
Abstract
Although the inhibition of acetylcholinesterase remains the primary treatment of Alzheimer's disease, little is known of the results of increased acetylcholine levels on muscarinic receptor occupancy or function. Using N-(2-[18F]fluoroethyl)-4-piperidyl benzilate ([18F]FEPB), a moderate affinity (Ki = 1.7 nmol/L) nonsubtype-selective muscarinic receptor antagonist, the authors examined the sensitivity of equilibrium in vivo radioligand binding in rat brain with changes in endogenous acetylcholine levels produced by treatments with acetylcholinesterase inhibitors. Phenserine administration 30 minutes before resulted in a dose-dependent increase in N-(2-[18F]fluoroethyl)-4-piperidyl benzilate binding to muscarinic cholinergic receptors, reaching a maximum increase of 90% in the striatum at a dose of 5 mg/kg intraperitoneally. Constant infusion of physostigmine at a dosage of 250 μg/kg/min produced an identical increase in radioligand binding. This agonist-induced increase of in vivo mAChR radioligand binding offers a new method for monitoring of the efficacy of acetylcholinesterase inhibitors or other drugs to enhance acetylcholine actions at the muscarinic receptors.
Alterations in the number of central muscarinic acetylcholinergic receptors (mAChR) in brain disorders such as Alzheimer's disease (AD) have been reported (Rodriguez-Puertas et al., 1997; Fisher, 1999). Moreover, AD also involves the loss of synaptic acetylcholine (ACh) (Fisher, 1999) and activity of the synthetic enzyme choline acetyltransferase (Wu and Hersh, 1994). These observations have led to the hypothesis that enhancement of the cholinergic system using mAChR agonists or acetylcholinesterase (AChE) inhibitors can lead to amelioration of the symptoms of AD. Although myriads of cholinergic drugs (AChE inhibitors, ACh-releasing agents, and cholinergic agonists) have been developed for treatment of AD, relatively little is known regarding how these drugs affect receptor occupancy by ACh. A method to measure changes in mAChR receptor occupancy by ACh could lead to much improved pharmaceuticals or dosage regimens.
In vivo imaging of muscarinic cholinergic receptor availability, using positron emission tomography (PET) or single photon emission computed tomography (SPECT), offers a method for evaluating ACh occupancy of mAChR, provided a radiopharmaceutical with demonstrated sensitivity to ACh levels were to be available. Although a large number of mAChR radioligands have been developed for PET and SPECT imaging in the human brain (Eckelman et al., 1985; Dannals et al., 1988; Wilson et al., 1989; Dewey et al., 1990; Wilson et al., 1991; Frey et al., 1992; Koeppe et al., 1994; Mulholland et al., 1995; Farde et al., 1996; Carson et al., 1998), most have been aimed at measuring the numbers of binding sites and not towards measuring changes of endogenous ACh levels. In this study, the authors evaluated the sensitivity of N-(2-[18F]fluoroethyl)-4-piperidyl benzilate (4-[18F]FEPB)—a moderate affinity (Ki = 1.7 nmol/L), reversibly-binding, nonsubtype-selective mAChR antagonist (Skaddan et al., 2000)—to changes in brain ACh levels induced by treatment with the AChE inhibitors physostigmine and phenserine.
MATERIALS AND METHODS
Radiochemical
N-(2-[18F]Fluoroethyl)-4-piperidyl benzilate (4-[18F]FEPB) was prepared in a two step procedure as previously described (Skaddan et al., 2000). Specific activity of the radiotracer at time of injection was >100 Ci/mmol.
In vivo regional brain distribution studies
All studies were performed on male CD rats (Charles River Laboratories, Wilmington, MA, U.S.A.) using a protocol approved by the University of Michigan Committee on Care and Use of Animals. Regional brain distributions of radioactivity were determined using a bolus plus infusion protocol. Under sodium pentobarbital anesthesia, catheters were surgically inserted and secured in one or both femoral veins. Rats were restrained (plastic tubes) and allowed to awaken. For the study of the dose-dependency of AChE inhibition, 2, 3, or 5 mg/kg phenserine ((−)-phenylcarbamoyleseroline) was administered intraperitoneally as a bolus in saline solution 30 minutes before beginning the 4-[18F]FEPB infusion. For the physostigmine study, a solution of the drug was infused through a catheter in the opposite femoral vein at a constant rate of 250 μg kg−1 hr−1 beginning 30 minutes before and continuing throughout the 4-[18F]FEPB infusion. Radiotracer infusions were performed using a Harvard programmable infusion pump, which administered 1.5 mL 4-[18F]FEPB (50 to 60 μCi, as solution in 10% ethanol/saline) as a bolus of 1 mL administered over 1 minute followed by a constant infusion of the remaining 0.5 mL over 59 minutes. Control animals were done simultaneously with the physostigmine and phenserine animals, and all control animals received sham injections of saline (phenserine studies) or an infusion of saline (physostigmine studies). At the end of the infusion, animals were killed with an intravenous administration of a sodium pentobarbital overdose, and the brains were removed quickly and dissected into regions of interest (striatum, whole cortex, hippocampus, thalamus, hypothalamic region, and cerebellum) (Iversen et al., 1967). Tissue samples were immediately counted for fluorine-18 using an automated gamma-counter and then weighed. Radioactivity retention for each region was calculated as % injected dose/gram tissue, and regional distribution volume ratio (DVR) was calculated as the ratio of radioactivity in a region of interest to the radioactivity concentration in the cerebellum.
