
Abstract
Human kynurenine 3-monooxygenase (KMO) is emerging as an important drug target enzyme in a number of inflammatory and neurodegenerative disease states. Recombinant protein production of KMO, and therefore discovery of KMO ligands, is challenging due to a large membrane targeting domain at the C-terminus of the enzyme that causes stability, solubility, and purification difficulties. The purpose of our investigation was to develop a suitable screening method for targeting human KMO and other similarly challenging drug targets. Here, we report the development of a magnetic bead–based binding assay using mass spectrometry detection for human KMO protein. The assay incorporates isolation of FLAG-tagged KMO enzyme on protein A magnetic beads. The protein-bound beads are incubated with potential binding compounds before specific cleavage of the protein–compound complexes from the beads. Mass spectrometry analysis is used to identify the compounds that demonstrate specific binding affinity for the target protein. The technique was validated using known inhibitors of KMO. This assay is a robust alternative to traditional ligand-binding assays for challenging protein targets, and it overcomes specific difficulties associated with isolating human KMO.
Introduction
In mammals, tryptophan metabolism is catalyzed by the enzymes of the kynurenine pathway. 1 The kynurenine pathway produces several biologically active molecules that have been implicated in various inflammatory disease states and in the regulation of the immune response.2,3 Kynurenine 3-monooxygenase (KMO; EC 1.14.13.9) is an important enzyme in the pathway and catalyzes the hydroxylation of kynurenine (L-kyn) to cytotoxic 3-hydroxykynurenine (3-HK). 1 The enzyme is an NADPH-dependent flavoprotein hydroxylase that is localized to the outer mitochondrial membrane via a C-terminal targeting sequence. 4 In recent years, KMO has been identified as an important therapeutic target for Huntington’s disease 2 and sterile systemic inflammation. 3 Therefore, there has been renewed interest in developing tractable in vitro assays for the identification of selective inhibitors.5,6
Recombinant protein production of human KMO is problematic, with a number of solubility and purification difficulties reported in the literature. A large membrane targeting domain at the C-terminus of the human enzyme (amino acids 385–486) is understood to account for these difficulties. 4 This domain, however, localizes KMO to the outer mitochondrial membrane and facilitates the formation of protein–protein complexes. This region of the protein renders it insoluble and difficult to purify following recombinant expression. 7 Although the C-terminal region does not form part of the active site, 8 it is required to maintain activity of the enzyme because truncated proteins possess no enzymatic activity. 7
Several in vitro assay techniques for the determination of KMO activity have been reported in the literature. Quantifi-cation of the product, 3-HK, by liquid chromatography–mass spectrometry (LC-MS) analysis has been commonly used as an assay to establish the enzymatic activity of KMO. 5 This method has been demonstrated both in isolated protein extracts and in a cell-based context using KMO from several species. Oxidation of NADPH measured by spectrophotometry has also been applied to measure KMO activity for rat (Rattus norvegicus) and bacterial (Pseudomonas fluorescens) enzymes. That assay requires purified protein, however. A competitive binding assay using anisotropy to measure the displacement of fluorescently labeled adenosine diphosphate substrate by compounds binding to KMO has also been reported. 6 That assay, however, also requires purified enzyme and has only been validated using bacterial (P. fluorescens) KMO, which can be produced in high yield.
Magnetic bead–based assays have been used as sensitive diagnostic tools when conjugated to various probes, such as oligonucleotides and antibodies, and can be used to detect DNA, proteins, or antigens from clinical samples. In a drug-screening context, magnetic bead assays have been applied in a functional assay of protein kinase activity and in a high-throughput one-bead–one-compound screening system, among other assay systems.9,10 Importantly, magnetic beads have been used for isolation of target proteins from crude bacterial and mammalian cell lysates, then incorporated in a specific assay system.
Here, we report a magnetic bead–based drug binding assay with LC-MS readout that represents a new method for specifically targeting human KMO enzyme. This assay can be used as an alternative screening technique to the commonly used biochemical method for quantification of 3-HK production, particularly because the novel assay uses tools and instrumentation that are widely available. The assay does not require purified protein and uses functionally intact human enzyme that has been expressed in a crude lysate from Escherichia coli (E. coli). Using human KMO as the exemplar, this assay is time and resource efficient, and it avoids the laborious technical challenges associated with the handling and preparation of difficult target proteins.
