
Abstract
Friedreich’s ataxia is a neurodegenerative disease caused by deficiency of the mitochondrial protein frataxin. This deficiency results from expansion of a trinucleotide repeat in the first intron of the frataxin gene. Because this repeat expansion resides in an intron and hence does not alter the amino acid sequence of the frataxin protein, gene reactivation could be of therapeutic benefit. High-throughput screening for frataxin activators has so far met with limited success because current cellular models may not accurately assess endogenous frataxin gene regulation. Here we report the design and validation of genome-engineering tools that enable the generation of human cell lines that express the frataxin gene fused to a luciferase reporter gene from its endogenous locus. Performing a pilot high-throughput genomic screen in a newly established reporter cell line, we uncovered novel negative regulators of frataxin expression. Rational design of small-molecule inhibitors of the identified frataxin repressors and/or high-throughput screening of large siRNA or compound libraries with our system may yield treatments for Friedreich’s ataxia.
Keywords
Introduction
Friedreich’s ataxia (FRDA) is an autosomal recessive disease resulting from repression of the iron-binding protein frataxin (FXN). 1 Reduced FXN expression results from an expansion of a trinucleotide repeat in the first intron of the frataxin gene 2 and provokes a spectrum of neuropathological defects in patients with FRDA. FRDA is the most common recessively inherited ataxia disorder across Europe, and several epidemiological studies have estimated the prevalence of FRDA as two to three cases per 100,000 people in Caucasian populations.2–4 Despite years of research, FXN gene regulation remains poorly understood, and treatment options for FRDA are limited.
Therapeutics that reactivate frataxin expression are expected to be beneficial to FRDA patients. 5 The long noncoding GAA triplet repeats found in intron 1 of the FXN gene in FRDA patients impede transcription elongation. Therefore, overcoming this blockade and reactivating FXN expression is an attractive therapeutic strategy. In addition, approaches that enhance transcription rates or increase FXN protein or messenger RNA (mRNA) stability could also correct the FXN protein deficiency in patients with FRDA.6–8
Cell-based assays are essential for the identification of druggable regulators of FXN gene expression and low molecular weight compounds that alleviate FXN deficiency. Previous attempts at developing high-throughput screening (HTS)–friendly assays have used artificial intronic sequences with extended GAA tracts or cell lines containing FXN reporter gene fusions randomly integrated into the genome.9–14 A potential drawback of these systems is that they do not assess the activity of the endogenous FXN gene in the native chromosomal context. This may limit the identification of modulators of FXN expression, because the endogenous chromatin structure and/or nuclear organization of the human genome are major determinants of FXN gene activity.15–18 Thus, more advanced reporter systems that enable the monitoring of gene expression from the endogenous FXN locus are desirable.
Here we report the use of genome-engineering tools to establish a novel drug discovery system that combines genetically accurate reporter systems and high-throughput biology to identify novel therapeutics for Friedreich’s ataxia. We used zinc-finger nucleases (ZFNs) to generate a cellular model in which a luciferase reporter gene is introduced into the endogenous FXN locus. Using this system in a high-throughput genomic screen, we discovered novel repressors of FXN gene expression. Using our system in future drug discovery efforts may finally yield treatments for FRDA.
Materials and Methods
Cell Culture
Dermal fibroblasts from patients affected by FRDA (GM04078) were obtained from Coriell Cell Repositories (Camden, NJ). GM04078 cells were immortalized by lentiviral delivery of BMI1 and hTERT as described previously, 19 resulting in the cell line FRDA-4078iBT. GM04078 and FRDA-4078iBT were maintained in Eagle’s minimal essential medium (Sigma, St. Louis, MO) supplemented with 10% non–heat-inactivated fetal bovine serum (FBS; Sigma) and 1× L-glutamine (Life Technologies, Carlsbad, CA) at 37 °C in 5% CO2. HEK293T cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% heat-inactivated FBS (Sigma), 100 U/mL−1 penicillin, 100 µg/mL−1 streptomycin, and 1× L-glutamine (Life Technologies) at 37 °C in 5% CO2.
