
Abstract
The identification of initial hits is a crucial stage in the drug discovery process. Although many projects adopt high-throughput screening of small-molecule libraries at this stage, there is significant potential for screening libraries of macromolecules created using chemical biology approaches. Not only can the production of the library be directly interfaced with a cell-based assay, but these libraries also require significantly fewer resources to generate and maintain. In this context, cyclic peptides are increasingly viewed as ideal scaffolds and have proven capability against challenging targets such as protein-protein interactions. Here we discuss a range of methods used for the creation of cyclic peptide libraries and detail examples of their successful implementation.
A surprisingly high number of drug discovery projects begin with a peptide as the initial hit.1–4 These starting molecules typically need significant chemical development as linear peptides have poor pharmacokinetic properties (e.g., oral bioavailability and stability to peptidases).5,6 Cyclic peptides, however, are far less susceptible to proteolysis 7 and often have increased biological activity because of their conformational rigidity, which decreases entropic loss upon binding.8,9 In contrast to (and possibly as a result of) their relative underutilization in industry, cyclic peptide libraries have been extensively employed in academia, with multiple approaches developed for their generation. Such work has demonstrated the suitability of the cyclic peptide scaffold for the identification of inhibitors against some of the most challenging targets, including protein-protein interactions (PPIs). 10 Macrocyclic peptide scaffolds appear to be optimal for this purpose, and with genetically encoded cyclic peptide libraries in particular,11,12 there is potential for the straightforward creation of large sequence diversity (e.g., 6.4 × 107 members for six randomized amino acids). For synthetic cyclic peptide libraries, there are several approaches for identification of hits, including mass spectrometry, or by sequencing an associated genetic tag (incorporated during library synthesis). 13
For the purpose of this review, we have divided cyclic peptide libraries into three main categories: biologic, semisynthetic, and fully synthetic. Some of these libraries are constrained to the canonical peptidogenic amino acids, whereas others are able to include nonpeptidogenic amino acids such as β- or γ-amino acids, D-amino acids, or nonnatural amino acids.14–16
The majority of biologically produced cyclic peptide libraries are formed using phage/phagemid display8,17 or by split-intein cyclisation of peptides and proteins (SICLOPPS).11,18,19 The latter uses a selectively randomized library of split-inteins for the production of genetically encoded, backbone cyclized peptide libraries. Semisynthetic libraries are able to incorporate nonpeptidogenic amino acids while retaining ribosomal synthesis and an associated genetic tag. The most frequently used method for semisynthetic library production is mRNA display7,20; novel variants thereof include random nonstandard peptide integrated discovery (RaPID) 14 or protein synthesis using recombinant elements (PURE). 15 Both methods use promiscuous enzymes to expand the amino acid library encoded into peptides by reprogramming codons. 16 Another semisynthetic technique has been used for the creation of macrocyclic organic-peptide hybrids (MOrPHs)21,22 and bicyclic organo-peptide hybrids (BOrPHs), 23 which produce geometrically constrained peptide libraries. Fully synthetic libraries have access to higher sequence diversity (e.g., 107), 24 but synthesis is more challenging, as is deconvolution/hit identification. 25
