optimization of gene target identification in screening applications. 23
Modifying or adding to the complexity of future screening methods will help increase their effectiveness; however, it is a dangerous notion to suggest that it would justify omission of stringent postscreening validation of their top-hit candidates. On the contrary: With the awareness of past shortcomings, better methods to challenge top-hit gene candidates post screening have gained in importance, to consolidate findings as early as possible within the drug development process and base subsequent developments on solid ground.
To address this need for top-candidate validation, we have developed a two-step method that relies on a full-scale shRNA validation (RNAiONE) and application process, followed by an ON-target control experiment (VariCHECK) to verify that phenotypic changes are not linked to OTEs.
With our method, we are able to define highly efficient (>85% knockdown) shRNA sequences against a gene of interest (GOI) and translate this efficiency into an in vitro cell model via lentiviral (LV) vectors. Exploiting the advantages of advanced, multicistronic, and all-in-one inducible expression systems, we can later rescue the shRNA-driven GOI knockdown by parallel ectopic expression of a knockdown-resistant form that recues the gene product. By using reversible expression methods, our strategy adds critical information to candidate genes from RNAi screens and avoids potential side effects that may be caused by other, irreversible gene-editing methods.
Materials and Methods
Cell Lines
Human embryonic kidney cells HEK293 and HEK293TN and murine fibroblast cells NIH-3T3 were grown in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 2 mM l-Glutamin and 10% fetal calf serum (FCS; Biochrom, Berlin, Germany). U87-MG cells were cultured in modified Eagle’s medium (MEM) supplemented with Earl salts + 10% FCS (Biochrom) + 2 mM l-Glutamin + 1× NEAA + 1 mM Na-Pyruvat. Cultivation of all cell lines occurred at 37°C, 5% CO2, and 100% humidity.
shRNAmir Template Cloning
A set of 10 shRNAmir (the microRNA backbone carrying the defined shRNA sequence) template oligonucleotide cassettes were cloned into a shuttle plasmid under the control of the human EF1a promoter. The correctness of all cloned oligonucleotide cassettes was verified by sequencing.