Genome Editing
Plasmids encoding FXN-ZFN-L, FXN-ZFN-R, and donor constructs (sequences available as supplementary material) were cotransfected into HEK293T cells with the polyethylenimine (PEI) transfection method. Briefly, exponentially growing cells were trypsinized and seeded in a 10-mm tissue culture dish 1 day before transfection. A solution containing 60 µL of 1 mg/mL PEI and 15 µg DNA (two ZFN plasmids + donor 1) in a 1:5 molar ratio (ZFNs/donor) was prepared in 1 mL of sterile DMEM lacking serum. The solution was briefly vortexed and left for 15 min at room temperature before pipetting dropwise to adherent HEK293 cells. Cells were cultured under standard conditions for 2 to 3 days, followed by culturing the cells in the presence of puromycin-containing growth media for 2 weeks. Single puromycin-resistant clones were picked and expanded for further analysis. Sequences of plasmids used in this study are provided as text files.
Luciferase Assays
Luciferase assays were performed with the Promega (Madison, WI) Dual-Luciferase Reporter Assay system according to the manufacturer’s protocol. Briefly, lysis buffer was added directly to confluent cell monolayers. Culture dishes were incubated at 25 °C for 15 min and rocked several times to ensure complete lysis. Cell lysates were centrifuged at maximum speed in an Eppendorf microcentrifuge for 10 min at 4 °C. Luciferase measurements were performed with lysate supernatants in duplicate. All measurements were performed on a Centro LB 960 luminometer (Berthold Technologies, Bad Wildbad, Germany). Bradford assays were carried out to determine total protein concentration, which was used for normalization.
Real-Time RT-PCR
For quantitative reverse transcriptase PCR (qRT-PCR) experiments, total RNA was extracted from GM04078, FRDA-4078iBT, and HEK293T-FF2AP cell lines with the Absolutely RNA Microprep Kit (Stratagene, La Jolla, CA). A 1-µg aliquot of total RNA was reverse transcribed with an AffinityScript enzyme (Stratagene) using the oligo(dT) primer according to the manufacturer’s instructions. qRT-PCR was performed on a CFX96 Real-Time PCR System (Bio-Rad, Hercules, CA) using the SsoAdvanced SYBR Green Supermix (#172-5264; Bio-Rad). Relative RNA levels were calculated from CT values according to the ΔCT method and normalized to GAPDH mRNA levels. Primer sequences are provided in
RNA Interference Screen
A synthetic small interfering RNA (siRNA) library targeting the druggable genome (4835 genes with 9670 constructs or two constructs per gene) was screened in a duplicate 384-well plate format with two gene-targeting siRNAs per well to identify genes that upregulate frataxin-luciferase fusion expression. The library was reverse transfected into the HEK293T-FF2AP reporter cell line via high-throughput transfection20,21 using a fully integrated genomic robotic system (GNF Systems, San Diego, CA). Each plate contained transfection controls targeting cell viability (AllStars Cell Death; Qiagen, Valencia, CA), as well as a nontargeting (AllStars Negative Control; Qiagen) and a frataxin-targeting siRNA (Frataxin-12; Qiagen). Data from each plate were normalized to the plate mean and processed for fold activity. Duplicate readings for each well were geometrically averaged to generate a single activity score. A nonlinear transformation was applied to remove plate variation. 22 Putative hit wells were rescreened as single siRNAs per well for validation. In addition to the first-round hit siRNAs, all siRNA constructs targeting genes of interest from other available libraries (Qiagen; Integrated DNA Technologies, Coralville, IA) were included in the validation screen. An additional 96 nontargeting controls were spotted into each 384-well plate of the validation assay (Qiagen). Data analysis included evaluation of individual and multiple-well RNA interference (RNAi) activities for each gene (Redundant siRNA Activity Analysis). 22
Short Hairpin RNA Delivery
Short hairpin RNA (shRNA) constructs were delivered to FRDA-4078iBT cells by lentiviral transduction. Lentivirus was produced by calcium phosphate transfection of HEK293T cells.
