
Abstract
For approximately a decade, biophysical methods have been used to validate positive hits selected from high-throughput screening (HTS) campaigns with the goal to verify binding interactions using label-free assays. By applying label-free readouts, screen artifacts created by compound interference and fluorescence are discovered, enabling further characterization of the hits for their target specificity and selectivity. The use of several biophysical methods to extract this type of high-content information is required to prevent the promotion of false positives to the next level of hit validation and to select the best candidates for further chemical optimization. The typical technologies applied in this arena include dynamic light scattering, turbidometry, resonance waveguide, surface plasmon resonance, differential scanning fluorimetry, mass spectrometry, and others. Each technology can provide different types of information to enable the characterization of the binding interaction. Thus, these technologies can be incorporated in a hit-validation strategy not only according to the profile of chemical matter that is desired by the medicinal chemists, but also in a manner that is in agreement with the target protein’s amenability to the screening format. Here, we present the results of screening strategies using biophysics with the objective to evaluate the approaches, discuss the advantages and challenges, and summarize the benefits in reference to lead discovery. In summary, the biophysics screens presented here demonstrated various hit rates from a list of ~2000 preselected, IC50-validated hits from HTS (an IC50 is the inhibitor concentration at which 50% inhibition of activity is observed). There are several lessons learned from these biophysical screens, which will be discussed in this article.
Keywords
Introduction
With the initiation of high-throughput screening (HTS) and the ease of its implementation and application in drug discovery, the necessity to classify and characterize low-molecular-weight hits was created not only to validate the results of HTS but also to cull the hit number to a digestible sum for candidate follow-up. This has led to the creation of orthogonal screening and counter-screening procedures to prevent false positives from being promoted to the next step of hit validation.
1
Methods in medium- to high-throughput (HT) biophysics were then developed to validate, confirm, and in addition profile these lead candidates for their specificity and selectivity toward the target. The main purpose of medium-throughput to HT biophysics is to provide such hits an expedient, focused, and decisive entryway into lead optimization.
Common Biophysical Methods Used for HTS Hit Validation
As one can see in
Generic Biophysics Screen Flow Chart
As
To illustrate and answer these questions, several screening approaches in biophysics will be discussed in this article, using a format in which the screen aim will be presented, and focusing on the hit-finding goal of the project and the technologies used to validate and deliver the desired set of compounds. Because the main purpose of this article is to discuss the biophysics validation approach with respect to the biochemistry of the target, details about the target itself or its mode of action in disease pathology, in some cases, will not be revealed.
Screen Approach: Validation of HTS Hits from a New Epigenetic Target Family Using Biophysics
A target from a new family of proteins thought to be involved in a particular disease pathology requires full “biophysical” attention to understand and design a screen that not only respects the biochemical profile of the target but also fulfills the aim of the screening campaign. While developing a screening assay, it is important that any potential caveats hidden within a chosen screen approach are revealed, thereby ensuring that the screening format itself does not falsify the hit rate or create nonlogical trends in the scaffold profiles of the hits, for example the misalignment of SPR affinity data with the time-resolved fluorescence resonance energy transfer (TR-FRET) results. Because the purpose of this article is to demonstrate how to construct a logical biophysics hit-validation scheme, an in-depth examination and discussion of dissecting or troubleshooting the misalignment of data with other approaches such as TR-FRET will not be presented here.
Once an understanding of how the target behaves in certain screening formats is cataloged, one can then approach the next class of similar targets in a streamlined manner, that is, leveraging certain technologies early and quickly to get the most relevant data and promote these hits to the next level of characterization. In the supporting examples, we will discuss the aim of a screening campaign for target A, a new target family, its related family member, and target B. The screening strategy for target A and target B, and how their results can be used to shape the next screening campaign for other epigenetic targets, will be presented. A summary of their outcomes quantified by the percentage confirmation of the HTS hits, the number of hit scaffolds promoted to the next level of characterization, and the number of scaffolds deemed worthy of further medicinal-chemistry efforts will be discussed.