Materials and Methods
Cloning
The full-length human KMO gene (GenBank Accession No. NM_003679, Arg 452 variant) was codon optimized for bacterial expression and constructed with a 12 poly-histidine tag (CATCATCACCATCACCATCATCATCACC ATCACCAT), TEV protease site (GAAAACCTGTATTT TCAGGGT), and 3xFLAG™ epitope (ATGGACTACAAAGACCATGACGGTGATTATAAAGATCATGATATCGATTACAAGGATGACGATGACAAGTGA) at the C-terminus. This gene was synthesized by GenScript in vector pUC57. The gene incorporated a NheI restriction site at the N-terminus and a NotI restriction site after the stop codon at the C-terminus. The gene was ligated using these restriction sites into vector pET24b (Novagen, Bornova, Turkey) for expression in E. coli cells.
Expression
Double-tagged KMO was expressed in E. coli competent cell strain BL21(DE3) pLysS (Invitrogen, Carlsbad, CA). Bacterial cells were grown in LB broth (Melford, Chelsworth, UK) with antibiotics kanamycin and chloramphenicol at 37 °C, with rotation at 250 rpm until the optical density at 600 nm was 0.8. Cells were then induced with 1 mM isopropyl β-D-1-thiogalactopyranoside (IPTG; Sigma-Aldrich, St. Louis, MO) overnight at 18 °C. Bacterial cells were collected by centrifugation at 6000 rpm for 10 min at 4 °C. Cell pellets were frozen at −20 °C until the preparation of lysate.
Preparation of Lysate
Bacterial cell pellets were thawed and resuspended in 20 mM HEPES pH 7.0 with ethylenediaminetetraacetic acid (EDTA)-free protease inhibitors (Roche, Basel, Switzerland). Cells were sonicated on ice to lyse and centrifuged at 20,000 rpm for 45 min at 4 °C. Total protein concentration in the supernatant fraction was determined by standard protein assay procedure using the Bio-Rad reagent and bovine serum albumin protein standards (Sigma-Aldrich). The crude lysate was run out on a sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) protein gel and stained with simplyblue (Invitrogen). KMO-FLAG protein was found to account for approximately 27% of the total protein in the crude lysate by gel analysis using ImageJ software. The lysate was diluted in the same buffer as above to 2 mg/ml total protein concentration, divided into 1 ml aliquots, and frozen at −20 °C. Bacterial cell lysate expressing KMO protein was previously shown by us to demonstrate enzymatic activity using the steady-state kinetic assay. 11
Magnetic Bead–Binding Assay
PureProteome Protein A magnetic beads (Millipore, Billerica, MA) were mixed well to resuspend. Beads were prepared as a master mix for all samples, using 5 µL of beads per assay sample. The beads were extracted from the storage buffer using the PureProteome magnetic stand (Millipore). After resuspension in PBS with 0.1% Tween 20, the beads were vortexed for 10 s to wash thoroughly and then were pulled down by magnet, and the wash buffer was removed. The beads were resuspended in PBS with 1 µL of anti-FLAG M2 mouse monoclonal antibody isotope IgG1 (Sigma-Aldrich) per assay sample and incubated at 25 °C for 40 min with gentle agitation. Protein A magnetic beads are reported in the manufacturer’s protocol to demonstrate strong affinity for mouse-derived antibody isotope IgG1. Following antibody conjugation, the bead–antibody complexes were washed once in PBS with 0.1% Tween 20. Human KMO lysate was added to the bead–antibody complexes at 100 µL lysate per assay, corresponding to an approximate concentration of 9.8 µM of human KMO according to gel analysis (ImageJ), to allow interaction between the FLAG tag on the protein and the on-bead anti-FLAG antibody. Each assay sample was aliquoted into 1 well of a U-bottom Co-Star 96-well plate. Assays were performed in duplicate. The incubation time was 1 h at 25 °C, with rotation at 400 rpm. Control assays consisted of bead–antibody complexes incubated with assay buffer 20 mM HEPES pH 7.0 only and were also carried out in duplicate. After 1 h incubation, 10 µL of 1 compound per well was added to the bead–antibody–protein complexes and to the control samples to a final concentration of 10 µM. Samples were incubated for 1 further hour at 37 °C, with rotation at 400 rpm. The bead complexes were isolated from the assay sample using the magnet and washed twice in PBS with 0.1% Tween 20. After the second wash, the bead complexes were resuspended in 30 µL of 20 mM HEPES pH 7.0, containing 1 µL TEV protease (1 mg/ml; Sigma-Aldrich) per well. One hour of incubation at 30 °C with rotation at 400 rpm allowed cleavage of the fusion protein between the 12 poly-histidine tag and the 3xFLAG epitope, and the dissociation of the protein–compound complex from the bead. It should be noted that the poly-histidine tag at KMO’s C-terminus does not serve a purpose in this particular assay. Following proteolytic cleavage, the beads were pulled down by magnet, and the supernatant was transferred to LC-MS vials and frozen at −20 °C prior to LC-MS and MS analysis ( Fig. 1 ).