Phage/Phagemid Display
Phage display uses modified virions that contain a fusion protein on their surface, typically a polypeptide library fused with a bacteriophage coat protein ( Fig. 1 ). 26 These recombinant bacteriophage particles are typically F-specific filamentous (Ff) phages,17,27 as their life cycle is non cell-lytic; however, T7, 28 λ, 29 and T4 30 phage display libraries have also been created. Phages are passed down a column containing the target of interest, which binds phages containing an active peptide motif. This technique has been employed for the identification of affinity31,32 or avidity binding proteins for antibody-antigen interactions31,33 and sequences that inhibit PPIs. 34 Phages displaying inhibitory proteins are then amplified and reintroduced to the column under more stringent conditions (e.g., fewer binding sites, competitive inhibitor; known as “biopanning”). This allows the identification of stronger binding partners and may provide a basic insight into potential structure-activity relationships.
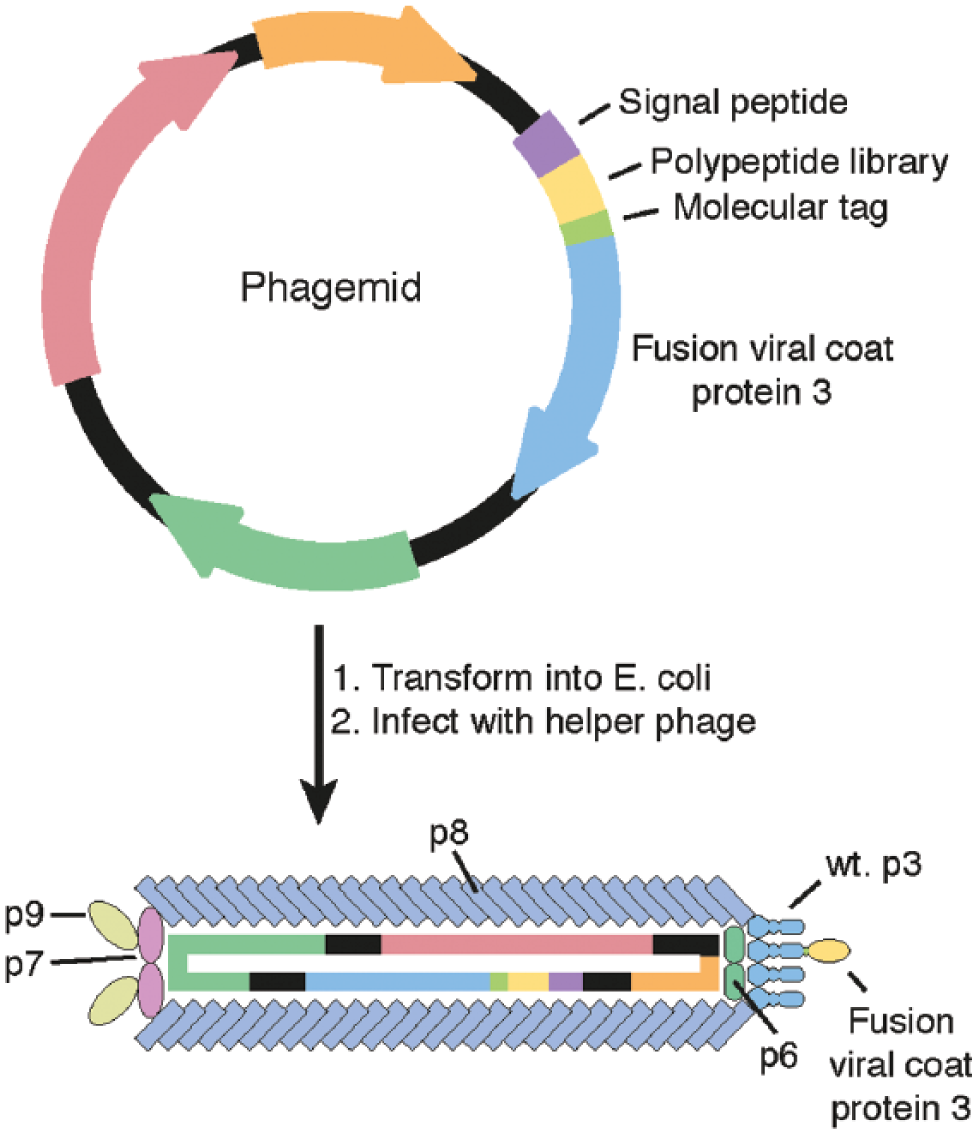
Phage display library creation with phagemid. A plasmid encoding the viral coat protein 3 (p3) fused to a randomized peptide sequence is transformed into Escherichia coli and subsequently infected with a helper phage that encodes the rest of the proteins required for phage assembly. Members of the resulting phage library are then assessed for binding to a given protein in vitro.
Phagemids are Ff phage (usually M13 variants)–derived vectors that encode a viral coat fusion protein (typically of p331,32,34 or p8 in M13). When present in a cell infected by a helper phage, the phagemid is packed into viral and fusion proteins and released from the cell, where they can be used to identify fusion proteins with affinity.
There are a number of benefits of phagemid display over phage display; the former contains a smaller genome, which allows a larger fusion protein gene fragment. Phagemids also transform more efficiently than phages, allowing libraries of high diversity to be created. Finally, as phagemids contain ds-DNA, they are more resilient to multiple propagations than recombinant (ss-DNA) phages. 35
M13-based phages are also resilient to various harsh conditions, including extreme pH, presence of DNase or proteolytic enzymes, and nonaqueous solutions. This additional tolerance allows sequences such as -CX5C- (where X = any peptidogenic amino acid) to be cyclized by disulphide cysteine oxidation. This has been used to create various phagemid disulphide-bonded cyclic peptide libraries. Examples of the successful use of this approach are the identification of ligands of tumor necrosis factor-α (TNF-α), 36 neutravidin and avidin, 8 and inhibitors of LEDGF/p75 PPI that prevent HIV replication. 17
More recently, bicyclic phage display libraries have been developed by expressing a fusion protein containing CXnCXnC (X = any amino acid, n = 3–6). This linear peptide is scaffolded by an aromatic core via reaction with a tris-(bromomethyl)benzene derivative to form a bicyclic peptide. 37 This methodology has advanced the utility of phage display screening and has recently been used to identify ligands for several proteins, including urokinase-type plasminogen activator. 38
SICLOPPS
SICLOPPS allows the intracellular production of cyclic peptide libraries of up to 108 members. 11 This genetically encoded approach uses the Synechocystis sp. (Ssp) PCC6803 DnaE trans inteins that have been rearranged so that their splicing results in cyclization of the extein; the C terminal intein (IC) is followed by a randomized extein and the N terminal intein (IN). Splicing of the resulting rearranged cis intein causes cyclization of the extein peptides ( Fig. 2 ). 12
