
Abstract
In the nuclei of hepatocytes, glucokinase regulatory protein (GKRP) modulates the activity of glucokinase (GK), a key regulator of glucose homeostasis. Currently, direct activators of GK (GKAs) are in development for the treatment of type 2 diabetes. However, this approach is generally associated with a risk of hypoglycemia. To mitigate such risk, we target the GKRP regulation, which indirectly restores GK activity. Here we describe a screening strategy to look specifically for GKRP modulators, in addition to traditional GKAs. Two high-throughput screening campaigns were performed with our compound libraries using a luminescence assay format, one with GK alone and the other with a GK/GKRP complex in the presence of sorbitol-6-phosphate (S6P). By a subtraction method in the hit triage process of these campaigns, we discovered two close analogs that bind GKRP specifically with sub-µM potency to a site distinct from where fructose-1-phosphate binds. These small molecules are first-in-class allosteric modulators of the GK/GKRP interaction and are fully active even in the presence of S6P. Activation of GK by this particular mechanism, without altering the enzymatic profile, represents a novel pharmacologic modality of intervention in the GK/GKRP pathway.
Keywords
Introduction
Glucokinase (GK) is an important sensor of blood glucose levels and plays a key role in glucose homeostasis.1,2 It belongs to the hexokinase family and is mainly expressed in hepatocytes, pancreatic β-cells, and glucose-sensitive cells of both the gut and the brain. 2 The unique characteristics of GK include a Km (S0.5) at physiological concentration (~8 mM) of glucose, a positive cooperativity (Hill coefficient at ~1.7), and no product inhibition.1–3 As the initial step in the synthesis of glycogen, GK catalyzes the phosphorylation of glucose to glucose-6-phosphate (G6P; Fig. 1A ), which in turn lowers blood glucose levels. Activating and inactivating mutations of GK that cause hypo- and hyperglycemic syndromes in humans have been well documented.1–3 Thus, a wide interest has been triggered in developing direct activators of GK (GKAs) for the treatment of type 2 diabetes.4–7 GKAs bind to an allosteric pocket on active GK, 8 which reduces the S0.5 of glucose and increases the maximum velocity (Vmax) of GK ( Fig. 1B ).4–8 As a consequence of this hyperbolic activity profile, increased GK catalytic activity can cause a more substantial decrease of blood glucose, independent of starting blood glucose levels. Therefore, the risk of hypoglycemia has turned out to be a significant obstacle in the development of direct GKAs. Some preclinical studies have indicated that partial activators of GK could reduce the hypoglycemic risk while maintaining efficacy. 7
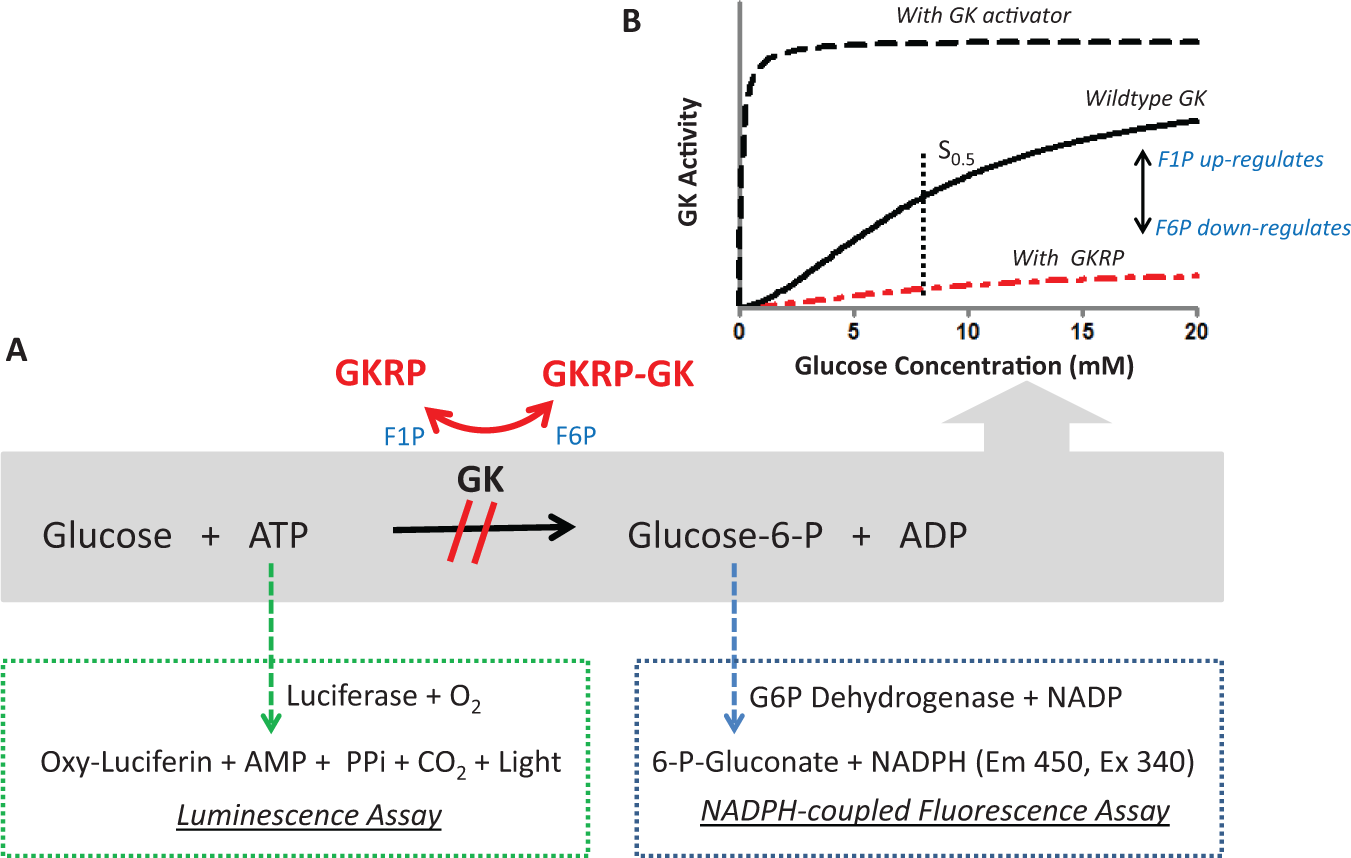
Glucokinase (GK) activity and the effect of glucokinase regulatory protein (GKRP) regulation. GK converts glucose to glucose-6-phosphate (G6P) with the consumption of adenosine triphosphate. In the presence of fructose-6-phosphate (or S6P), GKRP binds GK more tightly, which results in the inhibition of GK, whereas fructose-1-phosphate weakens the GK-GKRP interaction and releases GK to restore its activity. Current GKAs bind to an allosteric site on GK, causing an increase in Vmax and a decrease in S0.5 of glucose. Two different assay formats were used to monitor GK activity and test for GKRP modulators: the luminescence (green box) and the NADPH-coupled fluorescence (blue box).
