
Abstract
Methylated DNA binding proteins such as Methyl-CpG Binding Domain Protein 2 (MBD2) can transduce DNA methylation alterations into a repressive signal by recruiting transcriptional co-repressor complexes. Interfering with MBD2 could lead to reactivation of tumor suppressor genes and therefore represents an attractive strategy for epigenetic therapy. We developed and compared fluorescence polarization (FP) and time-resolved fluorescence resonance energy transfer (TR-FRET)–based high-throughput screening (HTS) assays to identify small-molecule inhibitors of the interaction between the methyl binding domain of MBD2 (MBD2-MBD) and methylated DNA. Although both assays performed well in 96-well format, the TR-FRET assay (Z′ factor = 0.58) emerged as a superior screening strategy compared with FP (Z′ factor = 0.08) when evaluated in an HTS 384-well plate format. Using TR-FRET, we screened the Sigma LOPAC library for MBD2-MBD inhibitors and identified four compounds that also validated in a dose-response series. This included two known DNA intercalators (mitoxantrone and idarubicin) among two other inhibitory compounds (NF449 and aurintricarboxylic acid). All four compounds also inhibited the binding of SP-1, a transcription factor with a GC-rich binding sequence, to a methylated oligonucleotide, demonstrating that the activity was nonspecific. Our results provide proof of principle for using TR-FRET–based HTS to identify small-molecule inhibitors of MBD2 and other DNA-protein interactions.
Keywords
Introduction
Epigenetic silencing of tumor suppressor genes via DNA hypermethylation has been established as a common hallmark of oncogenesis. 1 The methylation of CpG dinucleotides, particularly at gene promoters and regulatory regions, has been shown to induce epigenetic gene silencing via the recruitment of methyl-binding domain (MBD) proteins such as Methyl-CpG Binding Domain Protein 2 (MBD2), Methyl-CpG Binding Domain Protein 1 (MBD1), and Methyl-CpG Binding Protein 2 (MeCP2) and their associated chromatin remodeling/co-repressor complexes such as Mi2-NuRD. 2 These complexes are capable of remodeling the local chromatin and preventing the transcriptional machinery from gaining access to DNA, leading to transcriptional repression. 3 Recent pharmacologic interventions for reversal of epigenetic gene silencing in cancer have focused on inhibiting DNA methyltransferases (DNMTs), which establish the DNA methylation marks, and histone deacetylases (HDACs), which are part of transcriptional repressive complexes that signal chromatin compaction via removal of acetylation modifications on histone tails. 4 Importantly, such efforts have led to Food and Drug Administration approval of two DNMT inhibitors and two HDAC inhibitors for myelodysplastic syndrome and cutaneous T-cell lymphoma, respectively. 5
However, there has been little effort in developing inhibitors of the methyl binding domain class of proteins, despite the findings from several recent reports credentialing these proteins, particularly the MBD2 protein, as anticancer drug development targets. 6 At the molecular level, RNAi-mediated depletion of MBD2 led to reexpression of epigenetically silenced tumor suppressor genes with promoter CpG methylation. 7 In vivo, genetic disruption of Mbd2 in Apcmin mice, which are prone to developing dozens of intestinal tumors within 4 to 6 mo of age, 8 led to remarkably reduced tumor formation and increased survival. 9 Interestingly, although mice carrying homozygous disruption of Dnmt1 alleles show embryonic lethality, mice with homozygous Mbd2 disruption have a normal life span, size, and reproductive potential, suggesting a favorable toxicity profile for targeting MBD2. Taken together, these observations suggest that MBD2 has potential as an anticancer drug development target. 6
Development of MBD2 antagonists as molecular probes of epigenetic mechanisms and as anticancer epigenetic drugs would be greatly aided by the availability of a suitable high-throughput screening (HTS) assay. Several potential assay formats can be considered for screening for inhibitors of protein-DNA binding interactions.10,11 The most basic of these assay formats involves immobilization of either the protein or DNA to a surface and labeling of the nonimmobilized binding partner. After the binding reaction is complete, unbound molecules can be washed away, and the bound fraction can be detected by measurement of the label. Because such assays involve multiple steps and washes, they often have low signal-to-noise and are often not ideal for HTS. In contrast, “homogeneous” assays (separation free assays) can be developed by taking advantage of optical principles such as fluorescence resonance energy transfer (FRET), time-resolved FRET (TR-FRET), and fluorescence polarization to measure specifically the signal from the bound fraction even in a background of unbound molecules. 11 These technologies can exhibit high signal-to-noise even in high-throughput and miniaturized formats. However, one disadvantage is that molecules that interfere with the fluorescence readout and other assay components can lead to false-positive and false-negative results. 12 One way to overcome this disadvantage is to use label-free detection strategies such as surface plasmon resonance and nuclear magnetic resonance. 11 However, the major disadvantage of these assays is that they often require specialized equipment and/or may not be suitable for HTS because of inadequate parallelization.