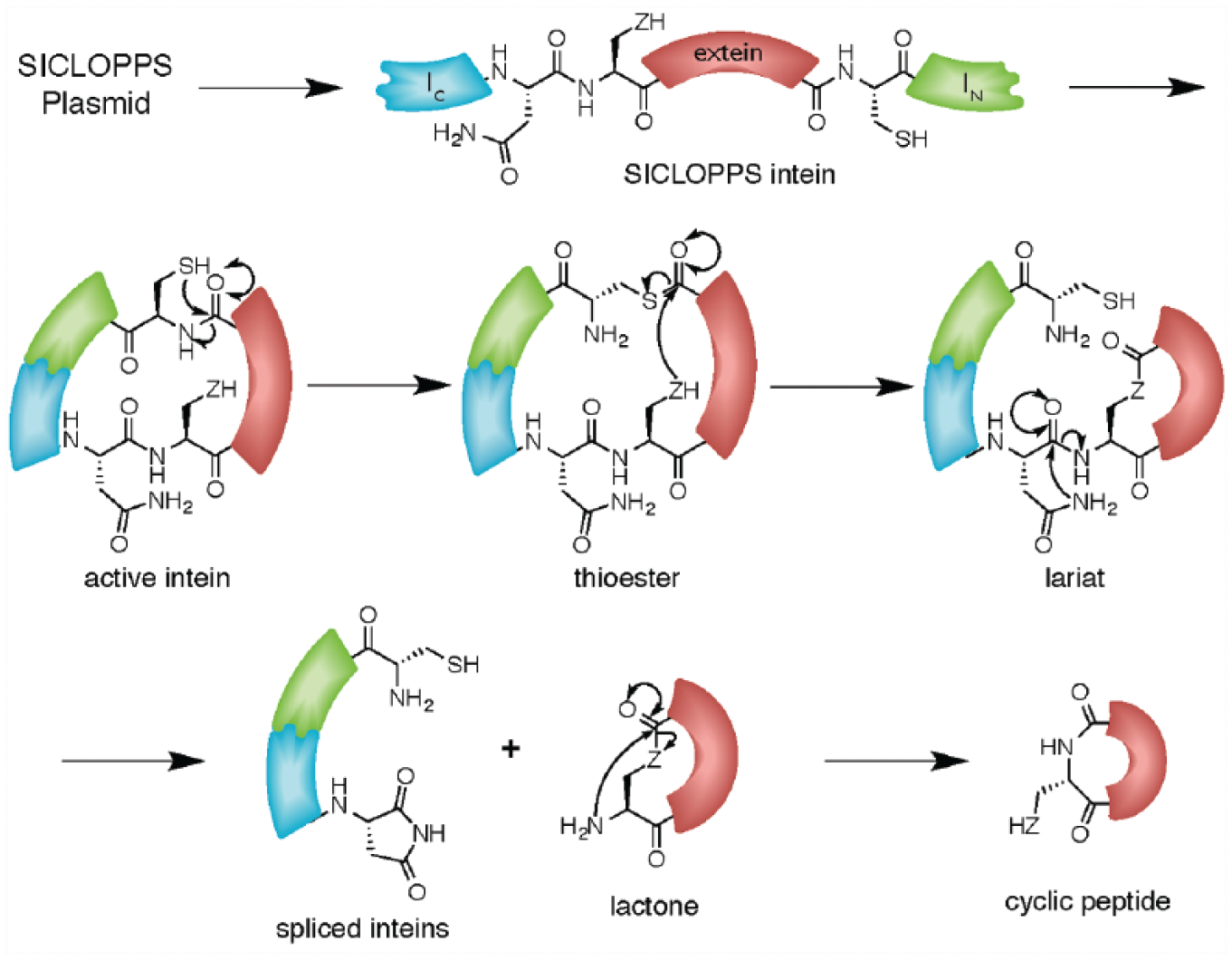
Mechanism of split-intein cyclisation of peptides and proteins (SICLOPPS). The SICLOPPS plasmid encodes the SICLOPPS split-inteins, which come together to form an active intein that splices to the target peptide sequence through a series of nucleophilic displacement reactions (Z = S or O).
SICLOPPS libraries have been shown to function in a variety of prokaryotic and eukaryotic organisms 10 and so may be used in any cell-based screen. However, they have been most widely employed in conjunction with a bacterial reverse two-hybrid system to enable the rapid identification of PPI inhibitors from cyclic peptide libraries of up to 100 million members.18,39 SICLOPPS screening has successfully identified inhibitors of difficult-to-target and previously undruggable PPIs on multiple occasions. In an early example, cyclo-CRYFNV was identified as an inhibitor of the homodimerization of AICAR transformylase/IMP cyclohydrolase (ATIC), a homodimeric enzyme with a 5000 Å2 interacting interface. 39 This approach to inhibiting ATIC was in contrast to previous efforts that relied on folate analogues 40 and is one of a handful of examples in which enzyme activity is controlled through modulation of its quaternary structure. The cyclic peptide ATIC inhibitor has more recently been developed into a more potent small molecule, 41 thus demonstrating the potential for using SICLOPPS libraries in early-stage screening. SICLOPPS screening has also proven successful for the identification of cyclic peptides that inhibit transcription factor assembly, generally considered to be a very challenging class of targets. These include a selective inhibitor of hypoxia inducible factor–1 (HIF-1) that targets the HIF-1α/HIF-1β PPI 42 and an inhibitor of the transcriptional co-repressor C-terminal binding protein. 43
SICLOPPS libraries have also been screened in mammalian cells. Retroviral vectors expressing variants of the DnaB intein from Ssp PCC6083 were delivered to human BJAB lymphocytes, and intein splicing in human cells was demonstrated with high efficiency and fidelity. 44 Once proof of concept had been established, a green fluorescent protein–tagged retroviral vector expressing an SX4 library with a potential complexity of ~1.60 × 105 members was screened for inhibitors of interleukin-4 induction of sterile ϵ-germ line transcription in the BJAB-derived cell line A5T4. The screen yielded 13 peptides that selectively inhibited germ line ϵ transcription. Interestingly, the 13 peptides identified did not contain a homologous motif, leading to the hypothesis that they act through independent targets to achieve the same altered phenotype. 44 Although this is the only example of using SICLOPPS libraries in mammalian cells, it is an excellent demonstration that functional screening of such cyclic peptide libraries is feasible in mammalian cells with relative ease.
A yeast-compatible SICLOPPS library has also been reported 19 and used within a yeast system to screen for peptides that inhibited α-synuclein toxicity, in an effort to identify a novel therapeutic for Parkinson’s disease. To maximize diversity, the library was designed with larger octameric backbones (CX7) with a diversity of 5 × 107. The screen yielded two cyclic peptides that displayed specific suppression of α-synuclein. The initial cyclic octamers were subjected to two rounds of rationally designed mutagenesis, generating cyclic hexamers with no observable change in their in vivo activity against α-synuclein toxicity. 19 This work demonstrated the potential for combining SICLOPPS libraries with yeast two-hybrid systems to identify inhibitors of PPIs not amenable to expression in a bacterial reverse two-hybrid system.