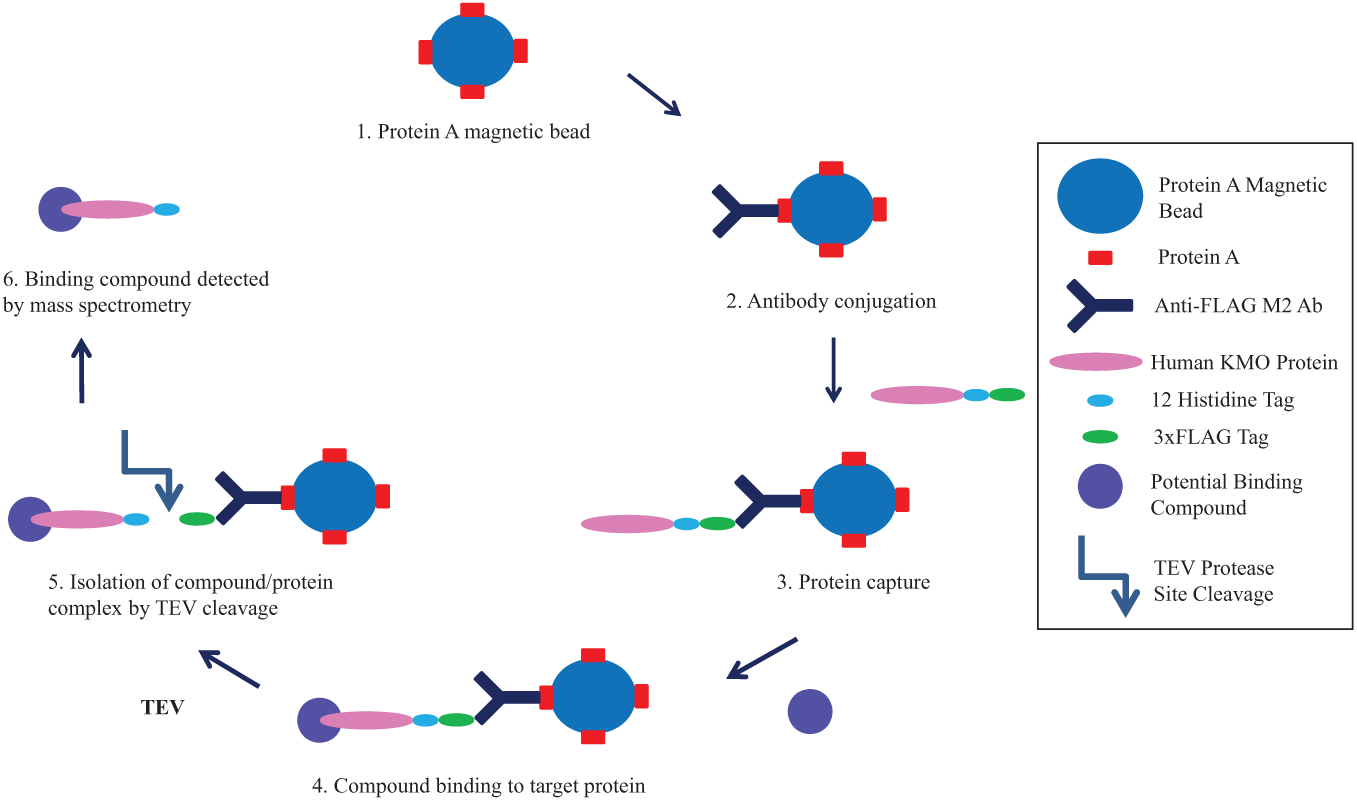
Assay scheme. Schematic representation of the novel magnetic bead–based kynurenine 3-monooxygenase (KMO) drug binding assay. The Fc portion of anti-FLAG M2 antibody is bound to Protein A on the magnetic beads. The anti-FLAG-specific domain binds the FLAG tag of the target protein, and magnetic separation is used to isolate bead-bound protein. Potential binder compounds are incubated with the enzyme-bound beads before a wash step. Binding compound–protein complexes are released from the bead by TEV-specific cleavage near the target C-terminus. The peak area of compound bound to the protein is detected by mass spectrometry analysis.