Cloning of Target cDNA into a Validation Vector pVal
The coding region of the full-length complementary DNA (cDNA) for a GOI was PCR-amplified and cloned into the pVal downstream of an enhanced green fluorescent protein (eGFP) coding region, resulting in pVal-GOI. Sequence identities of the exemplary gene targets presented in this publication (and, as a result, their corresponding shRNA sequences) are tied into ongoing R&D projects; as such, they are protected by confidentiality agreements and cannot be published at this point. General descriptions of the gene products’ biology and their role in medical research are provided.
Recombination of EF1a-shRNAmir Cassettes into pVal-GOI
The EF1a–shRNAmir regions of the shRNAmir shuttle plasmids, along with a shuttle plasmid encoding for a nontargeted (NT) shRNAmir (as control), were then transferred into pVal-GOI by recombination cloning. Each resulting validation vector contains three transcriptional units ( Fig. 1A ).
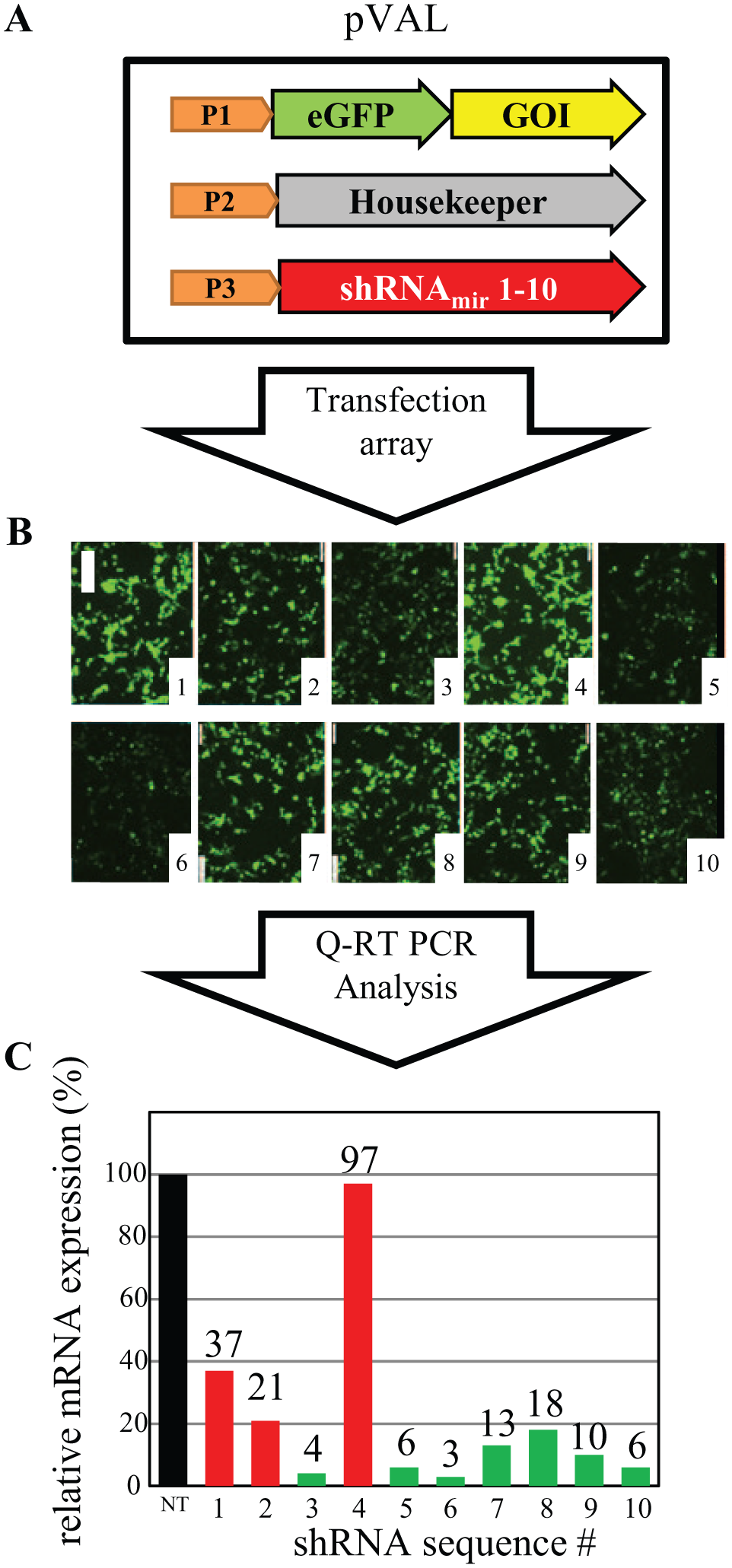
RNAiONE workflow to identify high-efficiency small hairpin RNA (shRNA) sequences. (
Cell-Based Quantification of Gene Knockdown by Quantitative Reverse Transcriptase (qRT)-PCR
NIH-3T3 or HEK293 (depending on the GOI species) cells that were seeded the day before on six-well plates were transfected at a confluence of 50% with 3 µg pVAL using JetPEI (Polyplus, Illkirch, France) as transfection reagent. After incubation of the transfected cells for 48 h under standard cell culture conditions (37 °C, 5% CO2, and 100% humidity), total RNA was isolated using the NucleoSpin RNA kit from Macherey-Nagel (Düren, Germany). Reverse transcription was carried out with 1 µg total RNA using the RNA to cDNA EcoDryTM Premix (double primer) system from Takara Clontech (Saint-Germain-en-Laye, France). The silencing efficiency of each shRNA was determined by quantification of the eGFP–target cDNA expression levels relative to those found in cells transfected with the NT-shRNA control vector using the vector-encoded marker transcript as the internal reference gene. qRT-PCR measurements were performed in duplicate to account for pipetting errors. In cases when two measurements would deviate more than 0.25 cycles, the experiments were repeated.