In hepatocytes, GK is primarily regulated by glucokinase regulatory protein (GKRP) via a reversible inhibitory mechanism in the nucleus, which depends on the fed state.9–14 Between meals, GKRP binds to GK, forming a GK/GKRP complex that sequesters GK temporarily in the nucleus. Upon postprandial glucose increase, GK rapidly dissociates from GKRP and translocates into the cytoplasm. In addition to glucose, fructose-6-phosphate (F6P), sorbitol-6-phosphate (S6P; an F6P analog), and fructose-1-phosphate (F1P) modulate the formation of the GK/GKRP complex ( Fig. 1A ). Both F6P and S6P enhance the formation of the GK/GKRP complex, whereas glucose and F1P promote the dissociation of GK from GKRP.8,9,12–14 GKRP binds to and inhibits GK in an allosteric manner that is competitive with respect to glucose, F6P and F1P.9,12–14 It is conceivable that targeting GKRP with a small molecule that mimics the effect of F1P would result in the translocation of GK into the cytosol without altering its kinetic profile, thus lowering blood glucose levels with minimal risk of hypoglycemia ( Fig. 1B ). The identification of a common GKRP variant (P446L) by genome-wide association studies supports this hypothesis. This variant’s effect is limited to hindering F6P-mediated GK/GKRP complex formation at physiological F6P concentration, resulting in a 0.06 mM (1.1 mg/dL) decrease in fasting plasma glucose levels.15,16
Targeting intracellular protein-protein interactions (PPIs) with a small molecule has generally been considered a challenge because the binding interfaces are often extensive. 17 A crystal structure of Xenopus GK/GKRP complex in the presence of F6P has recently been solved, and it reveals a hydrophobic interface of about 1913 Å 2 . 18 Interestingly, the F6P binding pocket on GKRP is distant from the actual PPI interface. The crystal structure of human GKRP in the presumably inactive form with F1P has also recently been solved. 19 These structures show that F1P (as well as F6P 18 ) binds a polar pocket buried deep within GKRP, which is probably intractable for small-molecule development because of high polarity. Nevertheless, here we present our effort in developing high-throughput screening (HTS) campaigns and hit triage strategies for identifying small molecules that will inhibit or weaken the GK/GKRP interaction, in addition to the traditional direct GKAs. This has led to the discovery of the first small-molecule GKRP modulator that binds GKRP in a novel allosteric pocket that is distinct from the sugar phosphates’ pocket and weakens the GK/GKRP complex even in the presence of S6P. This novel mechanism of GK activation provides an opportunity to circumvent the hypoglycemic risk, potentially leading to a viable treatment for type 2 diabetes.
Materials and Methods
GK Luminescence Assay
The enzymatic activity of GK was analyzed by monitoring the levels of adenosine triphosphate (ATP) in the GK reaction with the EasyLite-Kinase kit from PerkinElmer (Waltham, MA), which is based on luciferase luminescence detection. Within the linear ATP concentration range that is determined by the EasyLite kit, the GK activity is inversely proportional to the ATP concentrations that remain in the GK reaction. Our GK assay was performed in 50 mM Tris, pH 7.5, 4 mM MgCl2, 8% (v/v) DMSO, 8 mM DTT, 0.02% (w/v) bovine serum albumin (BSA), 0.002% (w/v) Brij-35 in either a Costar 96-well or 384-well plate at room temperature. The luminescence intensity was measured with an Analyst GT reader (Molecular Devices, Sunnyvale, CA; Fig. 1A ).
For the ATP Km determination, 8 concentrations of ATP from twofold serial dilutions ranging from 0.31 to 40 µM in assay buffer were incubated with 5 mM glucose and various concentrations of GK (0, 2, 4, 6, 8, and 100 nM). For the glucose S0.5 determination, eight concentrations of glucose from twofold serial dilutions ranging from 0.31 to 40 mM in assay buffer were incubated with 10 µM ATP and various concentrations of GK (0, 2, 4, 6, 8, and 100 nM). In the mechanism-of-action study of the Roche GKA (RO-28-1675), eight concentrations of compound from threefold serial dilutions in DMSO were mixed with 4 nM GK and subsequently reacted with 10 µM ATP and eight concentrations of glucose from twofold serial dilutions ranging from 0.31 to 40 mM. In all of the above assays, the reaction was stopped by the PE EasyLite kit at 0, 5, 10, 15, 30, and 60 min. The initial rate of each sample was obtained from a linear regression of the time course. The apparent S0.5 of glucose was calculated by fitting the initial rate data with the Michaelis-Menten equation using XLfit (ID Business Solutions Ltd., Surrey, UK).