Mass Spectrometry Method
Instrumentation
The chromatographic system used was a TurboFlow TLX Aria-1 system (ThermoFisher Scientific, Hemel Hempstead, UK), consisting of two Allegro pumps defined as the loading and eluting pumps, two valve-switching modules, and a CTC liquid autosampler. The detection was carried out using a TSQ Quantum Discovery triple quadrupole mass spectrometer (ThermoFisher Scientific).
Chromatography and Mass Spectrometry Parameter Optimization
Samples were subject to online extraction using a TurboFlow TLX Aria-1 system operated in focus mode. A 10 µL injection of the assay sample was loaded directly onto a C18PXL (50×0.5 mm; ThermoFisher Scientific, Hemel Hempstead, UK) TurboFlow column at a high flow rate, causing the proteinaceous material to flow to waste. A series of valve switches led to the elution of the extracted sample from the TurboFlow column directly onto the analytical column. Solvent A was water with 0.1% formic acid, Solvent B was methanol with 0.1% formic acid, and Solvent C was 45:45:10 acetonitrile–isopropanol–acetone. Conditions are shown in Supplemental Material 1.
Following TurboFlow extraction, the analytes were subsequently separated on a reverse-phase T3 Atlantis (2.1×150 mm, 3 µm; Waters, Manchester, UK) analytical column, protected by a Kinetex KrudKatcher (Phenomenex, Macclesfield, UK). The analytical column was maintained at 5 °C using a column chiller. Conditions are shown in Supplemental Material 1.
The online TurboFlow system Aria-1 was directly connected to a Quantum Discovery triple quadrupole mass spectrometer, operated in electrospray ion mode with polarity switching for positive and negative ion monitoring. The source temperature was 300 °C, the spray voltage 3 kV, and the skimmer offset 12 V. Argon, the collision gas in Q2, had a pressure of 1.5 mTorr. Automated tune settings were used to achieve the maximum ion signal for each analyte, optimizing on tube lens voltage, parent → product transitions, and collision energy for each transition. A quantifier and qualifier ion was determined for each analyte. An acceptable quantifier: Qualifier peak area ratios in assay samples were considered to be those that fell within 20% of the average ratio seen in standards. Monitoring for quantifier and qualifier ions adds additional specificity to the assay.
A scan width of m/z 0.5, scan time of 0.1 s, and unit resolution on Q1 and Q3 were applied. Data were acquired and processed using Aria 1.3, Xcalibur 1.4, and LC Quan 2.0 SP1 software packages.
Compounds
Three known and commercially available KMO inhibitors from the scientific literature12,13 and a non-active compound from our in-house library were used to validate the new binding assay ( Fig. 2 ). The inhibitory activities of these compounds against human KMO were verified in a separate kinetic assay. 5 The direct binding affinities of each compound for human KMO were generated using microdialysis, as described in the Microdialysis section. Compound 1 was assayed in the presence and absence of enzyme to generate the Z’ factor for the magnetic bead–based assay.