Here we describe the development of a modified TR-FRET 13 assay for measuring MBD2-MBD binding to methylated DNA ( Fig. 1 ). TR-FRET uses the long-lived fluorescence of lanthanide metals to monitor FRET after a time delay, when the auto fluorescent signal has decayed significantly. This translates into a robust signal-to-noise ratio when measuring the binding of two ligands. The TR-FRET assay was highly amenable to HTS of small-molecule libraries and showed significantly superior performance compared with a fluorescence polarization–based 14 assay format. We used this TR-FRET screening approach in a pilot screen of 1280 highly studied compounds, identifying small molecules capable of inhibiting MBD2-MBD binding to methylated DNA.
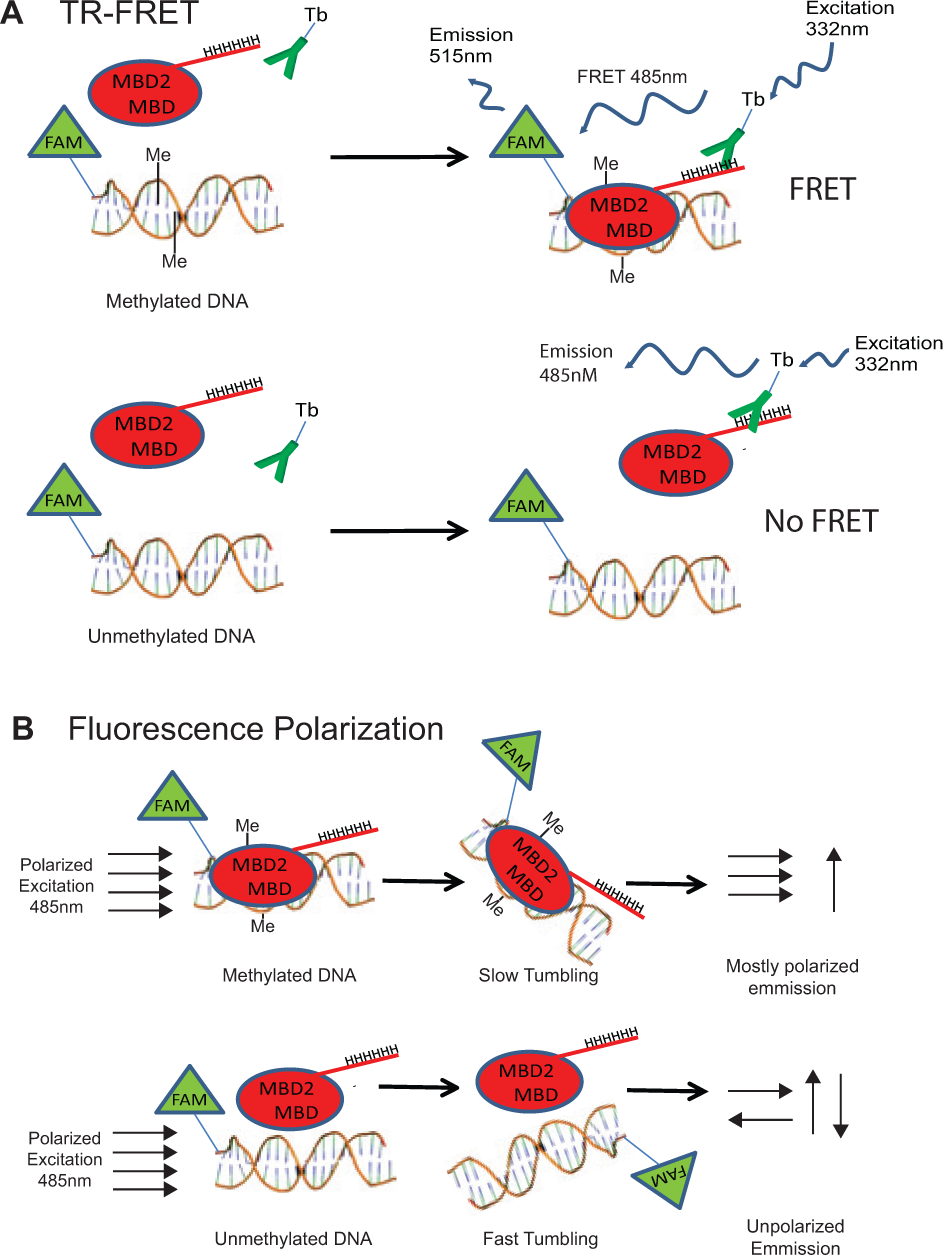
Overview of time-resolved fluorescence resonance energy transfer (TR-FRET) and fluorescence polarization MBD2-MBD DNA-binding assays. (
Materials and Methods
MBD2-MBD Production
A codon-optimized sequence for the MBD2-MBD polypeptide was synthesized and cloned into the pGSE6 vector (Genscript USA Inc, Piscataway Township, NJ) for expression in bacteria as a C-terminal hexa-histidine tagged fusion protein. Briefly, BL21 DE3 cells (Agilent Technologies, Westlake Village, CA) were transformed with this construct, allowed to grow to an OD600 of 1.0, and induced with 1 mM isopropyl β-D-1-thiogalactopyranoside (Cellgro, Corning, Manassas, VA) overnight in a shaking incubator at 220 rpm and 20 °C. Bacteria were lysed using a French press in equilibrium buffer containing 300 mM NaCl, 50 mM sodium phosphate pH 8.0, 5 mM imidazole (Sigma Aldrich, St. Louis, MO), and a protease inhibitor cocktail (Roche USA, Indianapolis, IN). Lysates were centrifuged at 17,000g for 30 min and then incubated with IMAC Nickel NTA beads (Bio-Rad, Hercules, CA) for 1 h at 4 °C. The beads were washed three times with equilibrium buffer containing 15 mM, 20 mM, and 25 mM imidazole, respectively, and affinity purified protein was eluted in equilibrium buffer containing 150 mM imidazole. The his-tagged protein was further purified by