In the GK primary screen and the follow-up single-point duplicate confirmation, 4 nM GK was reacted with 10 µM ATP and 5 mM glucose in the presence of 20 µM compound for 60 min. In the dose-response EC50 assay, 10 concentrations of compound from threefold serial dilutions in DMSO were mixed with GK and then were reacted with 10 µM ATP and 5 mM glucose for 60 min. Subsequently, the luminescence intensity of each test well was measured and was normalized to percentage of positive control (POC) using the following formula:

Positive control wells contained GK, ATP, and glucose, whereas negative control wells contained only ATP and glucose. Both positive and negative control wells had the same amount of DMSO. The EC50 values were calculated by fitting POC values at each concentration with a four-parameter nonlinear regression equation using Activity-Base software (ID Business Solutions Ltd., Surrey, UK).
GK/GKRP Luminescence Assay
For the IC50 determination of GKRP in the presence of F6P or S6P, 10 concentrations of GKRP from twofold serial dilutions in the above GK assay buffer were preincubated with 4 nM GK and eight concentrations of F6P or S6P from threefold serial dilutions for 10 min. The concentrations of GKRP, F6P, and S6P ranged from 0.78 to 400 nM, 0.82 to 1800 µM, and 0.069 to 150 µM, respectively. To initiate the reaction, 10 µM ATP and 5 mM glucose were added, and the luminescence intensity was measured after 60 min. The Kd value of F6P or S6P was determined by fitting the IC50 values of GKRP at each F6P or S6P concentration with a hyperbolic equation using XLfit (
In the GK/GKRP primary screen and the follow-up single-point triplicate confirmation, 4 nM GK and 50 nM GKRP were preincubated for 10 min to form a GK/GKRP complex. This complex was then mixed with 20 µM compound and subsequently reacted with 10 µM ATP and 5 mM glucose for 60 min. The raw data were normalized to POC using the above formula. However, in this case, positive control wells contained the GK/GKRP complex, ATP, and glucose, whereas negative control wells contained only ATP and glucose.
GK and GK/GKRP NADPH-Coupled Fluorescence Assay
Traditionally, the activity of GK is determined spectrophotometrically by an NADPH-coupled assay system with G6P dehydrogenase ( Fig. 1A ). 1 As β-NADPH has intrinsic fluorescence and the signal-to-background (S/B) ratio is superior compared with the absorbance readout, our GK and GK/GKRP NADPH-coupled assays were established as fluorescence readouts using the same assay buffers as the above described luminescence assays.
The ATP Km and glucose S0.5 determinations adopted the same reaction conditions in the NADPH-coupled fluorescence assays as those in the luminescence assays, except that the ATP concentration range was from 0.063 to 8 mM. In the follow-up single-point and dose-response EC50 GK and GK/GKRP NADPH-coupled fluorescence assays, 240 µM ATP and either 5 or 20 mM glucose were used. In all of the above assays, NADPH was generated in the reaction mixture by the addition of 1 mM β-NADP and 0.02 U/µL G6P dehydrogenase from Leuconostoc mesenteroides in 0.2 M Tris, pH 9.2, 8% (v/v) DMSO for 5 min. Subsequently, the fluorescence intensity of each well was measured with a Safire II reader (Tecan, Männedorf, Switzerland) with excitation and emission wavelengths of 340 and 450 nm, respectively. In the NADPH-coupled fluorescence assay, the POC was calculated based on the following formula:

Surface Plasmon Resonance Spectroscopy
Surface plasmon resonance (SPR) experiments were performed on a Biacore T200 instrument (GE Healthcare) at 25 °C. To imitate the conditions used in the GK/GKRP enzymatic assays, the SPR buffer consisting of 50 mM Tris, pH 7.5, 4 mM MgCl2, 150 mM NaCl, 0.002% Brij-35 (w/v), 20 mM D-(+)-glucose, 50 µM S6P, 5 mM DTT, 5% (v/v) DMSO was used for the initial hit assessment. Note that NaCl was added to the buffer to minimize potential electrostatic interactions with the carboxymethyl dextran on the biosensor surface. Biotinylated GKRP and GK at 100 µg/mL were captured onto separate flow cells at a density of ~4700 and ~3500 RU, respectively. Remaining biotin sites on SA were blocked with biotin. S6P was diluted to 10 µM from 50 mM stock (in water) and serially diluted (twofold) 10 times. Compounds were diluted to 5 µM from 10 mM stock (in DMSO) and serially diluted (twofold) 10 times. At a flow rate of 70 µL/min, S6P and the compounds were injected over GKRP and GK surfaces for 60 s, and dissociation was observed for 60 to 240 s depending on the off rate ( Fig. 2 ). The effect of S6P on AMG-6861 binding was investigated by performing the SPR measurements in the absence of S6P. The raw data were processed using Scrubber software (BioLogics), and the data were kinetically fit to a 1:1 binding model, which included a mass transfer limitation term. Standard deviations were based on at least two separate measurements.