23
All infections were performed at a multiplicity of infection of approximately 30 viral particles per cell. Cells were harvested 6 days after transduction for total RNA isolation. Lipofection was used to deliver shRNA constructs into HEK293T-FF2AP cells; ~200 cells per mm2 were seeded 1 day before transfection. Cells were transfected with plasmids using Lipofectamine 2000 (Invitrogen, Carlsbad, CA) according to the manufacturer’s protocol. shRNA sequences are provided in
Western Blotting
Protein samples were separated on NuPAGE-Novex Bis-Tris 4% to 12% gradient gels (Invitrogen) in MES buffer at 200 V for 40 min. Transfer to Immobilon-P PVDF membrane (Millipore, Billerica, MA) was performed at 25 V (constant) for 45 min in Towbin transfer buffer with 5% methanol and 0.05% sodium dodecyl sulfate (SDS). The membranes were blocked in 5% nonfat dry milk in tris buffered saline with 0.1% Tween-20 (TBST) for at least 30 min at room temperature and subsequently washed once in TBST for 5 to 10 min. The membranes were incubated with the anti-FXN antibody (1:1000, SKU 456300; Invitrogen) in 5% nonfat dry milk in TBST overnight at 4 °C. Membranes were washed in TBST and incubated with secondary antibody (anti–mouse horseradish peroxidase [HRP], 1:10,000) in 5% nonfat dry milk in TBST for 45 to 60 min at room temperature. Blots were developed with Immobilon Western HRP Substrate (Millipore).
Results and Discussion
We aimed at generating genome-editing tools that will enable the FRDA research community to knock in any given sequence tag into the FXN locus in different cell types. To this end, we chose a ZFN-mediated genome-editing approach to tag the 3′ end of the protein-coding region of the endogenous FXN gene with a firefly luciferase (FL) reporter gene.24,25 To induce a double-strand break approximately 1 kb downstream of the 5′ splice site in the last intron of the FXN gene, we designed a pair of ZFNs that bind to the FXN locus uniquely ( Fig. 1A ). For homologous recombination, we created a transgenic donor DNA fragment encoding the last FXN exon fused to the open reading frame of FL ( Fig. 1A ). To allow clonal selection after targeted insertion, we created donor constructs encoding either puromycin N-acetyl-transferase (Puro) preceded by 2A self-cleaving peptide linked to the FXN-FL fusion (donor 1) or an autonomous neomycin (Neo) selection cassette (donor 2). The left homology arm of the donor constructs was designed to introduce a functional branchpoint and 3′ splice sites to favor efficient splicing between exon 4 and inserted exon 5 ( Fig. 1A ).