Screen Aim for Target A
Globally, epigenetics and its role in disease pathology command tremendous interest in academia and in industry. The permeation of target A, an epigenetic modulator, was one of several chosen as an entryway into this particular field of epigenetic drug discovery. The screen aim was to validate reversible competitive inhibitors of target A, its domains, and target B (selected from an HTS screen performed using a TR-FRET assay). The desired inhibitors were predicted to have one of two profiles: (1) selectively inhibit target A via one of its domains, or (2) pan-inhibit several targets within the same target family, such as target B. Currently, target A is not well characterized biochemically; it is not known which domain of target A is pathologically relevant, whether these domains interact cooperatively, or if its binding pocket could be regulated allosterically. Thus, the HTS hit-finding strategy was designed to screen broadly to capture all possible molecules. It was known at the time that this particular target was not part of a multiprotein complex, and thus certain biophysical approaches were not used, such as AS-MS. Another interesting challenge about target A was discovered inadvertently using DSF, which revealed that target A was thermally stabilized by DMSO, a common diluent used for compound solubilization. DSF, at this moment in time in the project, was being used to screen a set of constructs and buffer conditions to optimize production capabilities and investigate construct robustness for certain biophysical assays. Traditionally, DMSO is routinely investigated to observe how well the target can handle this solvent, which is then used as a guide to know which concentration of compound or DMSO can be used in the biophysics assay. After this initial finding, DMSO binding was also confirmed via NMR to quantitate the KD of DMSO and in X-ray crystallography to understand where this solvent molecule was binding (data not shown); and, indeed, the molecule was found in the active site. Due to these unique factors, the biophysical characterization of target A received the utmost priority to guide screening assay development at all levels of the hit-finding flow chart and to streamline the subsequent screening campaigns for other family members.
Screen Aim for Set7/9
To illustrate how a biophysics validation strategy can serve as the basis to trim the hit-finding efforts for other targets, Set 7/9 (otherwise known as SetD7) will be used to exemplify this approach. Set7/9 is an epigenetic target of strategic importance to the modulation of DNA transcription. In this hit-finding approach, reversible competitive inhibitors were preferred that block the ability of Set7/9 to bind histone peptide tails. With the knowledge obtained from the epigenetic target-screening strategies, DSF was used as the primary screening approach for Set7/9 to quickly drill down to the most interesting scaffolds to expedite their promotion to chemical optimization.
Materials, Methods, and Target Background Description
These sections are located in the supplemental information.
Results
Pilot Screen for Target A
A pilot screen was performed using 18 selected compounds of various affinity ranges to determine the amenability of target A to various biophysical-screening formats.
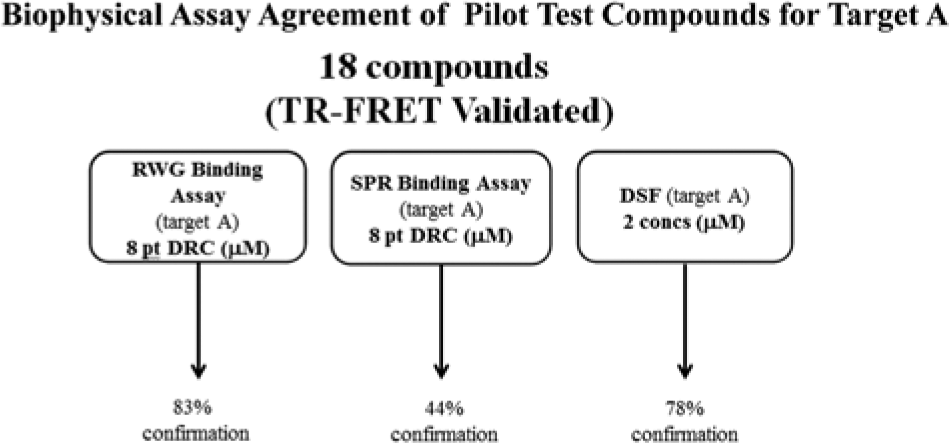
The confirmation rates (in %) between the different technologies used in the pilot screen of Target A. For this study, 18 compounds that were previously validated via time-resolved fluorescence resonance energy transfer were used to investigate the capability of Target A to be screened using these different biophysical approaches for high-throughput screening hit validation.