FIT and RaPID
Flexizymes are de novo tRNA aminoacylation ribozymes capable of charging a diverse range of amino acids onto tRNA. 45 Although not suitable for the intracellular production of cyclic peptides, flexizymes have been extensively employed for the in vitro production of cyclic peptide libraries that contain nonnatural amino acids. 16
Flexizymes have been most widely combined with the PURE in vitro transcription/translation (IVTT) system. As each component of the PURE system is individually purified and added to the IVTT mixture (as opposed to other approaches that use cell lysates), components that are incompatible with flexizymes such as the amino-acyl tRNA synthetases (ARSs) specific to the codon targeted for reprogramming are removed from the IVTT mixture. 46 The removal of the ARSs and their related amino acids generates an empty codon, which can then be targeted by the addition of a flexizyme-generated unnatural aa-tRNA displaying the relevant anti-codon. 47 The coupling of flexizyme-mediated tRNA charging with a modified PURE translation system has been termed the flexible in vitro translation (FIT) system ( Fig. 3 ). 16 The linear peptide undergoes cyclization via nucleophilic attack from the N-terminus to an electrophilic substituent on the C-terminus. 48 A variety of chemical reactions has been employed for this purpose; for example, incorporation of a C-terminal cysteine or proline leads to N-S and O-N acyl shifts, generating a thio-ester with which an N-terminal nucleophile will spontaneously react to form a cyclic product (Figure 4). 49
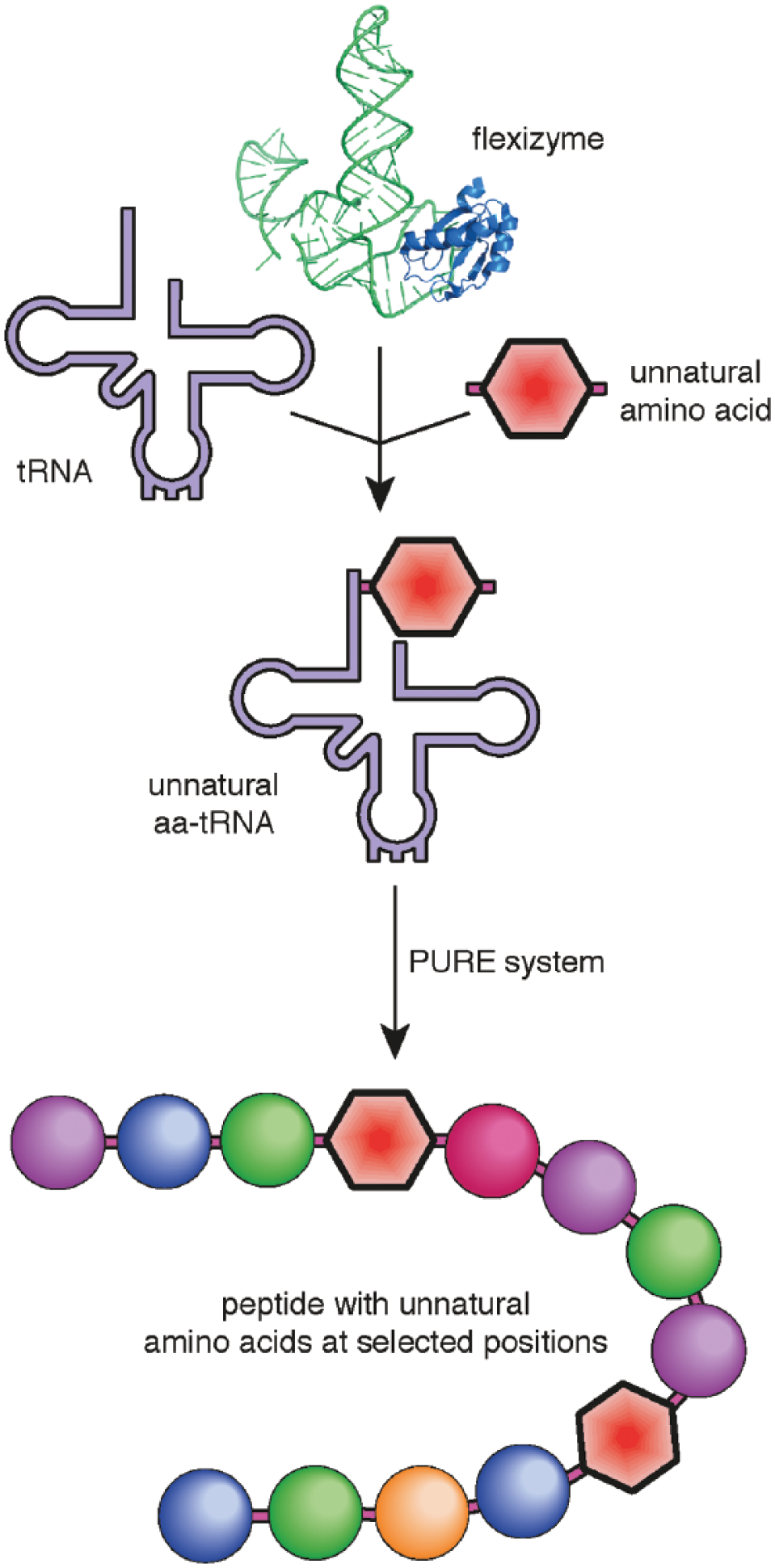
The flexible in vitro translation system. The flexizyme is used to generate an unnatural aa-tRNA with a desired anticodon, which is incorporated into a cyclic peptide using a modified PURE system lacking the corresponding native aa-tRNA.