Quantitative PCR Parameters
Total RNA used in RT: 1 µg
cDNA primer: Random hexamer/oligo dT
Real-time platform: Light Cycler 480 (Roche)
Detection method: SYBR I
Amplicon integrity verified by: Melting point analysis
Lentiviral Vector Generation
The best-performing shRNA cassette from the RNAiONE screen was cloned into the tetracycline (TET)-inducible all-in-one vector pLVi(3G)-MCS-Puro. For TET-inducible rescue of the cancer gene of interest (GOIc) the coding region of GOIcsilent was correspondingly cloned into pLVi(3G)-MCS-Neo. For the resulting vectors, cloning success was verified by restriction analysis and DNA sequencing. LV, HIV-based, VSVG-pseudotyped, self-inactivating transduction particles were produced by co-transfection of HEK293TN cells with the expression vectors and the third-generation LV packaging plasmid mix pPACKH1 (SBI, Mountain View, CA) following the instructions given by the manufacturer. Transfection reagent lipofectamine 2000 was used. Viral genomic titers were determined using the LentiX qRT-PCR kit (Clontech, Mountain View, CA). For subsequent cell transductions, infectious titers were calculated by assuming a ratio of genomic-to-infectious titer of 100:1.
Stable Cell Pool Generation
Stable HEK293 or U87MG cell pools were generated by transduction of the cells with LV transduction particles.
Transduction was performed with a multiplicity of infection (MOI) of 1 (293 cells) and MOI 5 (U87MG cells). Seventy-two hours after transduction, cells were cultivated for 2 weeks in the presence of puromycin or G418. Stably selected cells were then frozen in aliquots of 1.0E06 cells/vial.
Quality Control of Transduced Cell Pools
One cell aliquot per cell pool was thawed, and 1×105 cells were seeded in two wells of a six-well plate. Doxycycline was added to one well the next day, to a final concentration of 0.5 µg/ml. The cells were then cultivated for another 48 h. Isolation of total RNA and reverse transcription, as described in the “Cell-Based Quantification of Gene Knockdown by Quantitative Reverse Transcriptase (qRT)-PCR” section, were performed.
The relative expression levels of the target gene or the ectopically expressed gene were determined by qRT-PCR relative to noninduced cells and a reference gene.
Western Blot
Twenty micrograms of total cellular protein per lane were loaded onto a 10% sodium dodecylsulfate–polyacrylamide gel and separated by gel electrophoresis. Proteins were then transferred onto a 0.45 µm polyvinylidene fluoride membrane. Endogenous or ectopically expressed GOIs were detected by probing the blots with appropriate polyclonal primary and secondary antibodies, and subsequent visualization using the ECL Advance kit (GE Healthcare, Little Chalfont, UK).
Results
shRNA Validation in a Small Cell–Based Assay: RNAiONE
The success of any shRNA strategy is directly linked to its ability to silence the GOI efficiently. Several factors affect how well the expression of a target gene can be repressed in the final cell model. In silico calculations help predict the efficiency of shRNA sequences, and several algorithms have been made openly available for public use. These predictions help minimize the list of potential shRNA candidates when designing a silencing strategy against a defined GOI. The allure of these in silico predictions is to spring right into the application of the calculated top candidates in a full-fledged cell model. Our experience from more than 150 validation experiments has shown that even top algorithms cannot predict with certainty which sequences within a small set of 10–15 may be truly high-end (>85%) performers and which sequences might fail despite all odds (
Figs. 1C
To develop such a strategy, we base our RNAiONE technology platform on an in-house designed plasmid system, pVAL. pVAL is constructed to carry three separate expression units on one plasmid ( Fig. 1A ). GFP tagging of the GOI helps monitor both overall transfection efficiencies (the amount of cells with signal) and residual GOI expression levels measured by relative signal intensities ( Fig. 1B ). The Zeocin resistance gene, as the third expression unit, serves as an internal reference gene on pVAL to normalize GOI expression on the transcription level. Each plasmid is equipped with a single shRNA that was designed by using in-house algorithms similar to the publically available methods. In short, RNAi patterns against a defined target are ranked according to their conformity with the ideal Pei and Tuschl sequence; 24 their distance from the start codon; the overall GC content; their prevalence in other, openly accessible algorithms; and their homology frequency within a species’ genome [checked via a Basic Local Alignment Search Tool (BLAST) search]. The final RNAiONE experiment relies on a small transfection array of 10–15 pVAL vectors that carry the top picks from these rankings.
The RNAiONE validation array is performed by transfecting one of two possible cell types, Hek293 or NIH3T3 cells. The type of cells chosen for each RNAiONE experiment depends on the origin of the GOI: Human-derived GOIs are tested in murine NIH3T3 and vice versa, to avoid uncontrolled overlap of endogenous GOI expression levels. The final readout of the RNAiONE array is a qRT-PCR analysis to identify the top-performing shRNA candidate within the RNAiONE array ( Fig. 1C ; the top-performing shRNA is 6 with a 97% knockdown efficiency).