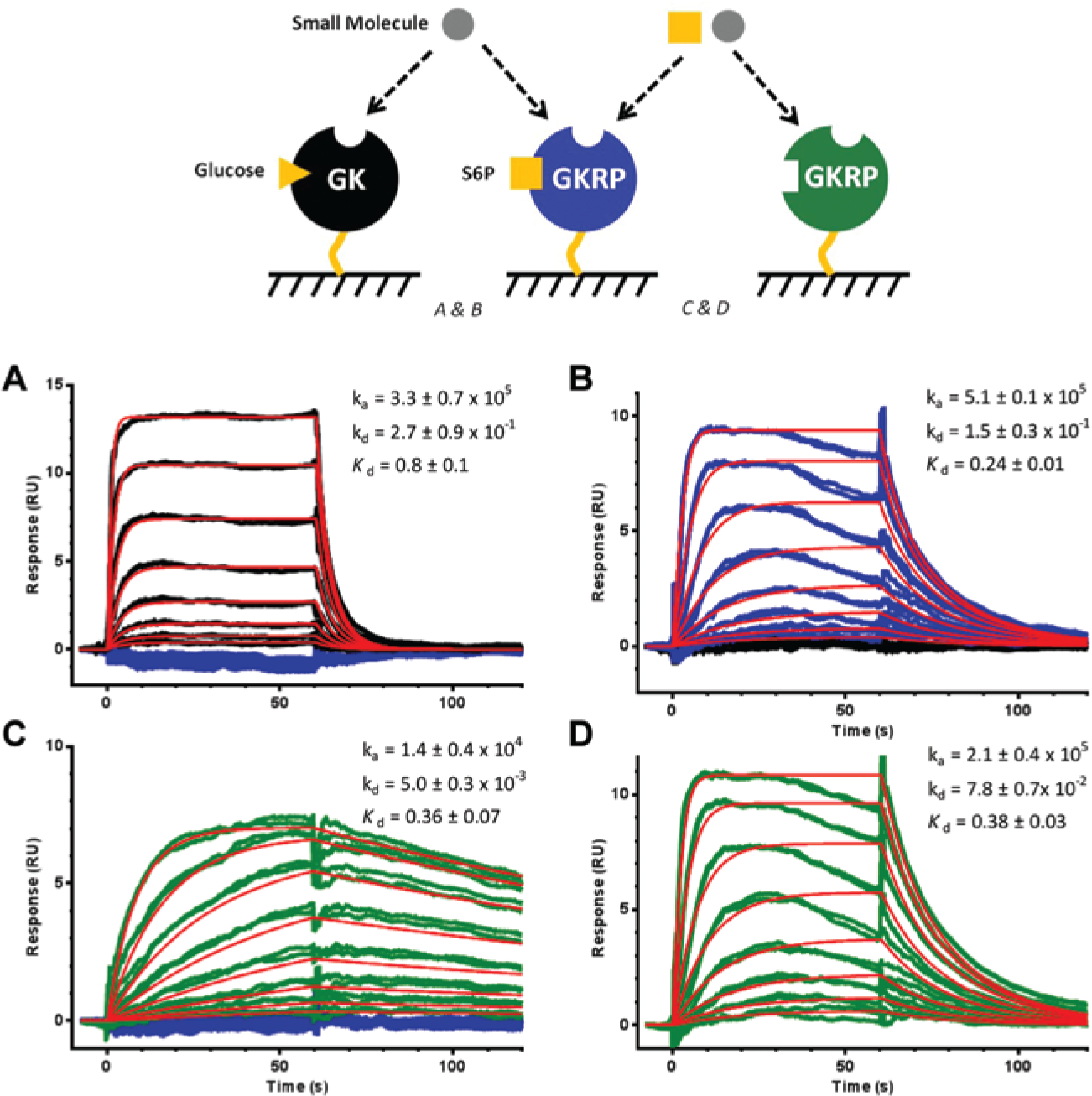
Small-molecule hit assessment by surface plasmon resonance spectroscopy. Analytes at 10 different concentrations in duplicate ([compound]: 5, 2.5, 1.25,…,0.01 µM and [sorbitol-6-phosphate {S6P}]: 10, 5, 2.5,…, 0.02 µM) were injected over the biosensor surface with high levels of immobilized glucokinase (GK) and glucokinase regulatory protein (GKRP). (
The GK/GKRP interaction in the absence and presence of S6P was measured by capturing biotinylated GK onto an SA chip at ~180 RU. GKRP (no biotin) was diluted to a top concentration of 1 µM and serially diluted (twofold) 10 times in the Tris-based buffer (no glucose, 2% DMSO, ±50 µM S6P). GKRP was injected for 2 min, and its dissociation from GK was observed for 10 min at a flow rate of 50 µL/min. The raw data were processed and binding parameters were established as described for the small molecules. To study the effect of AMG-6861 on the preformed GK/GKRP complex, the following steps were performed: (1) the GK/GKRP complex was allowed to form on the biosensor by injecting 1 µM GKRP (no biotin) for 2 min across the GK biosensor surface (in the absence and presence of S6P), and (2) during the GK/GKRP dissociation phase, either buffer, S6P (50 µM), RO-28-1674 (5 µM), AMG-6861 (5 µM), or a mixture of S6P (50 µM) and AMG-6861 (5 µM) was injected for 10 min.
Details regarding the expression and purification of GK and GKRP, SPR immobilization, differential scanning fluorimetry (DSF) and isothermal titration calorimetry (ITC) studies, as well as chemical characterization of HTS hits, are provided in the supplemental material.
Results and Discussion
Development, Optimization, and Validation of GK and GK/GKRP Assays in Luminescence and Fluorescence Formats
Traditionally, the activity of GK is analyzed in a spectrophotometric format, in which the formation of NADPH via G6P dehydrogenase coupling is monitored ( Fig. 1A ). To minimize compound interference in an HTS setting, we explored the feasibility of performing the GK assay in a luminescence format. The PerkinElmer EasyLite Luminescence kit is an ATP-monitoring system based on firefly luciferase for the evaluation of protein kinase activity. We have used this kit successfully in HTS campaigns and the subsequent lead optimization efforts for two protein kinases.
During the course of the luminescence assay development for GK, we optimized the buffer components and assay conditions, including the initial rate of reaction and the S/B ratio. We found that DTT, BSA, and Brij-35 significantly enhanced the activity of GK (data not shown). The apparent Km of ATP and S0.5 of glucose for the GK was determined to be 13 µM and 8.0 mM, respectively, in the optimized buffer (
Grimsby et al.