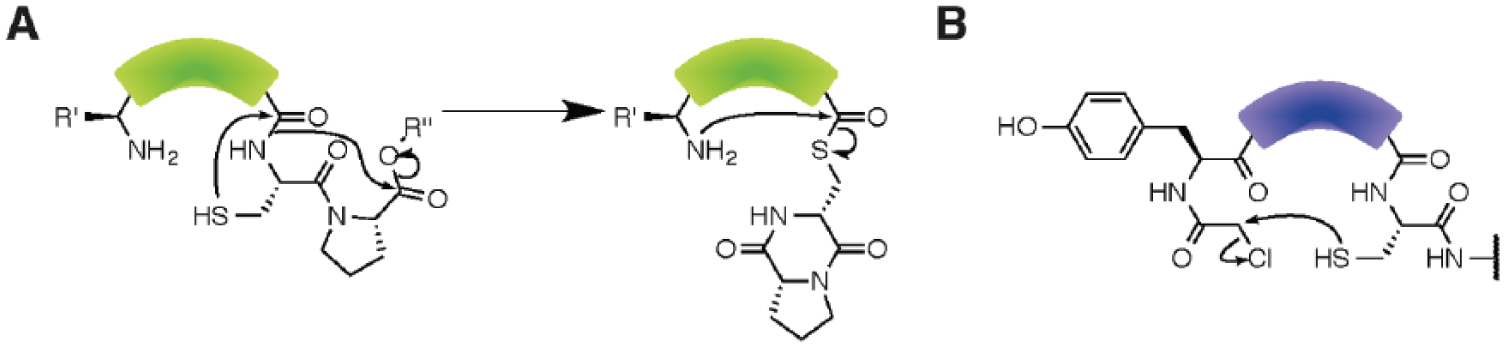
Mechanisms for flexible in vitro translation system (FIT)/random nonstandard peptide integrated discovery cyclization. (
Scaffolded and polycyclic peptides can also be designed through the incorporation of adjacent cysteine residues that react in situ to generate constrained ring structures.48,50 Incorporation of unnatural amino acids can also be used for the cyclization reaction, using techniques such as oxidative coupling, click chemistry, and Michael additions. 47 Such cyclization events can be programmed into the sequence by the addition of the relevant reactive species to the desired amino acid in the chain. These techniques are typically used for the generation of medium-sized (<105) cyclic peptide libraries, and the FIT system has been successfully used to develop and analyze several biologically active compounds, often involving rational design of a template. For example, sunflower trypsin inhibitor 1 (STFI-1) was used as a proof of concept, with a FIT system using spontaneous thioester rearrangement for cyclization to yield the STFI-1. 51 The FIT system has been used in screens by combination with mRNA display. This yields an in vitro technique for peptide generation that attaches the template mRNA to the peptide it encodes, allowing amplification of the mRNA-tag and identification of hits. 52 During mRNA display, a puromycin molecule is linked to the 3′ end of mRNA, which upon translation is incorporated into the ribosomal A site and added to the C-terminus of the peptide before release, thus covalently attaching the mRNA strand. 53 This has been used to screen cyclic peptides against an immobilized target, 54 and when used in combination with the FIT system, it leads to a platform for screening unnatural cyclic peptide libraries. This approach, called random nonstandard peptide integrated discovery (RaPID), has been used to screen the cyclic peptide libraries generated by the FIT system 47 of >1012 members. 49 After puromycin attachment, a pool of randomized cyclic peptides containing a selection of nonnatural amino acids is generated. Reverse transcription prevents mRNA from interfering with binding and precedes affinity selection for a target protein immobilized on beads. Those displaying affinity for the target can then be amplified by polymerase chain reaction (PCR) to generate an enriched library of initial hits that can be used in further rounds of screening and amplification. 16 Because of the in vitro nature of library production, screening is limited to affinity binding.
This methodology has been successfully used to identify cyclic peptides with high affinity for a number of targets including E6AP, an enzyme involved in the conjugation of polyubiquitins onto multiple targets. 14 A RaPID selection was used for the discovery of N-methylated cyclic peptide inhibitors of this enzyme, with the most active compound containing both a D-amino acid and N-methylation. 14
MOrPHs and BOrPHs
Another approach to the in vitro production of cyclic peptides uses a self-splicing N-terminal intein to create MOrPHs from biosynthetic and synthetic precursors. 55 The coupling of the two precursors is mediated by a chemoselective reaction that occurs either in the presence of a copper catalyst or catalyst free by an N-S acyl shift reaction ( Fig. 5 ). 21 This technique allows the creation of macrocycles of differing sizes that incorporate phenyl, biphenyl, and diphenyl structures. 55
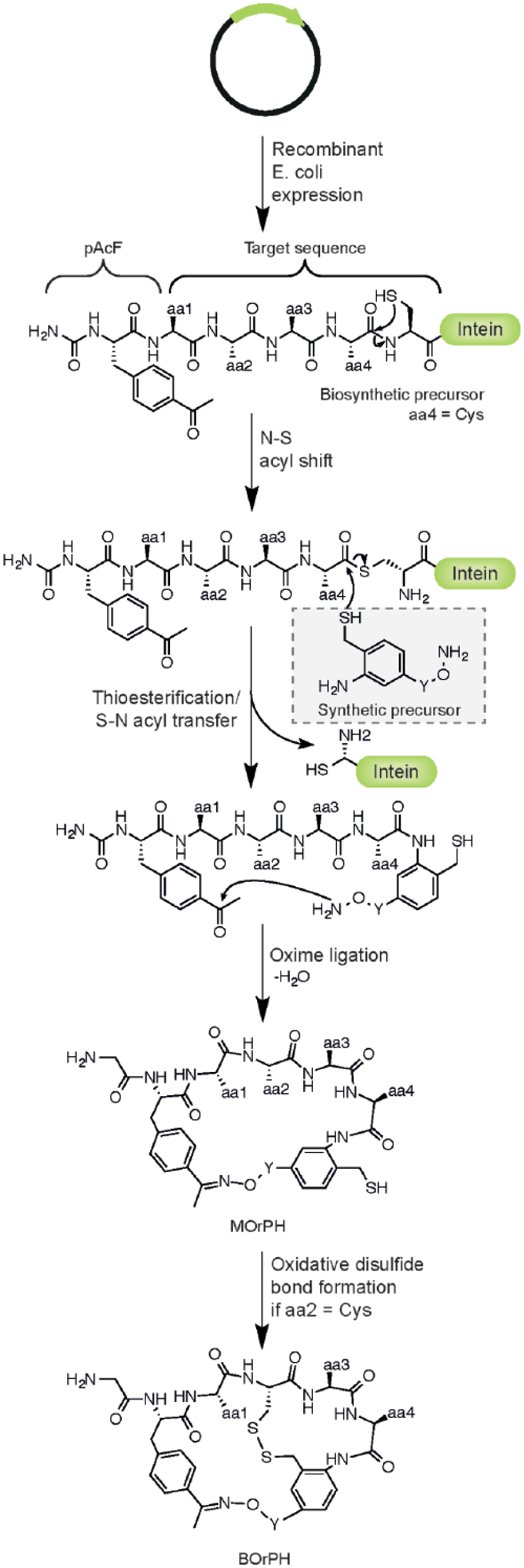
Mechanism for the creation of macrocyclic organic-peptide hybrids (MOrPHs) and bicyclic organo-peptide hybrids (BOrPHs). A plasmid encodes a linear peptide sequence with a ketone-containing nonnatural amino acid, fused to an intein. The intein processes to give a thioester, which is replaced by nucleophilic substitution, a synthetic precursor. The molecule undergoes an oxime ligation to give a MOrPH. If aa2 is a cysteine, a disulphide bond can form, resulting in a BOrPH. Y = variable linker.