Statistical significance was first assessed using a two-factor (groups and regions) analysis of variance. Drug-induced changes within each brain region were then evaluated using a single-factor analysis of variance. The dependency of regional distribution volume ratios on phenserine dose was examined using a simple linear regression analysis. P < 0.05 was considered significant.
RESULTS
As there were no differences between control animals whether using sham injections or infusions of saline, all control animals were combined into a single control group. Distribution volume ratios determined for 4-[18F]FEPB in the awake rat brain were highest in the striatum (3.46 ± 0.24), intermediate in the cortex (3.03 ± 0.34) and hippocampus (2.70 ± 0.23), and lowest in the hypothalamus (1.72 ± 0.17) and pons (1.18 ± 0.02) (Fig. 1). The rank order of specific binding correlated well with the known distribution of total muscarinic receptors in regions of the rat brain (Snyder et al., 1975; Lin et al., 1986; Lee et al., 1995).
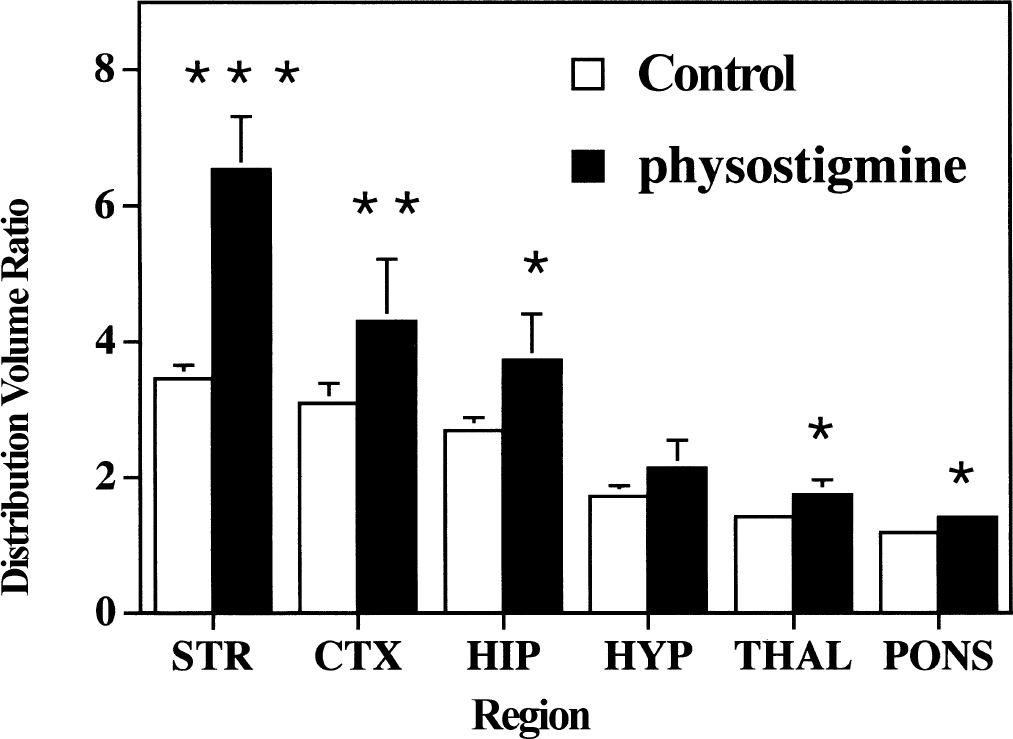
Comparison of regional brain 4-[18F]FEPB distribution volume ratios between control and physostigmine-treated male rats. Control rats (n = 7) were injected with 4-[18F]FEPB in a bolus plus infusion protocol for 60 minutes before death. Treated rats (n = 3) had physostigmine (250 μg kg−1 hr−1) coinfused with 4-[18F]FEPB for 60 minutes before death. Data presented as mean ± SD. *P < 0.05, **P < 0.025, ***P < 0.005 versus controls.