4
have demonstrated the ability of RO-28-1675 to alter both the S0.5 and maximum velocity (Vmax) of GK. We reproduced these observations in our GK luminescence assay. The data (
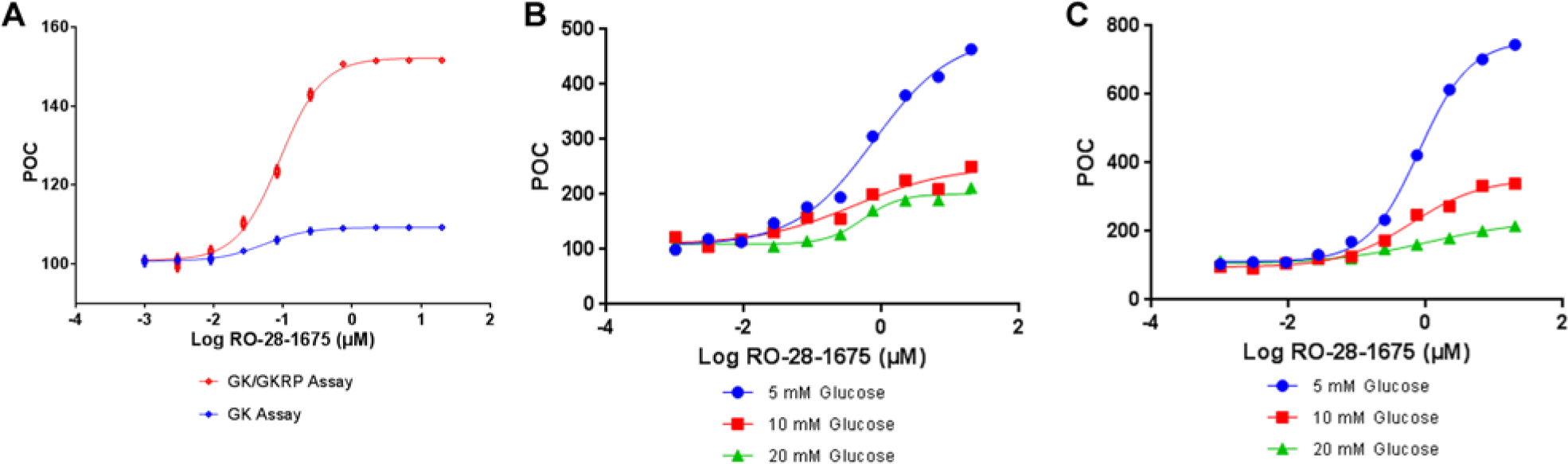
Studies of direct activators of glucokinase (GK) RO-28-1675. (
The next step was to optimize the concentration of GKRP in the luminescence assay. Physiologically, GKRP is in molar excess of the amount of GK in the liver.13,14 GKRP binds to GK, forming an inactive complex in the nucleus between meals. To maximize the sensitivity of the GK/GKRP assay for GK activation, the GKRP concentration was set at that which gives 50% GK inhibition. It is known that both F6P and S6P promote GKRP’s inhibitory effect.1,2 Thus, we first examined the IC50 values of GKRP in presence of either F6P or S6P (
In the NADPH-coupled assay (
Fig. 1A
), the final reaction product NADPH can be detected in either absorbance or fluorescence mode. We compared these two modes with an NADPH standard curve, a time course of G6P dehydrogenase, and a titration of GK. The data demonstrated that the fluorescence detection mode has a superior performance compared with the absorbance detection mode in terms of sensitivity and S/B ratio (
Discovery of GK/GKRP Modulator through GK and GK/GKRP Primary Screens and Hit Triage
The GK and GK/GKRP primary screens were performed independently to distinguish direct GKAs from potential modulators of the GK/GKRP interaction in our general compound library ( Fig. 4 ). 248,000 and 280,000 compounds were screened against GK and GK/GKRP in luminescence assay at 20 µM compound concentration, respectively, because the library was expanded by the time of GK/GKRP primary screen.
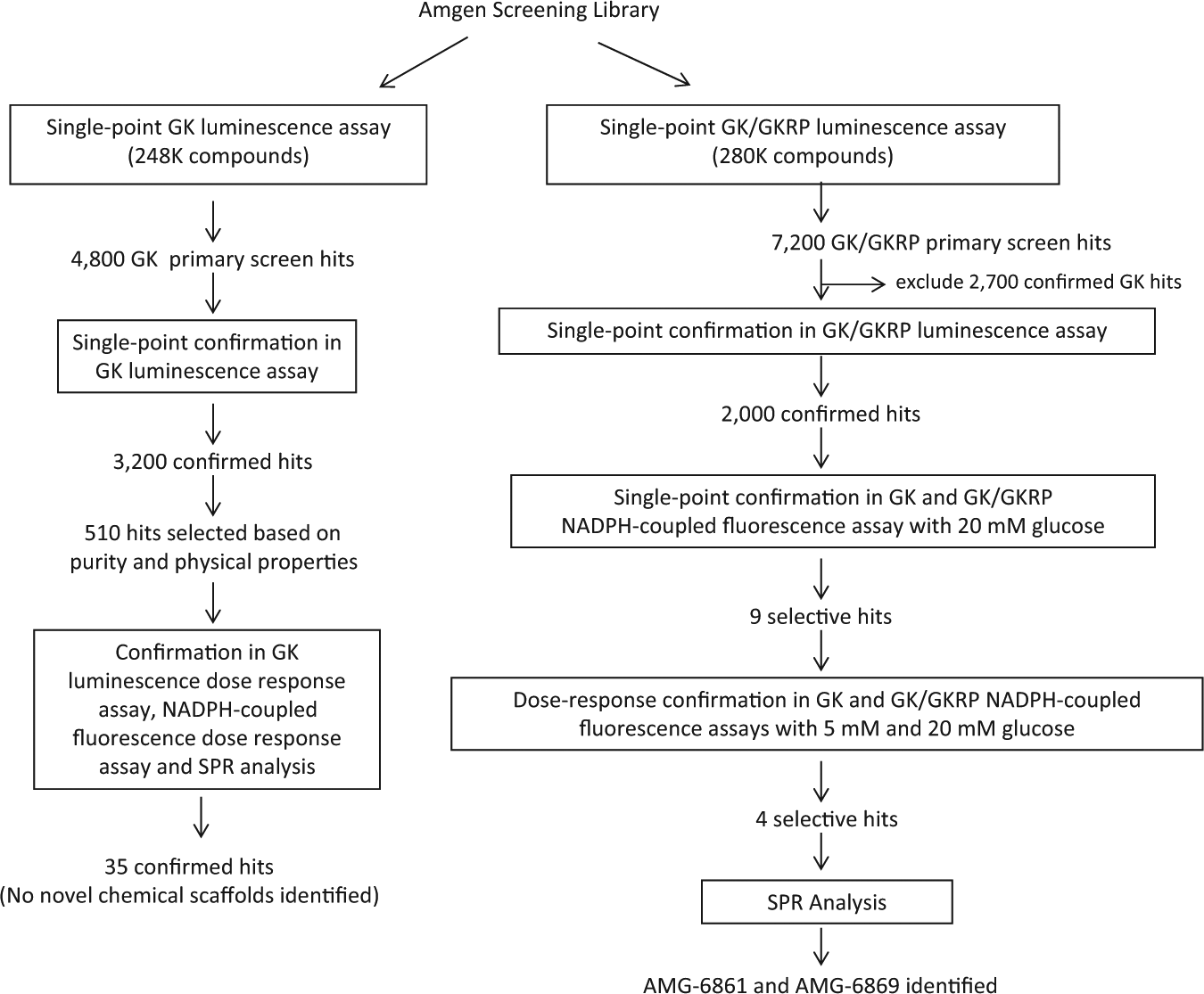
The flow scheme of glucokinase and glucokinase regulatory protein primary high-throughput screens and hit triage.