Translation of Validated shRNA Sequences into Inducible LV Vectors for Predictive Cell Modeling
Once a top-performing shRNA sequence is identified, it can be integrated into LV or adenoviral (AV) vectors and applied in pharmacologically relevant cell lines. An especially promising application is the construction of inducible LV vectors to generate stable knockdown cell pools for further target validation experiments.
We base our inducible LV shRNA system on the TET expression system. 25 We use a multicistronic LV construction vector to be able to include all integral elements of the TET system as well as an antibiotic selection marker on a single LV vector backbone ( Fig. 2A ). Using these “all-in-one” LV vectors, we avoid the necessity for repeated transductions, generating highly homogenous cell pools. Depending on the cell type, we can choose to add a poloxamere-based transduction enhancer to further increase transduction efficiencies and therefore homogeneity of the cell pool. 26 Figure 2B shows two exemplary transduction experiments using inducible all-in-one LV vectors expressing eGFP under control of a third-generation TET promoter. Fluorescence-activated cell-sorting analysis of the transduced cell pools demonstrates a quantitative shift of GFP-positive cells after application of 0.5 µg/ml doxycycline (1.0–99.1% for U87MG cells and 0.9–93.3% for HCT116 cells; see Fig. 2B ; dead cells are not gated out).
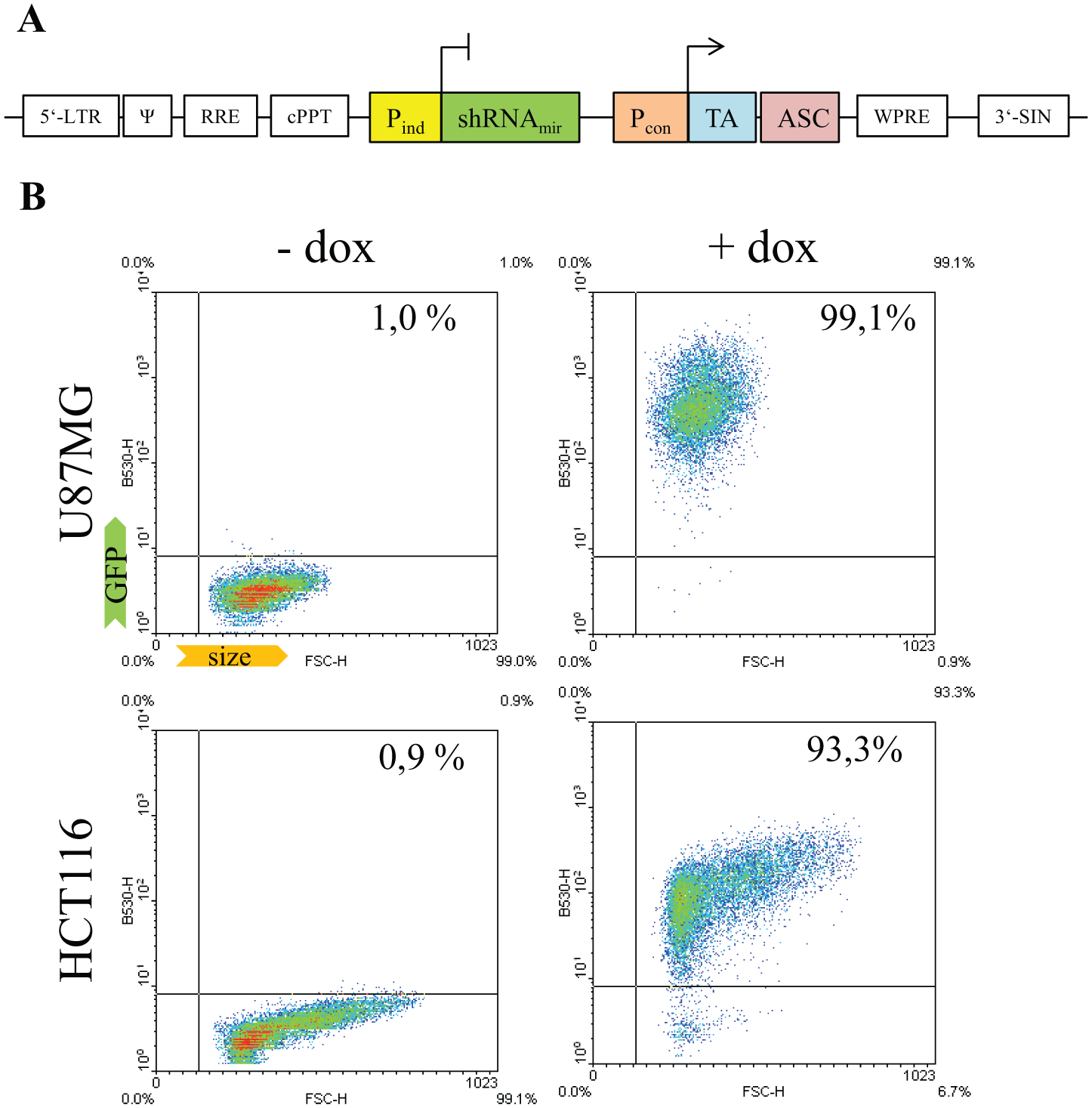
Lentiviral all-in-one inducible expression vectors enable highly homogenous gene-of-interest (GOI) expression in treated cell pools. (