The MOrPH strategy was recently applied to design and develop a macrocycle containing an α-helix that disrupts the interaction between p53 and HDM2. 56 A library of MOrPHs was created using a linear 12-mer peptide template identified via phage display. This library was then assessed for p53/HDM2 inhibitors using a surface plasmon resonance assay.
The MOrPH backbone was further rigidified by the formation of an intramolecular disulfide cross-link to create BOrPHs in a single-pot, two-step regioselective reaction ( Fig. 5 ).23,56
Split-and-Pool Synthesis
The generation of large linear peptide libraries by the repeated cycles of dividing the solid support (the resin beads) equally in portions, coupling each portion to a different amino acid, and then remixing the portions was reported. 57 This approach, termed split-and-pool ( Fig. 6 ) has been extensively employed in industry and academia for the formation of one-bead one-compound libraries,58–60 including cyclic peptide25,61 and bicyclic peptide libraries. 24 A key challenge, however, is uncovering the sequence identity of hits. In 1992, Brenner and Lerner 62 published a theoretical paper describing the possibility of creating a library of synthetic peptides bound to a solid support with an oligonucleotide coding tag. The library would be synthesized in a combinatorial manner, similar to split-and-pool approaches. The oligonucleotide would serve as a barcode that would allow the peptides to be identified at a later date. The first practical implication of this proof of concept was realized soon after in 1993.63,64 A decade later, libraries were created that did not require the bead support and instead coupled the chemical ligands directly to the oligonucleotide.65–67
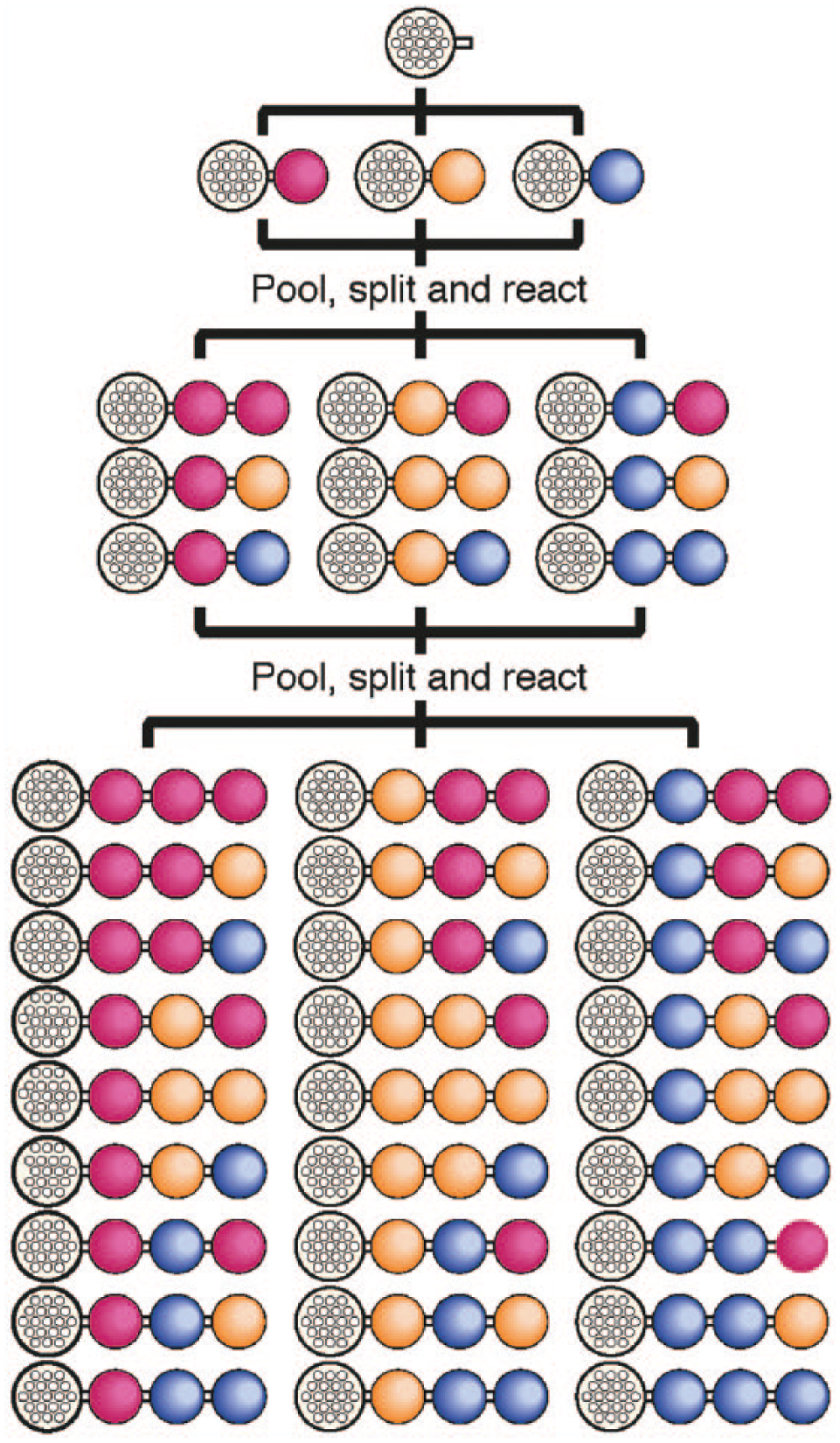
Generalized mechanism for split-and-pool synthesis of peptides. Amino acids are loaded onto a polystyrene resin. The pool is split, followed by coupling to an amino acid (a different compound used for each fraction). The fractions are combined to form a new pool, and the process is repeated several times to create a large library of peptides.