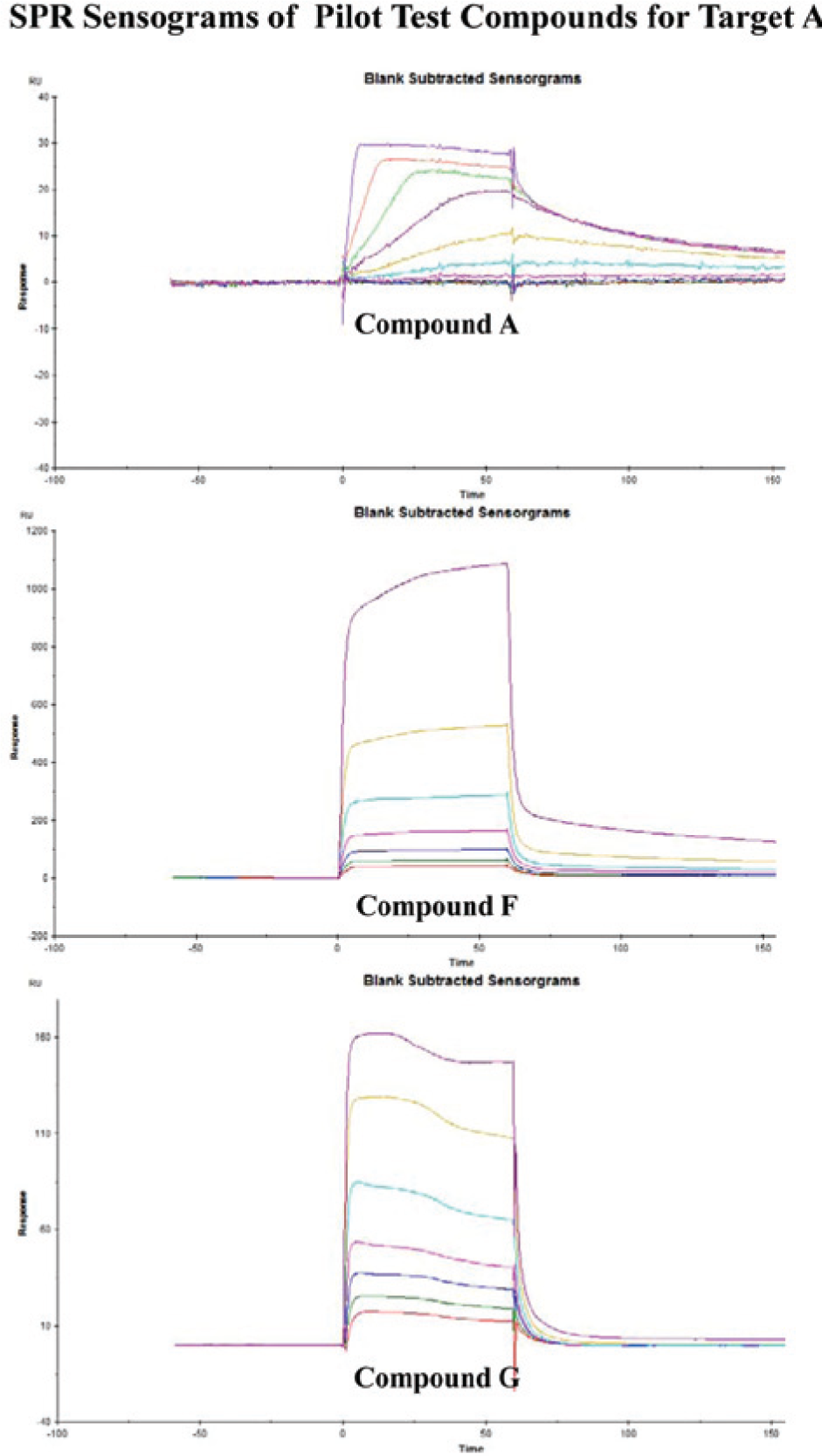
The surface plasmon resonance (SPR) sensograms corresponding to the affinity data listed in
At lower affinity ranges, RWG could detect binding, whereas DSF appears to have only a 50% confirmation rate in comparison to RWG. This did not reflect the incapability of the DSF technology to detect weak hits, but actually indicated that if weaker hits were desired by the project team to find new chemical space, a higher concentration of compound would be necessary so that target saturation is achieved and a DSF signal is observed (data not shown).