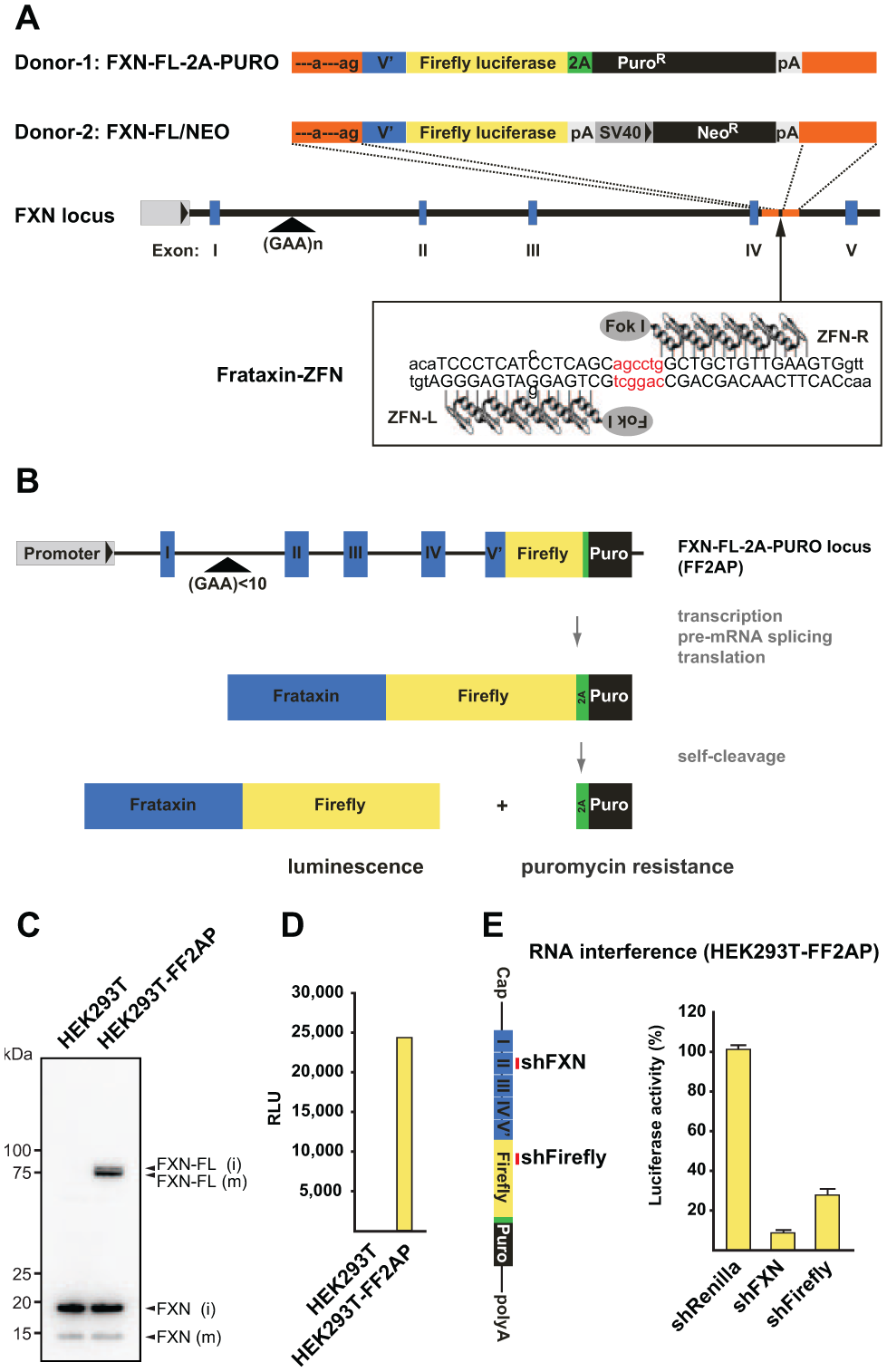
Generation of the HEK293T-FF2AP cell line. (
To provide proof of concept that the ZFN approach is working and to establish a novel reporter cell line that is suitable for high-throughput genomic screening, we used the ZFN system to introduce the luciferase reporter gene into the FXN locus of human embryonic kidney (HEK) 293T cells. This cell line was selected because it can be expanded to a very large scale at a relatively low cost. As a consequence, our FXN reporter system should be compatible with the technical and financial constraints of most academic and industrial screening platforms. We cotransfected human HEK 293T cells with expression plasmids encoding the two frataxin-ZFNs (ZFN-L and ZFN-R) and a plasmid encoding donor 1 ( Fig. 1A ), which confers puromycin resistance only after correct integration, proper pre-mRNA splicing, and translation of the FXN-FL-2A-PURO (FF2AP) fusion protein ( Fig. 1B ). At 2 to 3 weeks posttransfection, puromycin-resistant colonies were isolated and subjected to Western blot analysis. This revealed specific expression of a 75-kDa FXN-FL fusion protein in puromycin-resistant cells (hereafter referred to as HEK293T-FF2AP; Fig. 1C ). We also observed a FXN fusion protein of higher molecular weight than the mature FXN-FL fusion protein, demonstrating efficient cotranslational self-cleavage of the 2A peptide, release of the Puro protein, and mitochondrial processing of the FXN-FL fusion protein. Importantly, we detected firefly luciferase activity in lysates from HEK293T-FF2AP but not from parental HEK293T cells ( Fig. 1D ), an activity that could be abrogated upon expression of an shRNA targeting exon 2 of the FXN mRNA ( Fig. 1E ). Thus, we concluded that the firefly luciferase activity in HEK293T-FF2AP cells came solely from the targeted integration at the FXN locus and, thus, reliably reports expression of the FXN gene from its endogenous location.