The average Z′ value for the GK primary screen was 0.94, and the hit rate was ~1.9% based on the statistical analysis of the whole screening data set. Among the 4800 hits, only 3200 compounds were confirmed with the same POC values in single-point duplicate GK luminescence assay at 20 µM compound concentration ( Fig. 4 ). Based on purity and physical properties, 510 of the confirmed hits were further assessed in GK luminescence, NADPH-coupled fluorescence dose-response assays, and direct binding to GK by SPR spectroscopy. Of 35 direct GKAs that were confirmed, several compounds were successfully co-crystallized with GK and were found to occupy the same allosteric pocket as other known GKAs. 20
The average Z′ value for the GK/GKRP primary screen was 0.81. Based on statistical analysis, the hit selection criterion was set at POC >133, resulting in a hit rate of ~2.6%. Our overall strategy for finding GKRP-specific hits was to remove hits that can activate GK in the absence of GKRP from the GK/GKRP primary screen hit list. Among the 7200 preliminary GKRP primary screen hits, 2700 compounds that overlapped with GK primary screen hits were excluded from further follow-up. For the remaining hits, 2000 compounds were confirmed with the same POC values in single-point triplicate GK/GKRP luminescence assay at 20 µM compound concentration ( Fig. 4 ).
To enhance the physiological relevance of the third triage step, we exploited the tactic of screening GK/GKRP hits at a higher glucose concentration to mimic diabetic conditions. Normal human blood glucose concentration ranges from 5.6 to 6.9 mM (100–125 mg/dL) depending on the fed state. However in diabetic patients, the postprandial blood glucose can reach more than 11 mM (200 mg/dL). 21 We therefore studied RO-28-1675 ( Fig. 3B , C ), along with seven other direct GKAs (either benchmarks or internal GK screening hits; data not shown), at variable glucose concentrations. For this step, the reaction time for both the GK and GK/GKRP assays with 10 and 20 mM glucose was adjusted to 60 and 30 min, respectively, to confirm that the GK activity was within a linear range. As illustrated in Figure 3B and 3C , RO-28-1675 maintained a consistent potency across 5, 10, and 20 mM glucose concentration in both assays. However, some benchmark GKAs and internal GK hits lost their activation potency at high glucose concentrations. More significantly, we observed a general trend for all GKAs, including RO-28-1675, in which their efficacy (maximum response) demonstrated an inverse proportional relationship to the glucose concentrations. These results suggested that the 20 mM glucose assay condition would improve the chance of discovering GKRP-specific compounds because glucose at this concentration highly suppresses the GK activation by the direct GKAs.
Based on the above proof-of-concept experiment, we analyzed all 2000 confirmed GK/GKRP primary screening hits in the GK and GK/GKRP NADPH-coupled fluorescence assays with 20 mM glucose at 20 µM compound concentration. In combination with adopting the same hit selection criteria as in the GK/GKRP primary screen (i.e., POC >130 in GK/GKRP assay at 20 mM glucose), we set POC <120 in the GK assay at 20 mM glucose to exclude any direct GKAs. Note that in the presence of higher glucose, the threshold for removing GK-only hits had to be lowered because of the suppression of GKA efficacy. We defined that hits were GKRP-specific modulators if (1) they preserved the ability to activate the GK in the GK/GKRP assay and (2) they lacked any direct GK activation in the GK assay. Surprisingly, only 9 of the 2000 confirmed GK/GKRP primary screening hits met these criteria. The subsequent dose-response assays narrowed the list to four compounds, of which two were close analogs. The follow-up SPR analysis revealed that only these analogs were selective for GKRP, the best one (AMG-6861) with the chemical structure depicted in Figure 5A and an EC50 value of 0.21 µM ( Fig. 5B ).