Biophysics Screen for Target A
As one can see in
As described in the supplemental information, turbidometry data were then merged with the binding signal data to flag any potential problems in RWG or DSF prior to SPR. SPR itself is crucially liable to compound artifacts, because it is a method that serially injects each compound over the same target surface. Thus, if there is, by chance, a compound that binds in a superstoichiometric manner or aggregates and does not dissociate from the target surface, the ability for the next compound to bind and be detected may be compromised. To circumvent this, an SPR clean screen was performed on target A and its family members prior to the actual SPR binding screen. A clean screen is, as its name indicates, a screen in which one deselects compounds for nondesirable behavior in the SPR screen format. 13 Very simply, a target is immobilized on the SPR sensor surface, and the compounds are injected over the surface. The compounds are then annotated for their ability to bind irreversibly to the target or to the reference surface alone.
RWG confirmation results, in contrast, were reasonably challenged by SPR. This is evident by the fact that about 20% of these types of hits did not survive the SPR clean screen process. This failure rate was not due to the compounds sticking to the SPR sensor surface, but in fact it was caused by improper dissociation of the compound from the target. Figure 3 depicts an example of this type of result. The RWG assay read data of compound C17 displayed a healthy signal that was in agreement with its molecular weight and the amount of target immobilized. However, in SPR, C17 demonstrated a lack of dissociation from target A. This nonspecific binding issue was not revealed by the RWG screen format, in which binding data can be