Since their conception, DNA-encoded chemical libraries have fallen into two main types: single-pharmacophore or dual-pharamacophore libraries. Single-pharmacophore libraries are made up of a small organic molecule bound to an oligonucleotide and can be further defined into two subdivisions: libraries that are created combinatorially using a split-and-pool method as first imagined by Brenner and Lerner or libraries that are created using DNA-templated synthesis (DTS). The combinatorial split-and-pool synthesis approach is a robust method for creating DNA-encoded libraries of large magnitude (between 105 and 108) 68 and has identified inhibitors for tumor necrosis factor, 69 carbonic anhydrase IX, 70 polyclonal human IgG, 71 and trypsin. 72
DTS involves the DNA fragments not just as encoding tags but also as a means of directing the generation of the library. It was found that when DNA-linked reagents are brought into close proximity by Watson-Crick base pairing, a chemical reaction is promoted 67 ; therefore, when small organic molecules are linked to oligonucleotides, they can be transferred to the DNA fragment upon hybridization. The Lui group used DTS to create, first, a 65-member proof-of-concept library 73 and then a 13,000-member library of macrocycles, 74 assembled using a downstream Wittig macrocyclization reaction. The size and diversity of the library was increased by using 12 building blocks in each DTS step and eight different starting scaffolds, which lead to four different sizes of macrocycle. The 13,000-member library was screened against 36 kinase proteins in vitro, 75 leading to the identification of inhibitors of Akt3, MAPKAPK2, p38α, and Pim1 as well as a macrocycle that activated VEGFR2.
Another approach involves dual-pharmacophore libraries, which are made up of two small organic molecules bound to a single oligonucleotide. One method of creating dual-pharmacophore libraries makes use of a hybridization domain in two complementary oligonucleotides each containing a ligand, one bound on the 5′ end and one bound on the 3′ end. This strategy is known as encoded self-assembling chemical (ESAC) libraries, as the oligonucleotides combinatorially self-assemble to form a DNA heteroduplex containing the dual pharmacophore. A dual-pharmacophore strategy has also been implemented that is similar to the ESAC strategy but uses peptide nucleic acid (PNA)–conjugated molecules to self-assemble onto a DNA-template.76,77 PNA/DNA hybrids confer a higher stability and are compatible with identification by PCR/sequencing.
DNA-encoded libraries allow similar selection techniques to other display technologies (e.g., phage display and mRNA display), in that they can be panned across an immobilized target protein to identify hit compounds. In addition, they share the same relatively facile hit-identification procedure as genetically encoded libraries, as even one molecule of DNA can theoretically be PCR amplified and then decoded by high-throughput sequencing such as 454 technology 78 or Illumina. 79 DNA-encoded libraries have also proved effective at targeting protein-protein interactions,69,72 a historically difficult target. DNA-templated synthesis has also been proven to be compatible with many downstream reactions, including cycloadditions, Wittig olefinations, Heck couplings, reductive amination, and conjugate addition. 80 One disadvantage of this approach is the limited modes of binding available to the ligands tethered to the DNA tag, potentially preventing binding to targets with more inaccessible binding pockets, although it has been suggested that with a sufficiently large library diversity, this limitation might be overcome.
Other split-and-pool synthetic approaches have been reported for the preparation of cyclic peptide libraries that do not use a DNA tag. As other synthetic library production methods, these approaches allow the incorporation of nonnatural and D-amino acids. However, hit deconvolution is challenging compared with DNA-encoded and genetically-encoded libraries; cyclic peptides have a tendency to form complex fragment ions when analyzed by mass spectrometry and so have to be resynthesized and tested individually, which is time-consuming and expensive. To overcome this problem, the individual beads may be tagged with both the linear and cyclic form of the peptide to facilitate identification of active hits. However, this poses the problem of increasing nonspecific binding by weak ligands. This has been overcome by a one-bead two-compound library that uses spatially segregated beads, with the cyclic peptide expressed on the surface layer and the linear peptide in the interior as a coding tag. Once a hit has been identified, both forms are cleaved from the bead and are analyzed via matrix-assisted laser desorption/ionization time-of-flight tandem mass spectrometry 81 or Edman degradation/mass spectrometry. 61
Discussion
The increasing prevalence of cyclic peptides in early-stage drug discovery is a reflection of the utility and significance of this molecular scaffold; cyclic peptides and macrocycles have been successfully employed against the most challenging targets such as PPIs. Each of the approaches detailed above for the preparation of cyclic peptide libraries has its own merits and shortcomings ( Table 1 ), and the optimal method very much depends on the locally available resources (e.g., the use of chemical synthesis versus genetically encoded methods). Chemical methods allow the use of a large variety of nonnatural amino acids, but deconvolution of hits remains challenging, despite the development of several approaches for this purpose. The FIT system is a highly innovative genetically encoded approach that allows the incorporation of a similarly diverse set of nonnatural amino acids. However, the methodology is complex and requires multiple IVTT components to be individually prepared and purified. There is also little evidence that nonnatural amino acids are essential for the identification of peptide ligands in early-stage drug discovery; diverse chemical spaces may be explored in more focused, second-generation molecules. Both phage display and SICLOPPS are limited to canonical amino acids, although in both cases, a 21st amino acid may be added via an orthogonal tRNA/tRNA synthetase pair. Phage display has been extensively employed, and the recent development of displaying bicyclic peptides has further enhanced the utility of this technique by allowing access to more complex scaffolds.