Using our optimized validation system, we were able to validate shRNA sequences even against hard-to-silence targets such as G-protein coupled receptors (GPCRs). Using an extended array of 15 pVAL plasmids, we were able to identify two shRNA sequences that would reduce the expression of a cancer-related GPCR (GPCRx) by 96% and 95%, respectively, during validation ( Fig. 3A , shRNA sequences 5 and 12).
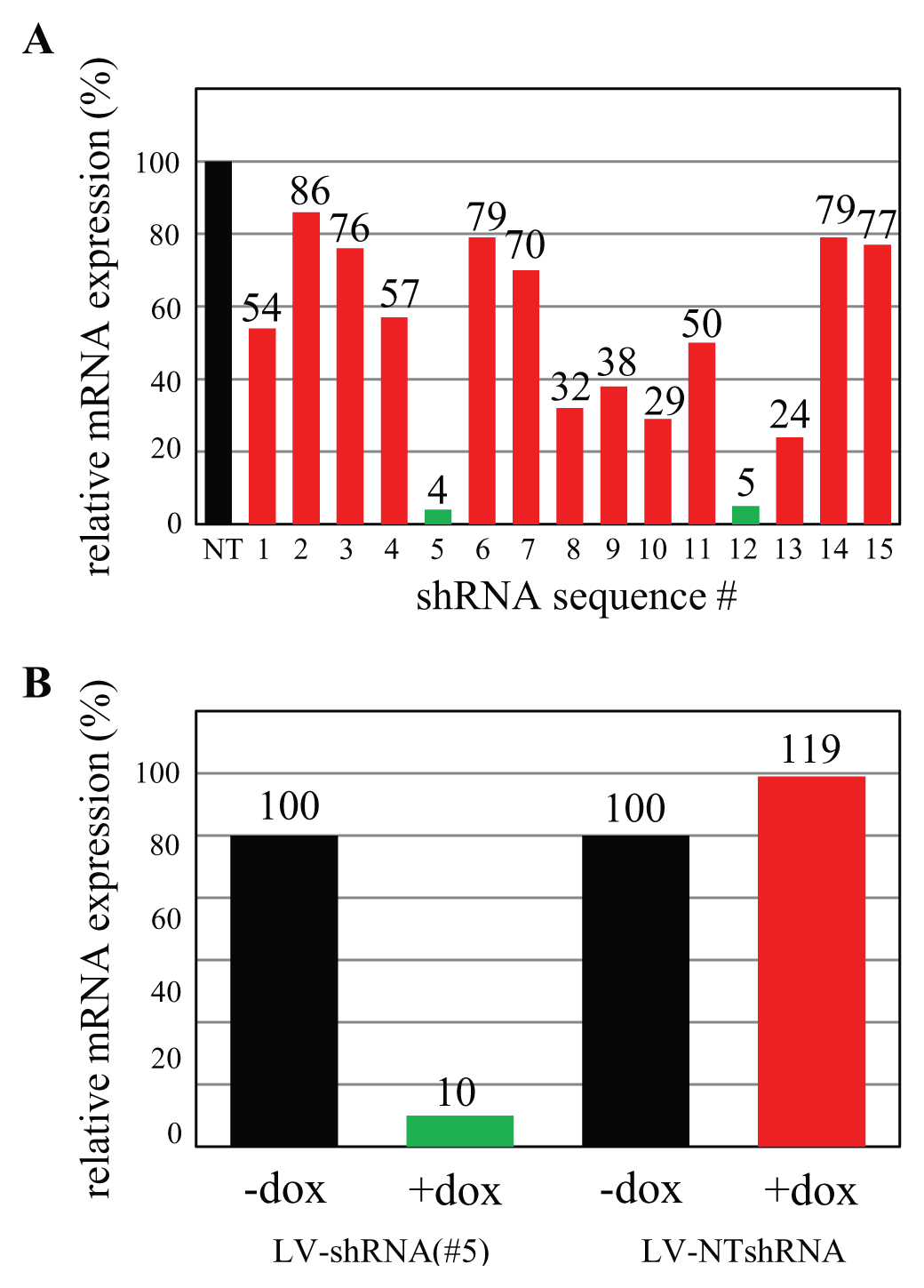
Translation of RNA interference (RNAi) efficiencies from validation platform. (
After RNAiONE validation, the top-performing shRNA sequence (sequence 5) was cloned into an all-in-one inducible LV vector. The resulting LV particles were used to transform Hek293 cells that endogenously express the GPCRx. The resulting cell pool retained high expression levels under standard culturing conditions but silenced GPCRx efficiently (90%) after application of 0.5 µg/ml doxycycline ( Fig. 3B ).
ON-Target Phenotype Verification by GOI Rescue
To control for functional OTEs on a knockdown phenotype, we devised a LV-GOI-rescue-vector that relies on the redundancy of the genetic code. We introduce silent point mutations in each codon triplet within the GOI sequence that is targeted by an RNAiONE-validated shRNA sequence. This renders the mutated GOI resistant to the knockdown and rescues its expression after induction of the shRNA ( Fig. 4A ).
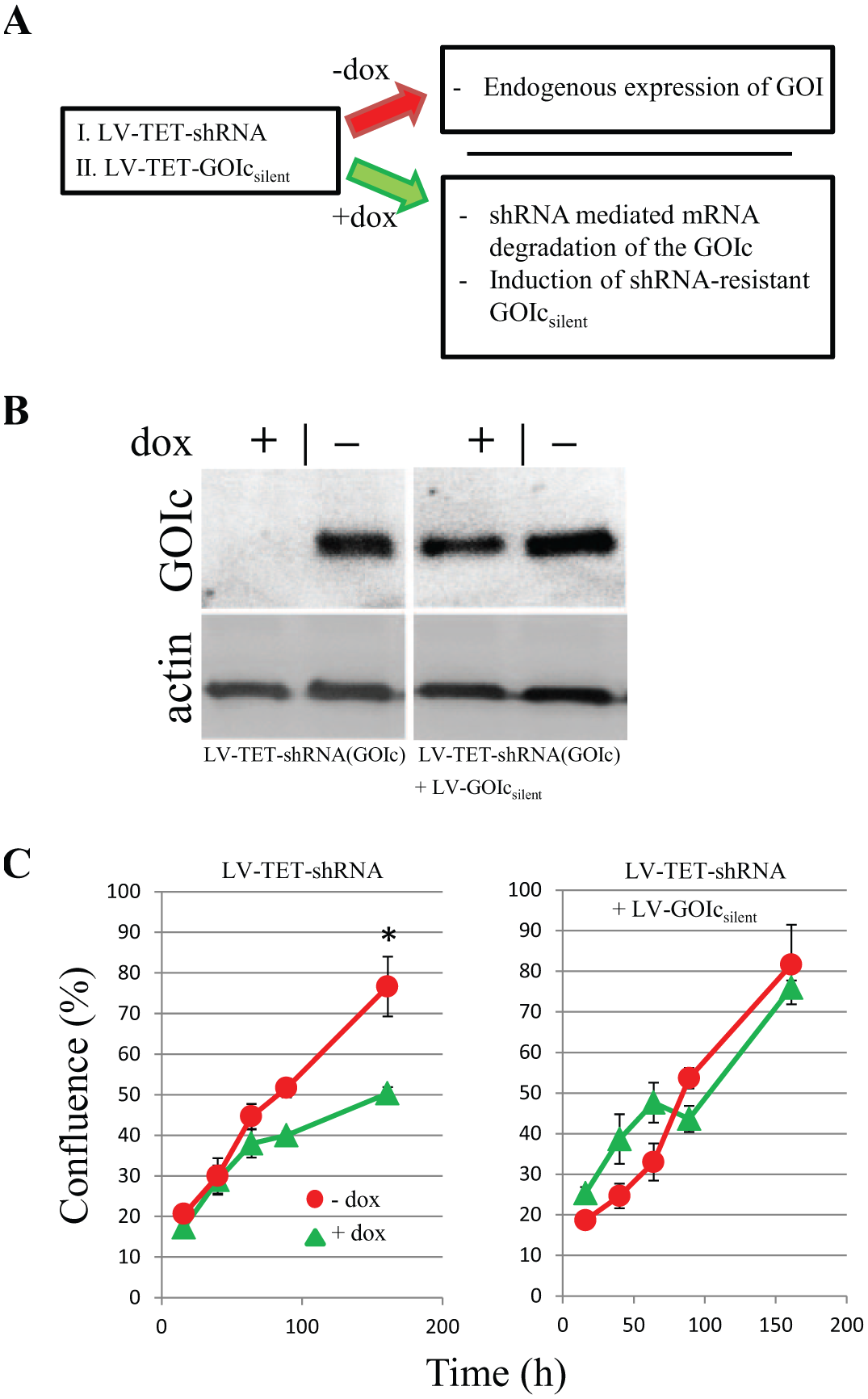
VariCHECK gene-of-interest (GOI) rescue as an ON-target verification method. (
In a proof-of-concept experiment, we were able to rescue the expression of a gene, GOIc (
Fig. 4B
), that is coding for a membrane-spanning protein indicated in the growth and survival of certain cancer types. To perform an ON-target control, we treated an established inducible U87MG clonal knockdown line (