rapidly detected and quantified. Due to the lack of a buffer injection and flow system in the RWG instrumentation, additional information about compound dissociation was not obtained. Due to its flow system, SPR can dissect the binding signal by looking at the stickiness of the compound or its dissociation rate as it comes off the target, without placing extra manual steps in the screen assay.
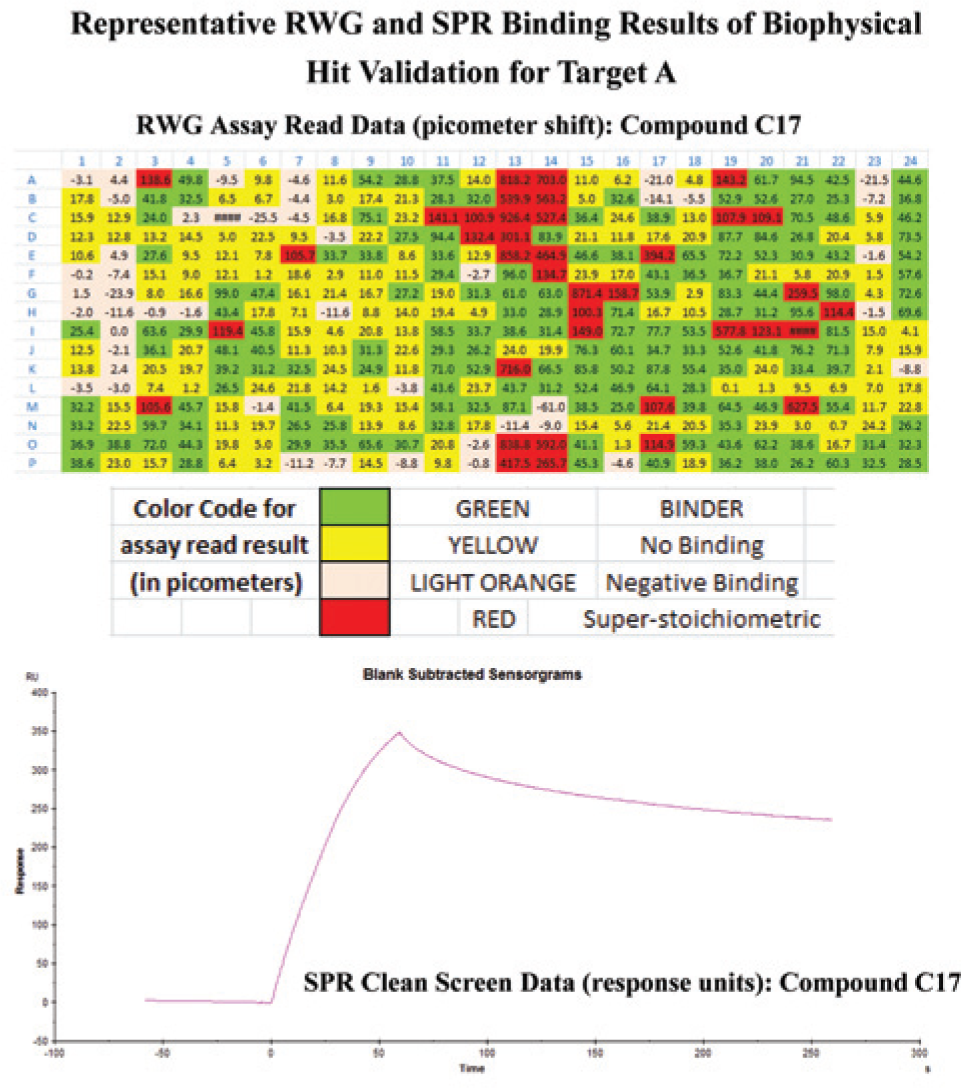
The misalignment of binding interaction data between resonant waveguide (RWG) and surface plasmon resonance (SPR) using compound C17 as an example. In RWG, C17 displayed a healthy RWG signal (a 38.9 picometer shift); however, in SPR, this compound exhibited the tendency to bind irreversibly to Target A.
The results displayed in
The SPR validation characterized the hits further and prioritized their placement for studies in X-ray and NMR, according to their binding affinity. 14 To aid in the prioritization, the SPR affinity hits were placed in categories according to such qualities that answered the following questions: did the compound behave specifically with the target, or did it bind and not dissociate in a timely manner? Were the curvature and time scale of compound association and dissociation in agreement with the affinity estimated from other approaches? Did the response reach equilibrium or saturation? If saturation was not reached, did a higher concentration series of compound help or cause problems in the SPR readout? Did the compound exhibit a dose response that was in accordance with its molecular weight? Could an affinity at equilibrium be assigned or just estimated? How did the SPR affinity measurement compare to the IC50’s or KD’s from other approaches? Did the compound show target selectivity or selectivity for a particular domain of target A?