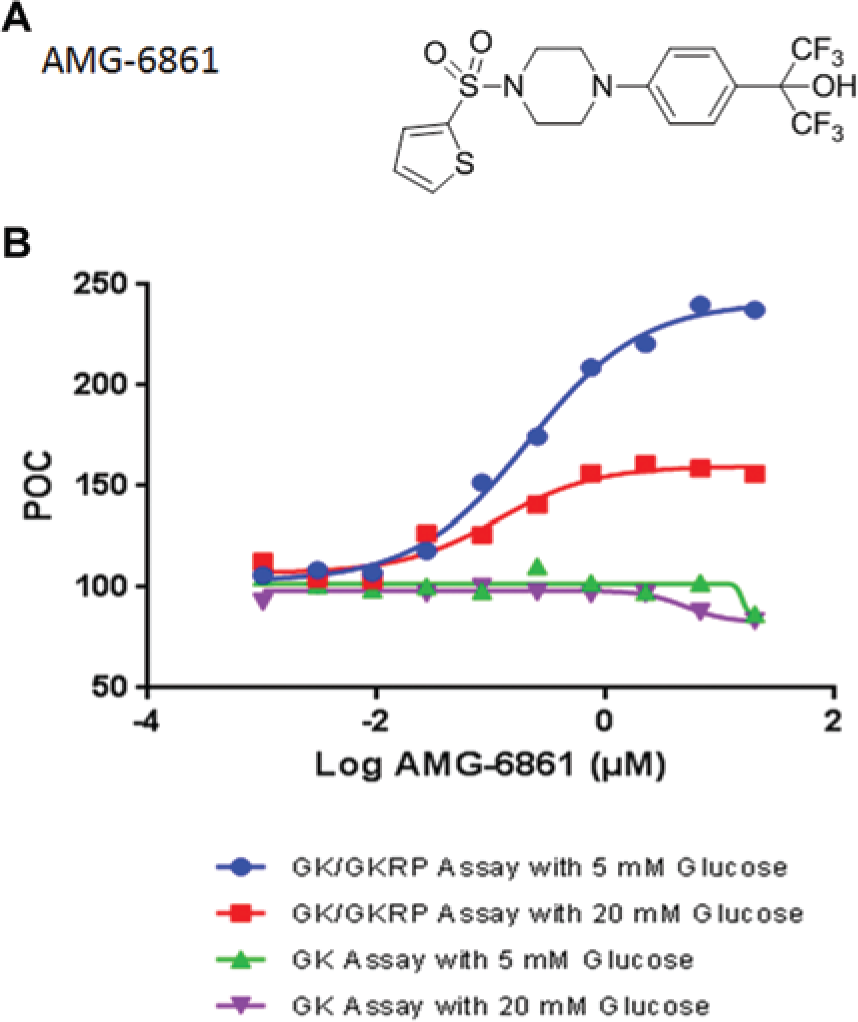
The chemical structure and enzymatic activities of the high-throughput screening hit. (
Characterization of HTS Hits by SPR, DSF, and ITC
To eliminate any false-positives due to the enzymatic detection formats ( Fig. 1A ), the screening hits were assessed for direct binding to GK and GKRP by SPR spectroscopy ( Fig. 2A , B ). In Figure 2A , the RO-28-1675 bound GK with a Kd value of 0.8 ± 0.1 µM, whereas it did not show any binding to GKRP. Only two of the hits demonstrated direct binding to GKRP and not to GK (data for AMG-6861 shown in Fig. 2B ). AMG-6861 had a Kd value of 0.24 ± 0.01 µM for GKRP and exhibited fast binding kinetics (ka = 5.1 ± 0.1 × 105 1/[Ms] and kd = 1.5 ± 0.3 × 10−1 1/s), which is similar to the binding kinetics profile of RO-28-1675. The measured Kd values for RO-28-1675 and AMG-6861 were within the range of the measured EC50 values in the GK/GKRP biochemical assay, which confirmed the correlation between protein binding and effect on GK activity.
The binding of AMG-6861 was also analyzed in the absence of S6P to determine if the hit’s interaction with GKRP was S6P dependent (
Fig. 2C
,
D
). Compared with AMG-6861, S6P binds GKRP with significantly slower binding kinetics (
Fig. 2C
) yet quite similar binding affinity (Kd = 0.36 ± 0.07 µM). The binding kinetics were consistent with the sugar binding pocket being buried deep inside the GKRP, as published recently.18,19 With a Kd value of 0.38 ± 0.03 µM and similar kinetic behavior, the binding of AMG-6861 to GKRP was largely unchanged in the absence of S6P (
Fig. 2D
). The data therefore suggested that AMG-6861 binds GKRP in a pocket separate from the sugar phosphate binding pocket; presumably, this pocket is more accessible based on the faster binding kinetics. To support the finding of another pocket on GKRP, DSF protein thermostability experiments were performed with GKRP (
AMG-6861 was also tested by ITC to confirm the binding stoichiometry and affinity (
AMG-6861 Is an Allosteric Modulator of the GK/GKRP Interaction
To investigate the effect of AMG-6861 on the GK/GKRP complex, SPR binding studies were conducted. First the GK/GKRP complex was measured in the presence and absence of S6P ( Fig. 6A , B ). S6P enhanced the GK/GKRP association and slowed down its dissociation, improving overall binding affinity 10-fold to 15 ± 3 nM. This Kd value is within the range reported with rat GKRP and human GK. 22 Next, the GK/GKRP complex was allowed to form on the biosensor in the presence and absence of S6P ( Fig. 6C , D , respectively). During the dissociation step, S6P, RO-28-1675, AMG-6861, or a mixture of S6P and AMG-6861 was injected. Because of the low density of GK/GKRP on the biosensor, the effect of the small molecules on the protein complex was observed rather than small-molecule binding to the proteins. In other words, S6P strengthened the GK/GKRP complex, whereas AMG-6861 weakened the complex quite dramatically considering the compound’s ~25-fold lower affinity compared with the GK/GKRP complex. More interestingly, for the mixture of S6P and AMG-6861, AMG-6861 seemed to abolish the strengthening effect of S6P even when the S6P concentration was tenfold higher than the compound concentration. This suggests another allosteric site on GKRP that can modulate GK/GKRP interaction. As expected, no effect was observed with RO-28-1675 because it binds GK only in the active, closed conformation with glucose present. 20