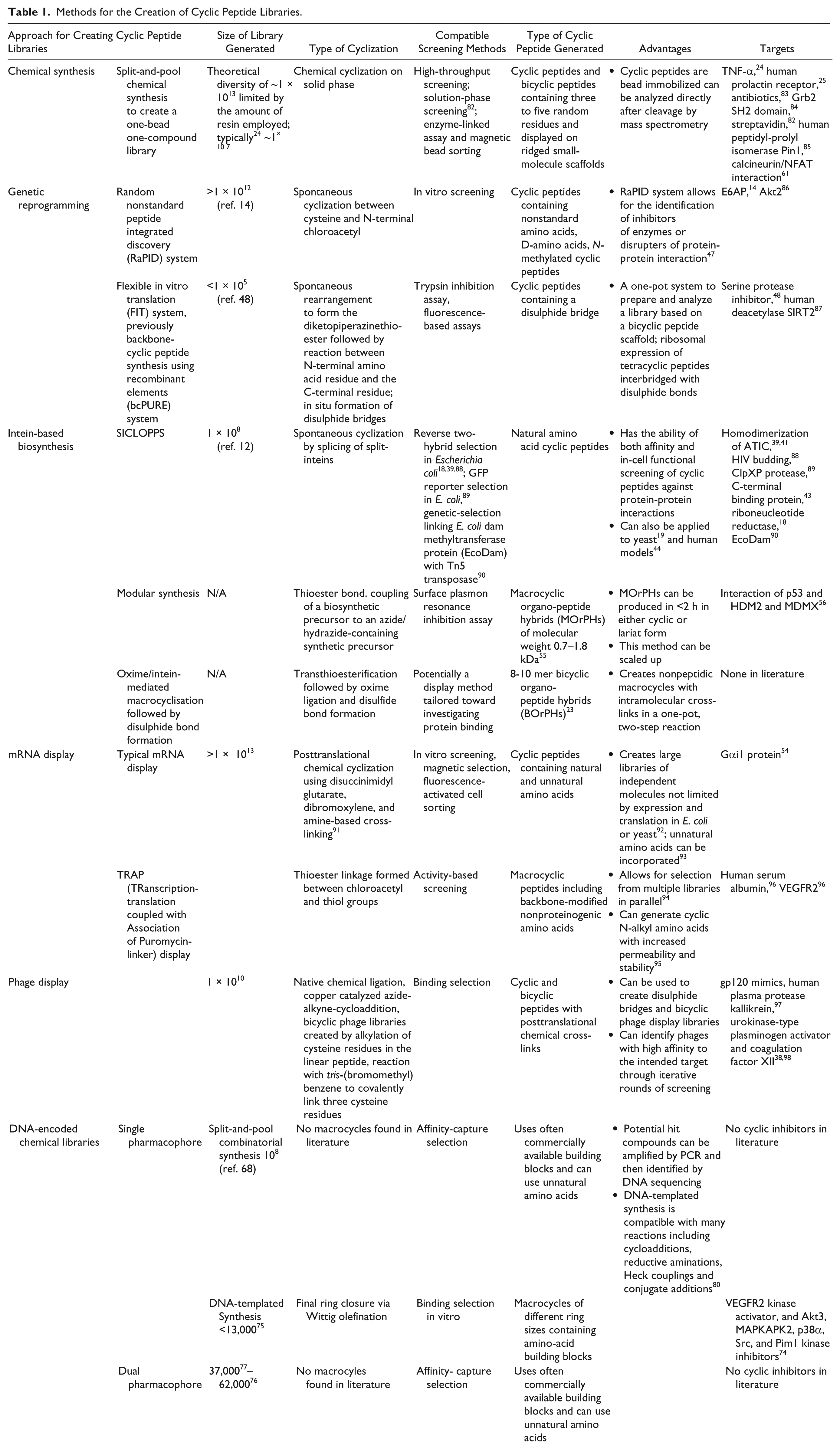
Methods for the Creation of Cyclic Peptide Libraries.
SICLOPPS allows the intracellular production of cyclic peptide libraries; this subtle difference has significant implications for screening, allowing functional and phenotype screens to be conducted (as opposed to binding assays typically employed with in vitro libraries.
Given the prevalence of available methods for cyclic peptide library production, it is surprising that these approaches, and more generally this molecular scaffold, are relatively underused by pharmaceutical companies. There are several reasons for this. First, peptides are traditionally seen by the pharmaceutical industry as un-drug-like and suffering from poor oral bioavailability and cell permeability. The counterargument that screening approaches, such as those outlined above, allow the discovery of first-stage ligands that may be used as the starting point for a drug-discovery campaign are slowly changing the mind-set of even the most traditional companies. Another challenge faced by cyclic peptides is that significant effort by several companies in the 1980s to develop peptidomimetic small molecules led to very little, resulting in overcautiousness toward new projects with peptides. Given the significant advances in medicinal chemistry (especially computational chemistry and in silico screening), it may be argued that we have many more, and more powerful tools at our disposal for this purpose. Again, successful examples are changing these dogmatic views, and given the prevalence of antibody therapeutics, peptides are increasingly being viewed by the pharmaceutical industry as the middle space between small molecules and biologics. The methods developed and screens conducted by academia have made it increasingly clear that macrocyclic peptides are a privileged scaffold that holds much promise for use in early-stage drug discovery.
Footnotes
Acknowledgements
The authors would like to thank GlaxoSmithkline (studentship to A.D.F), Pfizer Neusentis (studentship to J.D.I), AstraZeneca (studentship to E.K.L. and E.L.O.), C4X Discovery (studentship to K.R.L), and the Engineering and Physical Sciences Research Council (Doctoral Training Award to all above) for financial support.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.