Fig. 4B
, LV-TET-shRNA) with a LV-TET-GOIcsilent vector. The functional phenotype of the inducible GOIc knockdown was a significant reduction in cell proliferation after induction (confluence 161 h after cell seeding was 76.7% in doxycycline-free media versus 50.3% in media containing 0.5 µg/ml of doxycycline; p > 0.001 in Student’s t-test). After LV-TET-GOIcsilent treatment, the expression of GOIc product was restored, as shown by Western blot analysis in
Figure 4B
. We performed a qRT-PCR analysis of dox-treated and -untreated cells to demonstrate this switch from the endogenous GOIc to GOIcsilent expression on the transcript level (
Discussion
In light of the current movement within the RNAi screening community to increase the output of true positive hits on novel gene candidates, it becomes apparent that downstream validation of top-hit candidates needs to be improved as well. In comparison to the preceding cost-intensive, large-scale screening process, any postscreening validation strategy should fulfill the following criteria:
Time efficient
Adds credibility to top-hit candidates
Flexibly adaptable to future applications
RNAiONE: Basing Subsequent Experiments on Solid Ground
To fulfill these goals, we decided to devise a straightforward method for target gene validation that uses established technologies that are easily adaptable to a number of different end applications. Using a plasmid-based shRNA strategy as a first step enables us to quickly develop a small customized array composed of 10–15 calculated sequences against a defined target. Total time-to-success at this stage is only 3 weeks.