Initial comparison of all groups (two-factor repeated measures analysis of variance) showed a significant effect of group (F = 10.2, P = 0.002) and region (F = 708, P < 0.0001), and a significant interaction of group with region (F = 18, P < 0.0001). Further analyses then were conducted on a regional basis for each treatment group.
Infusion of physostigmine at a constant rate of 250 μg kg−1 hr−1 throughout the radiotracer infusion period produced a 88% increase in the distribution volume ratio for 4-[18F]FEPB in the rat striatum. More moderate increases were also observed in cortex (+39%) and hippocampus (+39%), with lower but still significant increases seen in thalamus (+22%) and pons (+20%).
The use of phenserine as a single administration produced measures of distribution volume ratios with lower variances. At the highest dose of 5 mg/kg phenserine, increases of specific binding of 4-[18F]FEPB in the striatum (+90%), cortex (+37%), and hippocampus (+29%) were very similar to those observed with physostigmine infusion. Phenserine-induced increases in 4-[18F]FEPB binding (Fig. 2) were dose-dependent in striatum (r2 = 0.81, P < 0.0001), cortex (r2 = 0.57, P < 0.001), and hippocampus (r2 = 0.50, P < 0.001), but not in the hypothalamus.
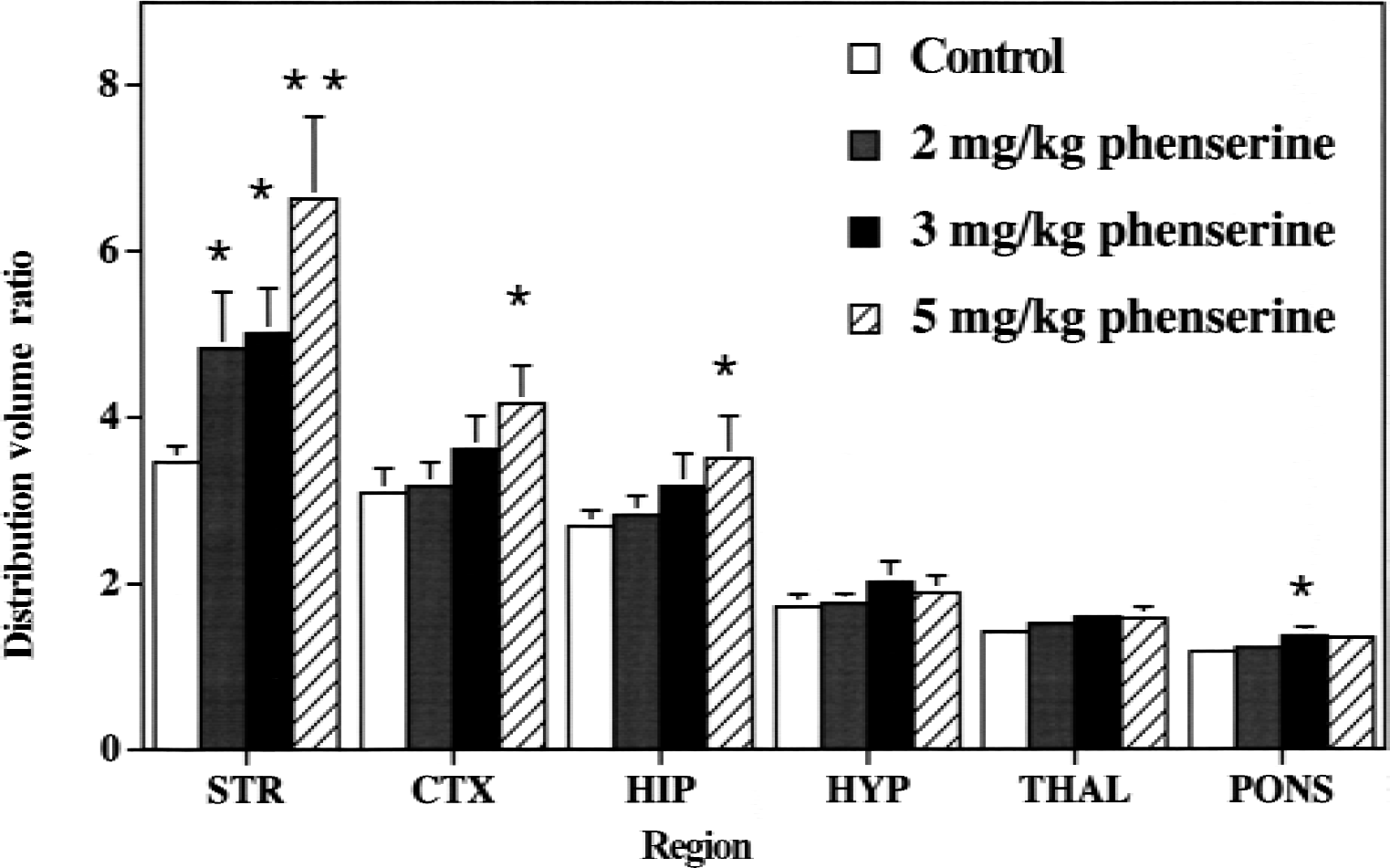
Dose-dependent increases in regional brain 4-[18F]FEPB distribution volume ratios between control and phenserine-treated male rats. Control rats (n = 7) were injected with 4-[18F]FEPB in a bolus plus infusion protocol for 60 minutes before death. Treated rats (n = 4 per group) had phenserine (2, 3, or 5 mg/kg) injected (intraperitoneally) 30 minutes before 4-[18F]FEPB infusion. Data presented as mean ± SD. *P < 0.05, **P < 0.005 versus controls.