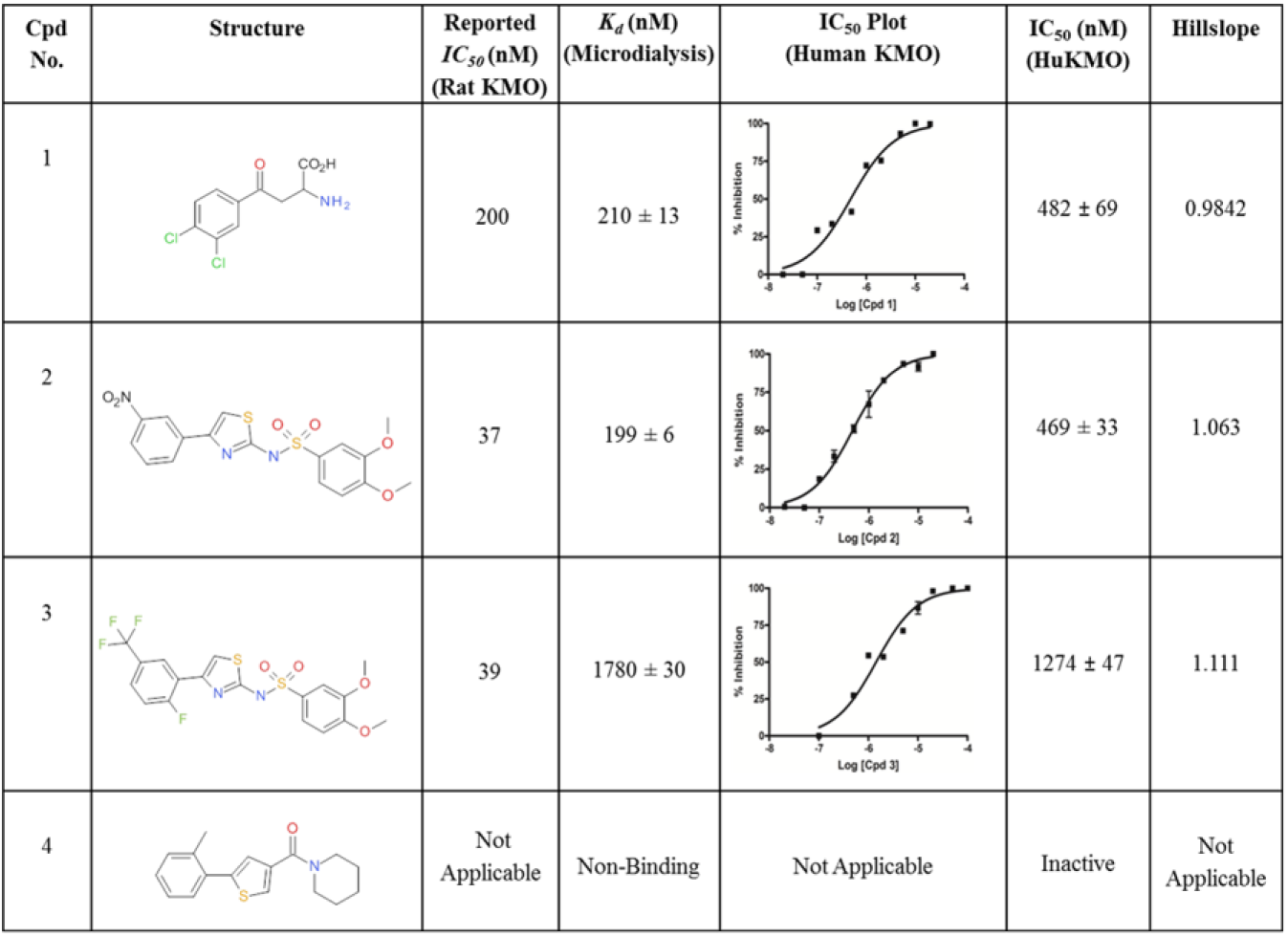
Assay validation compounds. Structures and activities of the compounds used to validate the new magnetic bead assay. The literature-reported activities (rat kynurenine 3-monooxygenase [KMO]) are shown, and so are the Kd and IC50 values for human KMO generated in the microdialysis and kinetic assays, respectively. For half-maximal inhibitory concentration plots, concentration of compound (log10) is plotted versus percentage inhibition and fitted to the 4-parameter equation {Y = Bottom + [Top − Bottom] / [1 + 10^ ({LogEC50 − X} * Hillslope)]} using GraphPad Prism4 software.