Any RNAi validation method should attempt to maximize knockdown efficiency while ensuring that it can be transported reliably into subsequent cell models. By choosing a cell line derived from a species that is different from the GOI source, we reduce background expression, making sure to quantitate only GOI transcripts expressed from the pVal vectors, not endogenous transcripts. By expressing GOI and shRNA from the same plasmid, both elements are expressed at comparable levels, negating the necessity to account for fluctuations of transfection efficiency and thus increasing confidence in the RNAiONE data set. We have adapted the RNAiONE-pVAL system to rely on the same RNA polymerase system as the subsequent viral vectors, to ensure that both the validation and final virus vector show comparable activities. With these optimizations in place, we are able to transport the high knockdown efficiency established in the RNAiONE stage into a pharmacologically relevant cell model by recloning the top shRNA candidate into a LV vector system (
Figs. 2A
,
3
,
Lentiviral TET Vector Construction for More Flexible Cell Models
Nearly any mammalian cell type is susceptible to LV transduction. Using novel LV transduction enhancers 26 and virion modification methods, 27 we can potentially increase the already-wide tropism of this virus family even further, making it a gold standard technology for cell-modeling applications. By using the LV transduction machinery, we generate stable shRNA cell pools that can later be used to generate clonal knockdown cell lines for drug-screening applications.