In contrast to previous work,
5
the HEK293T-FF2AP cell line allowed us to screen for modulators of FXN expression in its natural genomic context and in an HTS format. This cell line is easy to transfect and hence well suited for RNAi or complementary DNA (cDNA) overexpression screens. Therefore, to identify potential repressors of FXN expression, we performed an RNAi screen targeting 4835 human genes in the HEK293T-FF2AP cell line, using luciferase signal amplification as the readout (
Fig. 2A
). Two unique siRNAs were assayed in a pooled format (two siRNAs per well) at 72 h posttransfection (
Fig. 2B
). Due to the pooled construct format, we chose a baseline of 1.6-fold luciferase activation over the mean as a cutoff to capture phenotypes generated from wells in which only one siRNA may be active. Maximal luciferase expression was 5.1-fold over baseline (targeting hypothetical gene 1000292277), with an additional five siRNAs scoring between 4- and 5-fold. Minimal expressions were obtained with siRNAs targeting Qiagen AllStars Cell Death control (0.06-fold) and frataxin (0.09-fold), indicating that the siRNA transfection was successful and that the relationship between frataxin and luciferase expression was intact. In subsequent validation screens, positive hit siRNAs were spotted in an arrayed well format of one siRNA per well and screened in duplicate under identical conditions. Using the HEK293T-FF2AP cell line, we performed a reconfirmation screen of the primary screen hits (
Fig. 2C
). In parallel, we screened positive hit siRNAs for non-FXN-specific activators of luciferase activity with an HEK 293T cell line stably expressing firefly luciferase from a cytomegalovirus (CMV) promoter (HEK293-pGF1-CMV) (
Fig. 2D
). This allowed for thorough analysis of independent constructs via redundant siRNA analysis (RSA),
22
which enriched for genes where two or more independent siRNAs elevated the FXN-luciferase protein levels specifically (
Fig. 3A
). Complete ranked lists of hits after RSA are provided in the supplemental material (
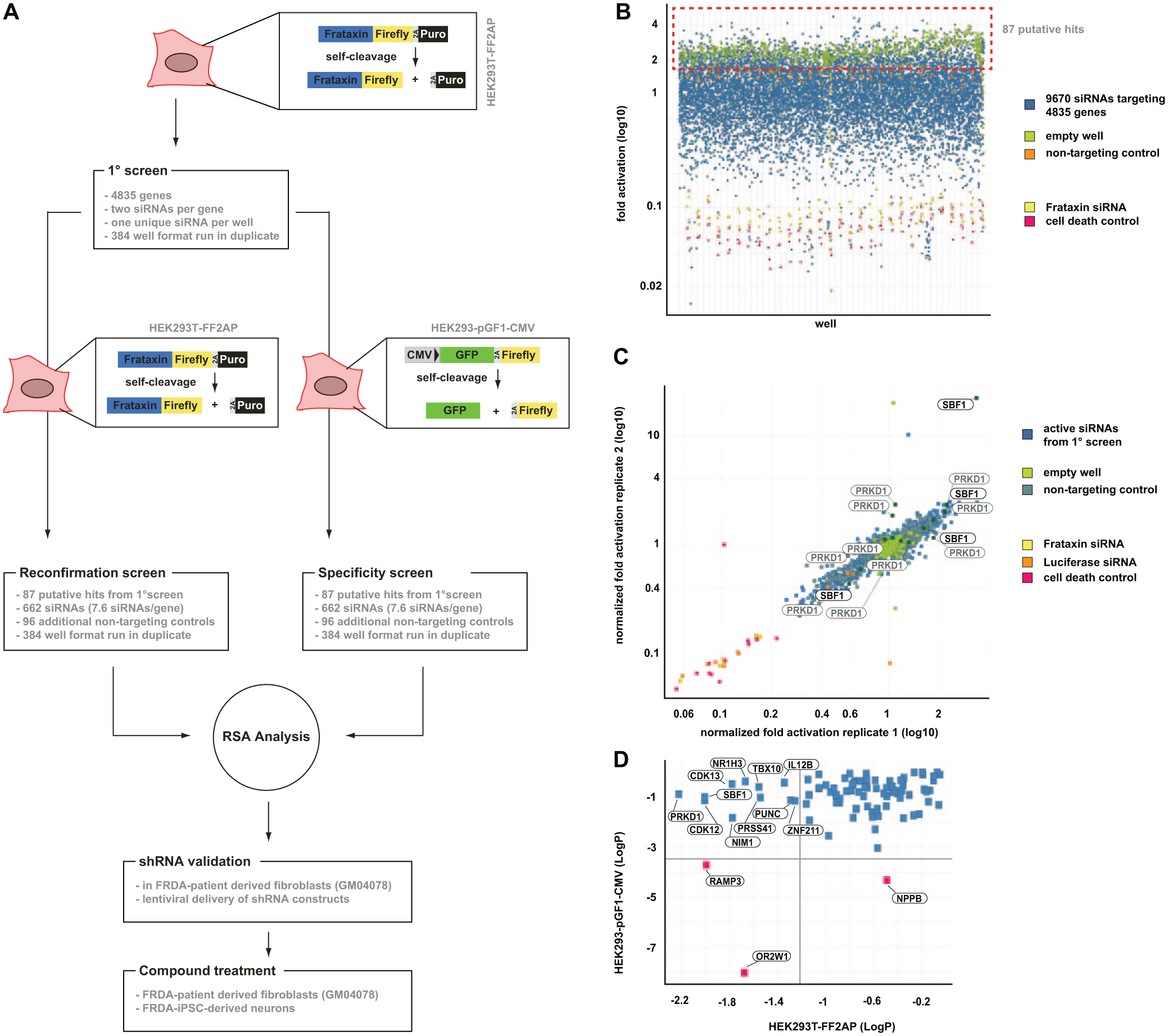
Small interfering RNA (siRNA) screen with the HEK293T-FF2AP cell line uncovers novel negative regulators of frataxin (FXN) expression. (
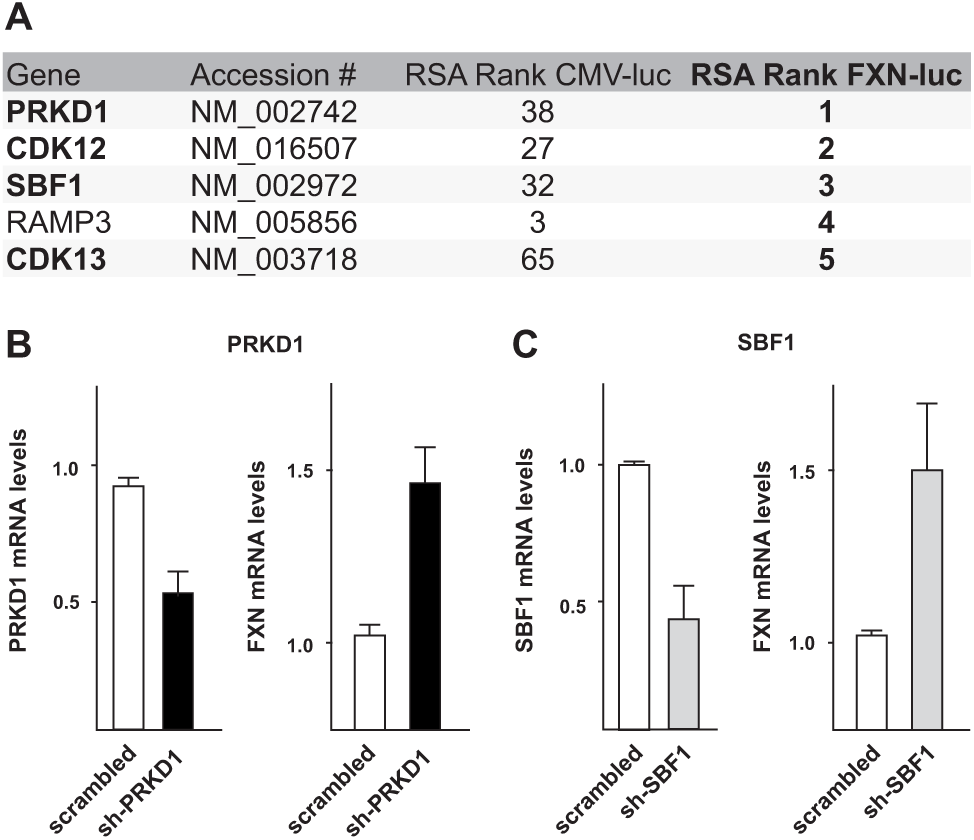
Validation of top five genes in Friedreich’s ataxia (FRDA) patient-derived fibroblasts. (