From the 60 compounds chosen for the SPR affinity screen, approximately 40% passed; that is, for these compounds, an affinity at equilibrium could be assigned using SPR that was in accordance with previous validation data. Of the 40% hits that passed, their affinity ranges were from 0.005 to 25 uM
Biophysics Screen for Set7/9
Using this knowledge, DSF was used as the main approach for gleaning chemical starting points for the next HTS screen. Figure 4 displays one such compound found in the Set7/9 DSF screen, displaying its thermal stabilization profile. 18 The first detail to notice in this plot is that the starting intensity (blue curve) was not elevated, indicating that the compound did not cause protein aggregation. As one can see from the clear, sharp hyperbolic shape of the melting curve (orange dashed line) and the magnitude of change in fluorescence intensity, a large thermal stabilization was obtained for this scaffold, equaling 3.2 °C. At higher temperatures, as shown on the right-hand side of the curve, a decrease in SYPRO Orange fluorescence intensity was observed. As the protein succumbs to aggregation, the SYPRO Orange disengages from the hydrophobic core of the protein and naturally loses its fluorescence. It was discovered that DMSO alone also causes a shift in thermal stabilization for Set7/9; therefore, all screening data were adjusted accordingly. The DMSO thermal stabilization contribution was approximately the same for both of these types of epigenetic targets (data not shown). Figure 5 shows the corresponding SPR affinity data for this compound. The curvature of the sensograms demonstrated that the compound’s interaction with Set7/9 was specific: the compound associated and dissociated in a timely manner from the target, reaching equilibrium after approximately 25 s. According to the molecular mass calculations, the predicted response signal should be 27.8 RUs, which is in close agreement to what was obtained at saturation, 22–26 RUs, indicating that the interaction was not superstoichiometric. The binding efficiency of the immobilized target was calculated to be 80–94%, indicating the surface was active, with the target’s binding pocket readily accessible for compound interaction. The curvature of the association and dissociation phases predicted that the compound would possess a midrange micromolar affinity. This was in alignment with its calculated value of 8.8 uM.
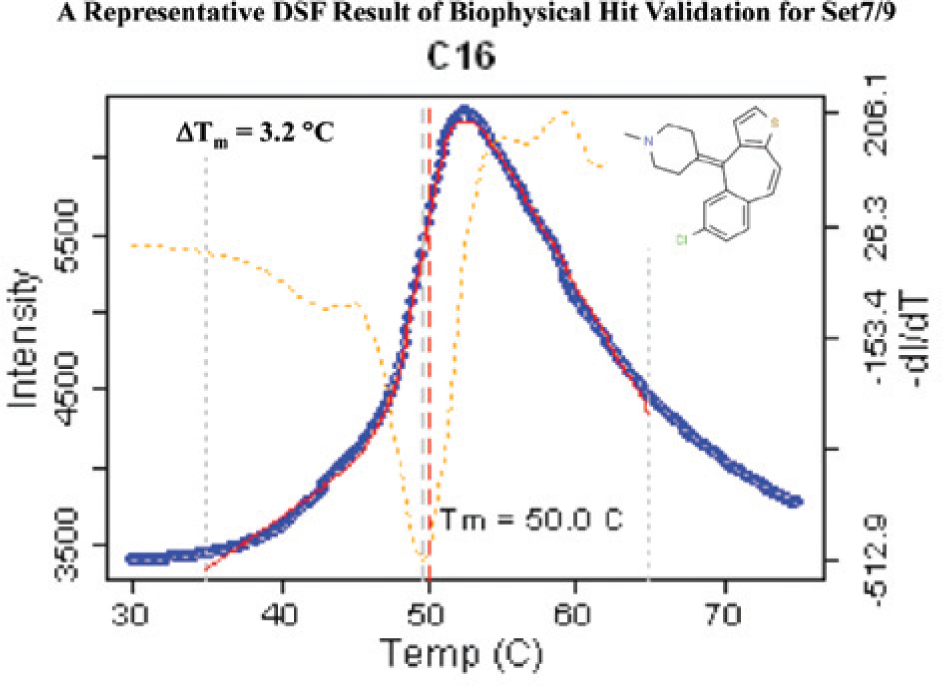
A representative differential scanning fluorimetry screening result for Set7/9. This particular heterocyclic scaffold demonstrated a 3.2 °C positive shift in thermal stabilization of Set7/9.