Continuous shRNA expression can reduce the viability of stable knockdown cells by affecting cell proliferation or inducing apoptosis in transduction-positive cells. Chronic changes of expression levels of cell cycle regulators such as p53, Myc, or Plk-1, undoubtedly clinically relevant gene targets, will change the overall genetic makeup of a cell line. Building an inducible vector platform ( Fig. 2A ) offers an elegant solution to this challenge.
We demonstrate that even difficult GOIs can be effectively silenced using inducible shRNA LV (
Figures 3
VariCHECK RNAi ON-Target Verification System
OTEs remain a main concern when working with RNAi methods. Especially, shRNA sequences have been reported to be affected to a greater extent, in part because minute changes in the shRNA vector design can lead to imprecise Dicer processing, effectively increasing the potential for mismatches with untargeted messenger RNA (mRNA) sequences. 21
Several methods have been devised to address the dilemma of RNAi OTEs. A very straightforward method is to introduce not one but multiple different RNAi sequences against one GOI to dilute the OTEs but maintain strong silencing. This method has been reported to work when using several siRNA sequences against a GOI at low concentrations; 29 however, it would be cumbersome to transport into a shRNA model. It would significantly increase the number of vector cloning steps and necessitate a complex control system to manage expression levels, making timing of the design unfeasibly long. In addition, it is not clear how many shRNA sequences would be necessary to dilute OTEs substantially, and the risk of introducing a strong OTE effector would grow with each additional sequence.
A very elegant approach to control for OTEs by Buehler et al. 30 uses the observation that most OTE effects can be averted by mutating bases from the middle section of a siRNA sequence to their complement bases. Comparing results from a standard siRNA sequence with its mutated brother can measure the relative degree between ON- and OFF-target effects on GOI expression. This c911 technique is a promising tool for siRNA-based methods but again poses additional challenges when used with shRNA sequences.
The limited applicability of these methods to shRNA sequences underlines the importance of a well-founded validation method to identify the best shRNA sequence among a list of top candidates. With the RNAiONE system, we can rely on the top-performing sequence against a defined GOI, minimizing the chance that perceived changes in phenotype are due to OTEs. To further increase the confidence in the performance of our validated sequences within a final cell pool, we use VariCHECK to rescue the silenced GOI product while maintaining shRNA expression. Using silent mutations, we are able to rescue the unaltered gene product while rendering its mRNA resistant to the shRNA expression.
As demonstrated in our proof-of-concept experiment ( Fig. 4 ), knockdown of a cancer-related GOIc led to a convincing phenotypic change, namely, a strong reduction in proliferation activity. This pharmacologically interesting finding would justify further analysis of GOIc function, but the chance for functional OTEs greatly reduced the feasibility of further investments. Reconstituting the original phenotype by rescue of the GOIc gene product in the presence of the knockdown sequence increased the credibility of the original knockdown experiment significantly, justifying further investigations of the role of GOIc in cancer biology.
Bringing all these elements together, we believe that the RNAiONE–VariCHECK approach is an ideal system to define and apply highly efficient shRNA sequences for postscreening GOI validation in a time-efficient manner. Relying on inducible, multicistronic expression systems, we increase the credibility of the data acquired and are able to generate flexible and predictive cell systems that can be included in subsequent stages of the drug development process. Both applications (knockdown and rescue) are established as highly homogeneous cell pools to maintain the cell line’s genetic background, avoiding genetic drift from the parent line. This also increases the range of options regarding how to carry both cell pools into subsequent processing stages (e.g., clonal selection and/or upscaling for drug-screening applications). Importantly, by using an inducible system, we are able to work with shRNA sequences that are targeted against GOIs that are essential for either cell proliferation (compare Fig. 4C ) or cell survival, a limitation that can affect other, irreversible cell modification techniques.
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.