Microdialysis
Microdialysis was used for assessment of in vitro drug to KMO binding for the generation of compound dissociation constants in a method similar to that reported by Weidemann et al. 14 Rapid equilibrium device (RED) inserts (MWCO 8 kDa) were placed in the wells of a compatible base plate (Thermo Scientific Pierce, Rockford, IL). KMO protein was diluted to 10 µM in concentration (protein concentration estimated using ImageJ gel analysis software) in 100 µL of assay buffer (20 mM HEPES, pH 7.0) and added to the sample chamber (indicated by the red ring) of the RED device insert in the base plate. Three hundred microliters of assay buffer were added to the buffer chamber (indicated by the white ring) of the RED device in the base plate for each sample. Compound was added to the starter volume in the sample chamber at a final concentration of 10 µM to start microdialysis. The plate was covered with sealing tape and incubated at 37 °C for 6 h with rotation at 250 rpm. After incubation, a 50 µL sample was taken from each part of the dialysis chamber, 10 µL of acetonitrile was added to each sample, and the samples were transferred to LC-MS vials. Samples were frozen at −20 °C prior to MS analysis. The MS method described in this article was used for analysis. Kd values were calculated according to the formula determined by Weidemann et al. 14
Results and Discussion
In this article, we describe the development of a novel drug binding assay using magnetic bead selection specifically targeting human KMO enzyme. This new magnetic bead–binding assay has a total incubation time that is shorter than 4 h. Several 96-well plates can be assayed simultaneously due to assay automation using multichannel pipettes for liquid handling. The MS method incorporates a run time of 10 min per sample; this includes on-line extraction of samples, which reduces sample preparation time and human error. Although purification of this human KMO construct has been demonstrated by us, 11 there is no requirement for purified protein in this simplified assay system. By isolating KMO from a crude lysate “in-assay” using a specific isolation technique in the form of antibody-conjugated protein A magnetic beads, the time and resources associated with protein purification are reduced. Because commercially available KMO antibodies have demonstrated poor specificity, we created a 3xFLAG-tagged fusion protein, which retained KMO enzyme activity, used in conjunction with an anti-FLAG (M2) antibody. This enabled highly specific isolation of the target protein on-bead. Enhanced specificity was achieved by incorporating a TEV protease site to allow cleavage of the KMO–compound complexes from the beads. By using specific protease cleavage, only KMO and associated or bound molecules should be eluted from the beads for MS analysis, in contrast to nonspecific acid elution whereby all material is eluted from the beads and subject to MS analysis. Protein from the lysate that is nonspecifically bound to the antibody or the bead should remain on-bead and thus be excluded from the MS analysis. Our results suggest that incorporation of a protease site in combination with an affinity tag such as the FLAG epitope to other recombinant target proteins may enable this methodology to be used as a generic tool for the identification of specific drug binding. Also, successful application of the assay to this challenging membrane protein target indicates that this method is not restricted to soluble target proteins.
Compounds 1, 2, and 3, known inhibitors of KMO, were selected from the scientific literature12,13 ( Fig. 2 ). Kinetic and in vitro microdialysis binding assays were used to confirm inhibition of and binding to human KMO, with IC50 and Kd values similar to literature values reported against rat KMO enzyme ( Fig. 2 ). Compound 4, a compound selected to act as a nonbinding negative control, was confirmed to be inactive in these assay methods. When assayed in the new binding method, the known KMO inhibitors (compounds 1–3) have increased detected peak areas when assayed with KMO enzyme compared to the control assays (no enzyme), as indicated by their high peak area ratios ( Table 1 ). This enrichment of compound in the positive sample suggests specific binding to KMO. Because samples were prepared for MS analysis by cleavage of the enzyme from the bead, the presence of compound in the sample indicates complexation with KMO. Detection of compounds 1–3 was significantly lower in the absence of KMO protein. Compound 4, a compound shown to be inactive against KMO, has a peak area ratio close to 1 ( Table 1 ), indicating little or no specific binding to the enzyme because the presence of KMO in the assay had little effect on the concentration of compound 4, which was detected by MS. Although the MS signal may vary among compounds due to differential ionization, and the response is not directly quantitative without the incorporation of a standard curve for each compound assayed, the results demonstrate significant enrichment of drugs binding specifically to KMO ( Table 1 ). The background signal detected in the control samples also varies among compounds. Nonspecific binding may vary for each drug, but specific binding to KMO is strongly indicated by the peak ratio for these validation compounds. Compound 2 demonstrated high nonspecific binding, which is binding of compound to the beads or antibody component of the assay, as demonstrated by the high MS peak intensity in the control (enzyme-free) sample. The high peak ratio for this compound still indicates binding to KMO, particularly when compared with the non-KMO binding compound (compound 4) that showed little difference in signal between enzyme-conjugated and enzyme-free magnetic beads and, therefore, no specific binding to KMO. In this format, the assay can be used as a fast qualitative primary selection process for the identification of novel compounds with specific binding affinity for the target protein.