These changes in distribution volume ratios (region/cerebellum) were entirely because of alterations in radioligand binding in these regions and not because of decreased uptake and retention in the cerebellum (the reference region), as radioactivity concentrations in that region of the brain remained unchanged throughout all of the studies (controls, 0.19% ± 0.03% injected dose/g; physostigmine, 0.19% ± 0.03% injected dose/g; 5 mg/kg phenserine, 0.22% ± 0.03% injected dose/g).
DISCUSSION
Infusion of a radioligand to equilibrium provides an excellent method for estimating in vivo specific binding to a high affinity site, such as the muscarinic cholinergic receptors. At equilibrium, the ratio of radioactivity in a region of the brain with high concentrations of receptors (for mAChR, striatum or cortex) to a region with low or no concentration of receptor (for example, cerebellum) provides the term known as a distribution volume ratio (DVR), which is directly proportional to the numbers of available binding sites (Carson et al., 1998) and is independent of changes in radiotracer metabolism or delivery (that is, blood flow). Simple competition of an endogenous neurotransmitter, such as ACh, for radioligand binding would then be expected to result in decreased in vivo specific binding (lower DVR) of a radioligand such as 4-[18F]FEPB. On the contrary, and quite unexpectedly, coinfusion of the AChE inhibitor physostigmine produced increased binding of 4-[18F]FEPB in the rat brain, particularly in the striatum, cortex, and hippocampus.
Because physostigmine has considerable peripheral toxicity and its inhibition of brain AChE is short-lived, the authors used phenserine ((−)-phenylcarbamoyleseroline) for the study of the dose-dependency of AChE inhibition. Phenserine is an analog of physostigmine and outperforms physostigmine in the areas of peripheral toxicity, duration of action, and enzyme selectivity (Grieg et al., 1995). This allowed the authors to inject phenserine as a bolus (2, 3, and 5 mg/kg) 30 minutes before infusion of 4-[18F]FEPB. As with physostigmine, phenserine induced significant increases in specific binding of 4-[18F]FEPB in mAChR-rich regions of the rat brain (Fig. 2). More importantly, these increases occurred in a dose-dependent manner.
These studies using physostigmine and phenserine support ACh-mediated increases of in vivo 4-[18F]FEPB binding to the muscarinic acetylcholine receptors in the awake rat brain. The dose of physostigmine used here has been shown previously to increase extracellular endogenous ACh levels by approximately 1100% in the rat cortex (Messamore et al., 1993). The corresponding data for phenserine is not available, but the authors previously have examined the ability of phenserine to inhibit AChE in vivo in the mouse brain using a second radiotracer method employing 4-N-[11C]methylpiperidinyl propionate ([11C]PMP), a substrate for the enzyme that accumulates in the brain through metabolic trapping of the hydrolysis product. Phenserine dose-dependently decreases AChE-mediated hydrolysis of [11C]PMP (Kilbourn et al., 1999) and the extent of inhibition of the enzyme measured in vivo correlates very well with the increased specific binding of the muscarinic cholinergic radioligand 4-[18F]FEPB observed in this study (r = 0.98, data not shown). Thus, the increased 4-[18F]FEPB binding to the mAChR appears directly proportional to the extent of AChE inhibition by phenserine.