The FXN alleles in the HEK293T-FF2AP cell line used in our primary RNAi screen do not contain pathogenic GAA triplet repeat expansions. The main reason why we designed our screening cell line that way was because high-throughput biology requires large amounts of cells, and FXN deficiency could prevent cell expansion. Moreover, the readily available FRDA patient-derived B lymphoblasts turned out to be difficult to use for genome engineering and are not really well suited for high-throughput RNAi screening approaches. We also encountered poor cutting efficiency/homologous recombination in immortalized FRDA patient-derived fibroblasts that we have generated (FRDA-4078iBT), preventing us from establishing a luciferase reporter cell line that would allow direct monitoring of FXN expression from an expanded GAA allele. Therefore, we performed secondary hit validation experiments in the FRDA-4078iBT fibroblasts by quantitative real-time RT-PCR to determine whether our primary hits could increase FXN gene expression also in cells that contain an expanded repeat tract. Expression analysis revealed that four of the top five candidate genes ( Fig. 3A ) are also expressed in FRDA fibroblasts (PRKD1, CDK12, SBF1, and CDK13) (data not shown). To knock down these genes, we designed lentiviral shRNA constructs and delivered them into fibroblasts by lentiviral transduction. mRNA levels of all expressed candidate genes were reduced significantly upon shRNA expression ( Fig. 3B , C and data not shown). Depletion of the cell cycle–related kinases CDK12 and CDK13 resulted in strong proliferation defects and were omitted from further analysis (data not shown). Even though knockdown efficiencies for PRKD1 or SBF1 were never higher than 60%, we consistently observed a significant increase in FXN mRNA levels ( Fig. 2B , C ). These results confirmed that reducing the levels of the serine/threonine-protein kinase PRKD1 or the putative pseudophosphatase SBF1 results in increased FXN expression even in the presence of an expanded repeat tract. Thus, monitoring gene expression from the endogenous FXN locus irrespective of expanded GAA repeats opens up new opportunities for target discovery and the dissection of molecular pathways involved in FRDA. We note that the roughly 1.5-fold increase in FXN mRNA levels after PRKD1 or SBF1 knockdown is quite modest and was hence not reliably detectable by Western blot. This demonstrates the enormous power of using the highly sensitive luciferase approach to monitor changes in FXN expression but also urges us to expand our screens to genome-wide siRNA libraries to identify hits that lead to more drastic changes in FXN expression.