Magnetic Bead Assay Results.
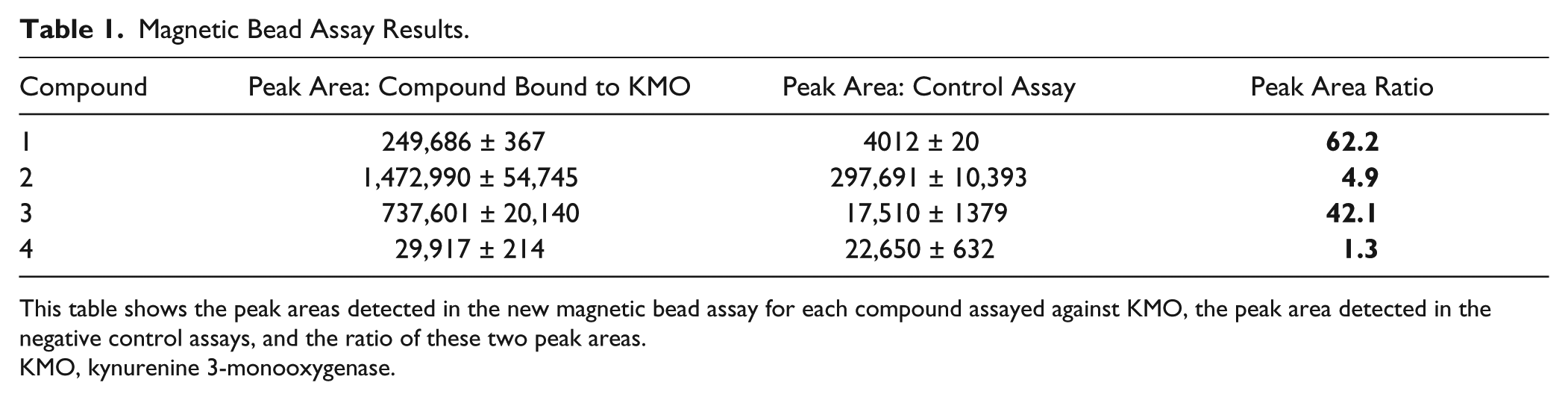
This table shows the peak areas detected in the new magnetic bead assay for each compound assayed against KMO, the peak area detected in the negative control assays, and the ratio of these two peak areas.
KMO, kynurenine 3-monooxygenase.
Compound 1 (di-chloro-benzoyl alanine), at 20 µM (10× Kd), was selected as the positive binding control for generation of the Z’ factor because it demonstrated a Kd of 210 nM for KMO in a separate microdialysis assay. The Z’ factor was calculated according to the formula described by Zhang et al. 15 The positive control sample peak area represented compound 1 detected by MS when assayed at a concentration of 20 µM with KMO protein, and the negative control sample peak area was compound 1 detected by MS when assayed at 20 µM in the absence of protein. The mean positive peak area signal (in arbitrary units) was 698,742 with a standard deviation of 58,021. The mean negative peak area was 3080 with a standard deviation of 2707. These peak areas detected for compound 1 when assayed in the presence and absence of human KMO enzyme enabled quantification of a Z’ factor value of 0.74 for this assay ( Fig. 3 ). Despite demonstrating a similar Kd of 199 nM for KMO in the microdialysis assay, compound 2 had a much lower peak area ratio than compound 1 in the new assay method. Consequently, the Z’ factor for compound 2 in the magnetic bead assay was evaluated, and the value found to be 0.60. The automated assay format and satisfactory range of Z’ values for this assay indicate that the assay is suitable for reliable use in high-throughput screens.