Although the agonist-mediated increase of in vivo 4-[18F]FEPB binding was surprising, unexpected increases of binding for in vivo radioligands have been observed in both the dopamine and muscarinic cholinergic receptor systems. Using [3H]quiniclidinyl benzilate ([3H]QNB) and simple measures of total radioactivity in brain regions at a single time point after bolus radiotracer administration, Boggan et al. (1988) and Pelham and Munsat (1979) have demonstrated increased [3H]QNB concentrations after treatments with morphine, amphetamine, and phencyclidine. Those studies, however, used an in vivo radiotracer method that can be sensitive to changes in radiotracer delivery to the brain tissues. Challenges of dopaminergic radioligand binding in vivo (both D1 and D2 receptor ligands) with dopamine-releasing drugs have produced changes of in vivo radioligand binding ranging from decreases (perhaps ligand-neuro-transmitter competition, seen with benzamides such as [11C]raclopride), to no change (no ligand sensitivity, seen with [18F]fallypride), to enhanced radioligand binding (agonist-mediated increases, seen with [11C]spiperone and [3H]pimozide) (Laruelle, 2000). To date, no clear explanation has been offered as to why dopamine D2 receptor radioligands of different structural classes should show such dramatically different behaviors in the face of increased dopamine concentrations. In the dopaminergic system, the agonist-dependent increases in spiperone binding have been postulated to represent agonist-mediated receptor internalization and ligand trapping, although this interpretation remains controversial. It is well-known that mAChRs, which like the dopamine receptors are G-protein linked receptors, also internalize in response to agonists such as ACh (Koenig and Edwardson, 1996; Krudewig et al., 2000; Tolbert and Lameh, 1996; Vögler et al., 1999), and it is possible that the same trapping mechanism could be occurring with 4-[18F]FEPB in response to increasing levels of ACh. However, the experiments performed here cannot distinguish between internalization and other possibilities, such as agonist-mediated changes in receptor affinity (possibly through phosphorylation and conformational changes), that might also produce altered in vivo radioligand binding. Changes in binding affinity, but not the numbers of receptors, has been recently suggested as the mechanism of cholinergic modulation of [11C]raclopride binding to dopamine D2 receptors (Tsukada et al, 2000).
Two aspects of the agonist-induced increases of 4-[18F]FEPB are different than that previously observed in the dopaminergic system. First, the magnitude of the increase seen here, nearly 90%, is considerably greater than the 30% to 50% increases seen for dopaminergic radioligand binding. Second, the changes in AChE inhibition effects are more dramatic for the striatum than the cortex. This difference could be the result of the fact that mAChR subtype distribution is not homogeneous and that the mAChR subtypes internalize and/or recycle to the neuronal membrane surface at different rates and by different mechanisms (Bernard et al., 1998; Edwardson and Szekeres, 1999; Hosey et al., 1999; Koenig and Edwardson, 1996; Krudewig et al., 2000; Vögler et al., 1999). Interestingly, in a study of the effects of physostigmine on in vivo binding of [18F]fluoropropyl-TZTP ([18F]FP-TZTP, a mAChR agonist radioligand), Carson et al. (1998) observed significantly less of an effect of AChE inhibition on striatal radiotracer binding as compared with the other regions of the brain, a regional effect that could not be adequately explained.
In summary, the current study has shown that the in vivo specific binding of 4-[18F]FEPB is sensitive to increases in endogenous ACh levels induced through the AChE inhibitors physostigmine and phenserine. In contrast to the expected decrease in radioligand binding predicted by simple competition of ACh for the mAChR binding site, 4-[18F]FEPB binding in mAChR-rich brain regions actually increased in response to elevated ACh levels. Although the exact mechanism of this enhancement of in vivo binding of radioligands needs to be determined, a method is now available to examine the relation between changes in ACh levels and muscarinic receptor occupancy induced by existing and new drugs intended to accentuate functioning of the cholinergic system. Applications of 4-[18F]FEPB together with measures of AChE inhibition (Kuhl et al., 1999), ACh-mediated changes in nicotinic cholinergic receptor binding (Ding et al., 2000), and mAChR agonist binding (Carson et al., 1998) provide exciting new means of examining the functioning of the ACh system in the living brain.
Footnotes
Acknowledgments:
The authors thank the cyclotron staff and radiochemistry staff of the Division of Nuclear Medicine at the University of Michigan Medical School for production of the radioisotope. Special thanks go to Kyle Kuszpit and Leslie Doherty for their assistance with the animal studies, Dr. Nigel Greig for a sample of phenserine, and Dr. Robert A. Koeppe for advice on statistical analyses.