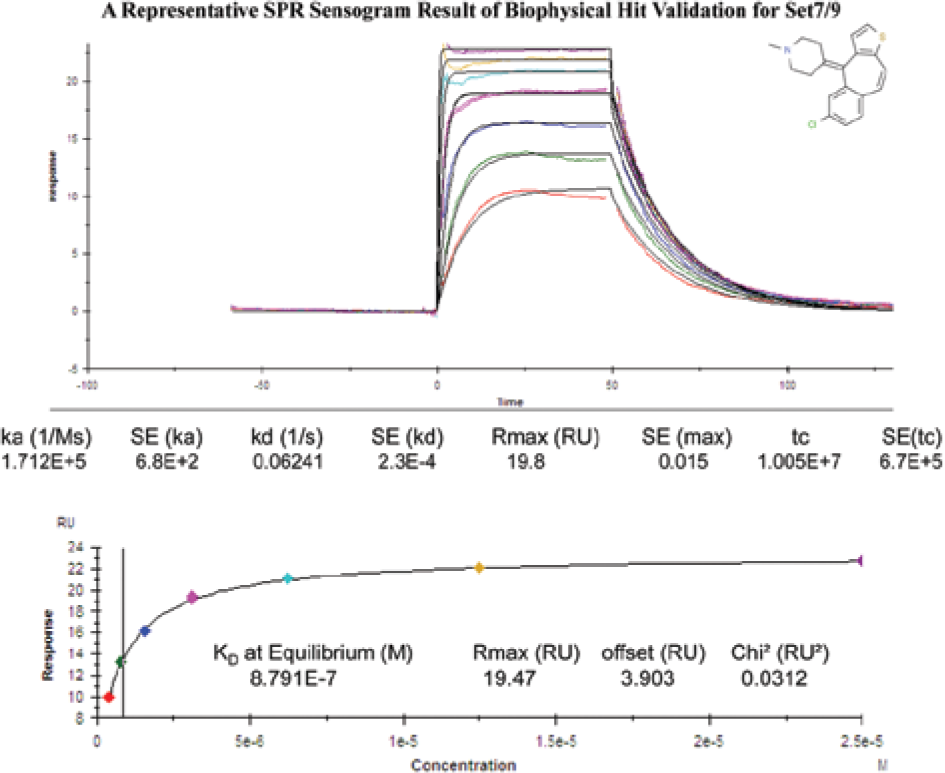
The respective dose–response curves for the differential scanning fluorimetry positive 3.2 °C hit on its interaction with Set7/9. This interaction was detected and quantitated via surface plasmon resonance.
Overall, approximately 25% of the scaffolds interrogated by the Set 7/9 HTS were confirmed DSF. Of those DSF hits, three major scaffolds were represented in 40% of the hits; thus, the most chemically attractive hits were chosen for further chemical optimization. For more details on how the DSF and SPR screens were conducted, please refer to the supplemental information.
Discussion
As shown in the various biophysics screening approaches presented, there are a number of different methods that can be implemented to validate hits garnered from HTS or any other large screening campaign. In addition, there are several ways one can place these methods to strategically obtain candidates that fulfill the necessary biochemical requirements for further lead-optimization efforts. With this collective knowledge gained from screening using biophysics, there are several points that are worthy to note and that should be considered when designing a biophysics flow chart for hit validation.
For target A, little was known about the manner in which it would interact with its binding partner; thus, a broader screening approach was used to ensure that hits could be obtained in either screening format: via target immobilization or in solution. Even between the immobilization approaches for RWG and SPR, there were differences in the content and in the type of affinity data obtained. The type of binding data from RWG was limited by the lack of an instrument flow system, without which the dissociation behavior of the inhibitor could not be monitored. Although RWG does not possess this functionality, it is highly valued for its ability to quickly determine binding interactions and assign an apparent KD. To understand the strengths and challenges for RWG and SPR further and where they diverge in their results to TR-FRET, an analysis of the KD results between RWG and SPR versus TR-FRET IC50’s was done for target A. Overall, it was observed that both RWG and SPR gave the same trend in KD affinities in comparison to TR-FRET IC50’s (data not shown). Because both technologies have the capability to confirm hits from TR-FRET, their placement in the screen approach was structured in such a way to emphasize their strengths and abilities to confirm hits in a swift manner. Both RWG and DSF technologies are relatively fast in their screening capabilities: RWG can scan a 384-well plate in 2 min (newer RWG-generation instruments are currently available that can scan 384 wells in 12 s), whereas DSF takes approximately 7 min for 384 wells; thus, they are very useful in screening expediently and culling the hits for SPR. Therefore, RWG and DSF were placed upstream of SPR in the biophysics hit-validation scheme.