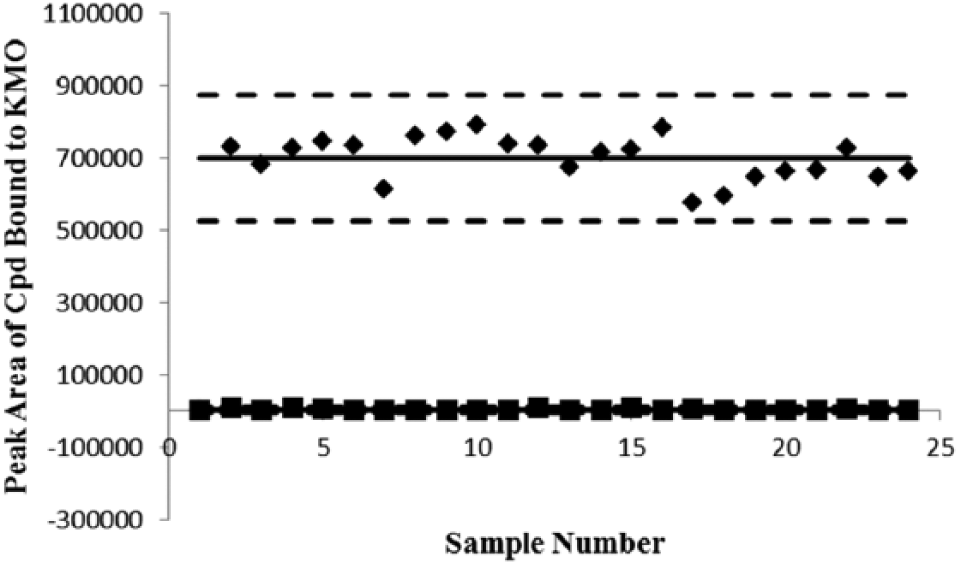
Z’ factor plot. Plot showing the mass spectrometry peak area responses for compound 1 assayed with (indicated by ♦) and without human kynurenine 3-monooxygenase (KMO) enzyme (indicated by ■) in the new assay plotted against the assay sample number. The solid horizontal lines represent the means of the positive and negative data, and the broken lines represent 3 standard deviations from the mean of each data set.
KMO is known to form complexes with other membrane proteins. 7 It is possible that the human enzyme used in this assay may aggregate with other co-expressed bacterial proteins because we previously identified two contaminant proteins that co-elute with this construct during purification. 11 Although this does not appear to have a detrimental effect on the specific binding of known KMO inhibitors, it may be necessary to validate hits in a separate KMO-specific assay when screening a library of new compounds. This assay could function both as a prescreening selection tool for the identification of novel binders and as a secondary validation assay demonstrating direct binding of hit compounds.
In summary, we describe a novel method for the identification of compounds with specific binding affinity for human KMO. Successful application of this method to compound screening for this particular drug target indicates that this technique may be suitable for targeting other challenging proteins, such as those that express poorly or are difficult to purify. In addition to its potential as a generic screening tool for both soluble and difficult-to-express proteins, the assay is efficient in time and resources. By using in-assay isolation of KMO followed by on-line extraction using the TurboFlow system, sample preparation time was minimized. To improve the throughput of this novel technique, the assay could be configured to test mixtures of compounds in each well, and the MS method altered to scan over a defined m/z detection range. This would remove the requirement for compound tuning prior to assay with the caveat that peak intensity will not necessarily indicate tight binding because this is dependent on the ionization efficiency of each compound within the mixture. We conclude that this assay is suitable for screening chemically diverse libraries for specific binding to protein targets, thereby accelerating and improving the development of novel drugs.
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: We are grateful to all supporters and funders. This work was supported by a Medical Research Council (MRC) studentship to KW. DJM holds a Health Foundation/Academy of Medical Sciences Clinician Scientist Fellowship.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.