In recent years, a significant body of literature has proposed that genome-engineering technologies and high-throughput biology each have the potential to revolutionize biomedicine and pharmaceutical pipelines. This study establishes that these emerging biotechnologies can be effectively combined to perform as proposed, exemplifying a new paradigm for drug discovery. The HEK293T-FF2AP cell line established in this study is the first luciferase reporter–based cellular model, which allows an accurate and effortless assessment of endogenous frataxin gene regulation and is compatible with high-throughput biology. HEK293T-FF2AP cells are easy to transfect and can be expanded to a very large scale at a relatively low cost. Thus, this system should be compatible with the technical and financial constraints of most academic and industrial screening platforms.
Using the HEK293T-FF2AP cell line in a pilot high-throughput genomic screen, we uncovered novel negative regulators of FXN expression, demonstrating the importance of monitoring gene expression from the endogenous FXN locus. Our results showing that FXN expression can be increased even in the presence of an expanded repeat tract also demonstrate that screening for general regulators of FXN transcription, irrespective of GAA repeat length, is an appropriate strategy. Thus, our approach opens up new opportunities for target discovery, and we anticipate that HTS of genome-wide siRNA libraries will reveal additional potential drug targets in the near future.
Finally, the genome-engineering tools designed in this study will serve as a powerful resource to the FRDA community. The ZNFs and donor constructs developed in this study will enable others to knock in any given sequence tag into the FXN locus in many different cell types, facilitating the dissection of molecular pathways involved in FRDA. Notably, our donor constructs can also be used in combination with the more recently introduced CRISPR/Cas9 technology, which may work more effectively in patient-derived cell lines than the ZNFs that we have used in this study.
In conclusion, we have developed new tools that will serve the FRDA research community as a valuable resource for future mechanistic studies and the identification of new therapeutic strategies for a so far untreatable disease.
Footnotes
Acknowledgements
We thank Yukiko Shimada and Nathalie Laschet for technical assistance, as well as the members of the Bühler laboratory for fruitful discussions and critical reading of the manuscript. We are grateful to Cécile Blaustein for sharing her expertise and providing helpful advice and to Ira Shulman for providing the HEK293-pGF1-CMV cell line.
Author Contributions
The ZFN targeting approach was designed by T.P. and M.B. in collaboration with Sigma-Aldrich. L.M., A.R., B.T., and T.O. conducted the RNAi screen; R.V., T.P., and P.K. performed the rest of the experiments. S.D. immortalized patient-derived fibroblasts. M.B. designed and oversaw the study, obtained funding, and assisted in data evaluation and interpretation. M.B. and R.V. wrote the manuscript with the help of L.M., T.O., and T.P.
Declaration of Conflicting Interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: All the contributors are employees of institutes (FMI and GNF) that are financially supported by Novartis. A patent application has been filed based on some of the results described in this contribution.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by funds to M.B. from the Gebert Rüf Stiftung, the Swiss National Science Foundation, and the EMBO Young Investigator Programme. The Genomics Institute of the Novartis Research Foundation (GNF) and the Friedrich Miescher Institute for Biomedical Research are supported by the Novartis Research Foundation. T.P. was supported by a postdoctoral fellowship from the Swedish Society for Medical Research (SSMF).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.