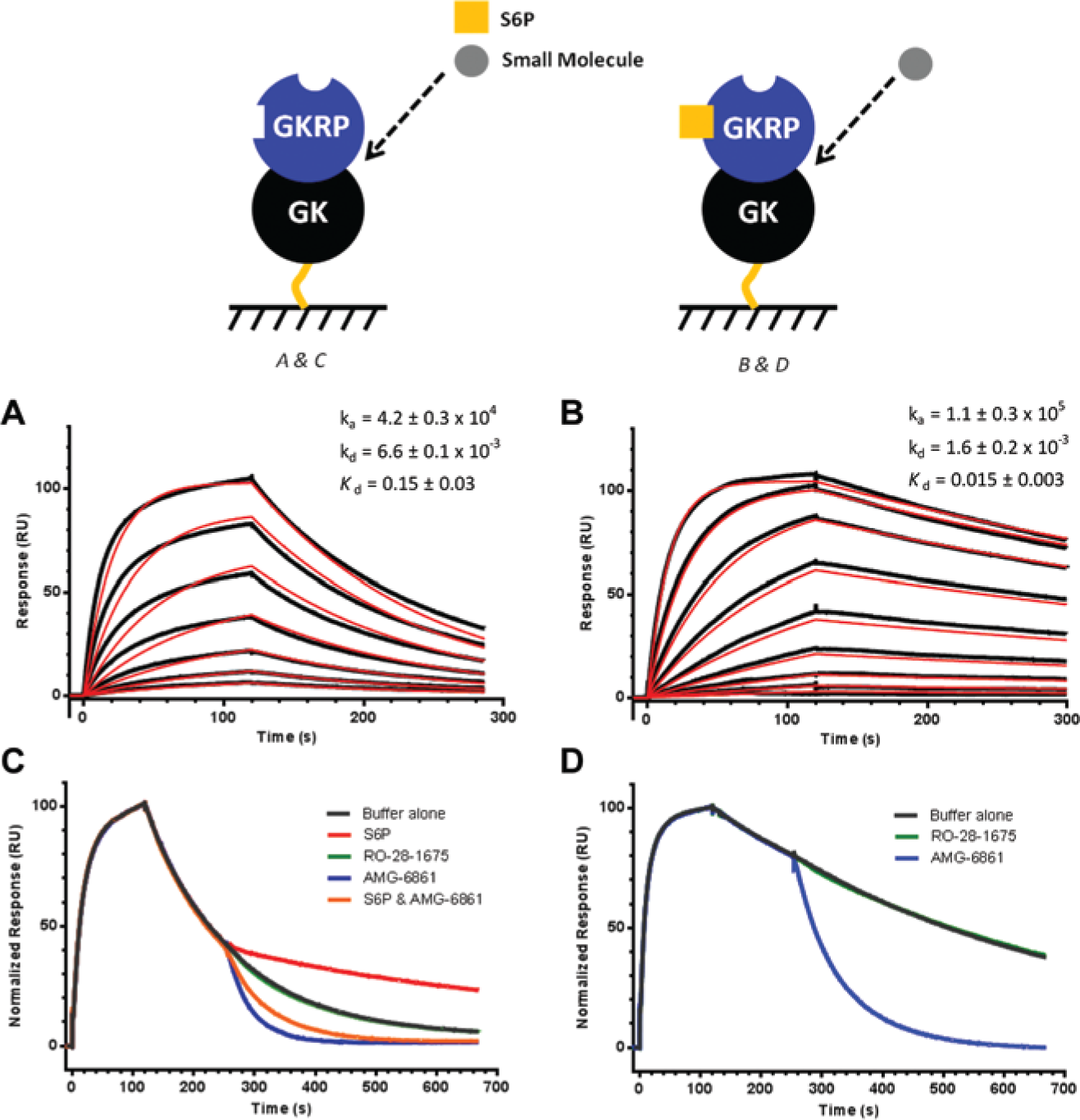
Glucokinase (GK)–glucokinase regulatory protein (GKRP) interaction and the weakening effect of AMG-6861 on the interaction. GKRP was injected over a biosensor surface immobilized with low levels of GK at 10 different concentrations in duplicate (1, 0.5, . . ., 0.002 µM) in the absence (
Through innovative design of the HTS campaigns and hit triage processes, we have discovered the first synthetic small-molecule GKRP binder that weakens the GK/GKRP complex. The two different assay formats complemented each other well. Compared with the traditional GK spectrophotometric assays, our luminescence and NADPH-coupled fluorescence assay formats are superior in terms of sensitivity and suppression of compound interference.
The GKRP modulator that we discovered binds GKRP in a pocket that is distinct from S6P, and it can still bind GKRP in the presence of S6P. Recently published structures18,19 show that F1P and F6P appear to bind within the same pocket on GKRP, yet they exhibit opposite effects on the GK/GKRP complex. Our GKRP modulator seems to bind a “master” allosteric pocket that completely abolishes the effect of the sugar phosphate on the GK/GKRP complex. Presumably, the GKRP modulator works by stabilizing the unbound, inactive form of GKRP. More interestingly from a diabetic perspective, the binding of a GKRP modulator, unlike the GKAs, is completely independent of the glucose concentration. By weakening the GK/GKRP interaction in the liver independently of the sugar phosphate and glucose status, GK should be released into the cytosol to lower glucose concentration without altering GK’s enzymatic profile. This represents a novel approach in terms of avoiding the hypoglycemia that is normally associated with the development of traditional GKAs for type 2 diabetes.
Many GKAs were filtered out in the process of finding the GKRP selective modulators. This points to the observation, not surprisingly, that it is significantly more challenging to find specific modulators of the GK/GKRP PPI. Despite finding only a single GKRP hit scaffold, this provided a starting point for a chemical series with sub-µM potency against GKRP 23 and efficacy in lowering glucose in an in vivo diabetic model. 24 Also, in-house co-crystal structures with GKRP plus S6P and AMG-1694 (a small-molecule disruptor or modulator of the GK-GKRP interaction) corroborated our intricate binding analysis. 24 The targeting of the GK-GKRP interaction serves as an example that certain PPIs can be targeted successfully with small-molecule modulators.
Footnotes
Acknowledgements
We thank Paul Andrews for discussion and input on the manuscript.
Declaration of Conflicting Interests
All authors are employees of Amgen, Inc.
Funding
Amgen, Inc,. sponsored the research, authorship, and publication of this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.