Not only is throughput important to a hit validation strategy, but also the screen setup is, that is, how each compound is introduced to the target to monitor their binding interactions. Both RWG and DSF are plate-based assays in which each compound is analyzed in a separate well, preventing secondary compound effects from interfering with the ability of the next compound to interact with the target protein. This is unlike an SPR setup, which involves the serial injection of compounds over the same target surface. SPR can lend high-content data to a screen approach with its ability to assign affinity and kinetic information to the interaction between the compound and target, albeit at the cost of processing time. One of the strengths of SPR is its low protein usage and its ability to screen in a multiplex manner, that is, several targets at once. Thus, SPR was better placed downstream to RWG and DSF, where it was used to confirm the hits and profile them for their ability to interact with various targets, for example target A, its individual domains, and target B, within the same screen without requiring tremendous protein-production resources.
In every hit-validation approach, it is equally important to look at the false-negative and false-positive hits and perform an analysis as to where and how these hits were created in the biophysics confirmation process. For target A, two compound characteristics were quantified to understand the origins of false-negative and false-positive hits. For these purposes, compound aggregation and characterization of the binding interaction were tracked. Using turbidometry, it was revealed that most compounds did not aggregate in the screen-assay buffer; hence, compound aggregation was not the main source of false positives in this validation strategy. In comparing the DSF results to RWG, it was discovered that RWG rescued 25% of DSF nonhits. Hence, DSF can generate false negatives. Thus, it is recommended to use DSF as an opportunistic approach to quickly find chemical starting points for optimization. By using the SPR clean screen, the compounds that passed either RWG or DSF were profiled for their binding stoichiometry, their ability to dissociate from the target, and their “stickiness” to the SPR sensor surface. Of those compounds that passed RWG and DSF but failed in the SPR clean screen, the principle reason for failure was their improper dissociation from the target, a parameter that is unquantifiable using RWG or DSF. Therefore, the follow-up of RWG or DSF using an SPR clean screen quickly confirmed the truly positive hits from these technologies.
In light of these biophysics validation results, it was theorized that a more streamlined approach in biophysics for another epigenetic target could be taken without a loss of hit material. In reference to target A, the DSF itself contributed robustly to the lead-finding efforts by promoting approximately 40% of the original HTS hits to the final biophysics hit list for medicinal-chemistry consideration. In the case of Set7/9, this streamlined approach was also shown to be robust in its output, generating scaffolds worthy of chemical optimization.
In summary, there are several biophysical approaches that can be used to flush out the real hits from an HTS campaign and prioritize compounds for further chemical optimization. As with every screen campaign and the validation of hits, one should consider what biophysical data are truly needed to promote a candidate to the next level of resource-demanding work. For example, if the project team wishes to have kinetic information on the hits at an early stage in drug discovery, then an SPR clean screen and SPR binding assay are essential to processing and profiling a large number of hits to prioritize their placement in an SPR kinetics assay. The amount of time needed for just the SPR segment of the validation process should be challenged with a question such as: what sort of data does the project team really need to choose a compound for chemical optimization? This question can be addressed by designing a biophysics flowchart that is tactful in its approach, placing certain technologies at the forefront and others at the characterization and optimization stages of drug discovery. It is during the validation phase of lead finding, after which a few high-quality scaffolds have been chosen, that more resources could be dedicated to profiling compounds by their on and off rates or their binding energies to fine-tune their efficacy.
Footnotes
Acknowledgements
The authors would like to thank the members of the Center of Proteomic Chemistry, the Medicinal Chemists, and the Disease Areas for their support throughout the various projects in lending their time for discussion and their technical support.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.