gel filtration using a Superdex g75 26/300 Column (GE Healthcare Life Sciences) in storage buffer (150 mM NaCl, 20 mM Tris pH 7.4, 1 mM tris[2-carboxyethyl]phosphine, P212121 LLC, Toledo, OH). Fractions containing the purified MBD2-MBD were concentrated with 3000 MWCO Amicon Ultra-4 Centrifugal Filter Units (Millipore, Billerica, MA) and flash frozen with liquid nitrogen and stored at −80 °C.
Fluorescence Polarization Assay
Fluorescence polarization binding assays were performed as previously described. 15 Briefly, MBD2-MBD protein was added in a dilution series to the reaction buffer containing 4% glycerol, 1 mM MgCl2, 0.5 mM EDTA, 0.5 mM DTT, 125 mM NaCl, 10 mM Tris-HCl (pH 7.4), and 0.2% Tween-20 up to a total working volume of 20 µL. Hairpin-forming oligonucleotides with the sequence 5′-fluorescein (FAM)-ATGCTCGTAGCACTTTTGTGCTACGAG-CAT-3′ (unmethylated) or 5′- fluorescein-ATGCTCmeGTAGCACTTTTGTGCTACmeGAGCAT-3′ (methylated) were annealed by heating to 95 °C and cooling on ice rapidly. They were then added to the binding reaction to a final concentration of 10 nM. Binding assays were carried out in 384-well black, flat-bottom plates (Corning) in quadruplicate replicates. The plate was incubated at 4 °C for 1 h with gentle shaking. Fluorescence polarization readings were performed on a Safire 2 (Tecan, Männedorf, Switzerland) instrument with excitation at 470 nm and monitoring emission at 525 nm. Anisotropy values were acquired and plotted relative to MBD2-MBD protein concentration and fitted using the open source software R version 2.12.1. 16 The optimal conditions of 10 nM oligonucleotide and 200 nM MBD2 protein were used for the high-throughput assay. Positive and negative inhibitor controls consisted of 5 µM of hairpin oligonucleotides (either methylated or unmethylated), with the same sequence described above but without a FAM label, and were analyzed in 32 replicates each. Z′ factors were calculated as described by Zhang et al. 17
TR-FRET
TR-FRET assays were performed in 384-well black low-volume plates (Greiner Bio One) with a total working volume of 20 µL. MBD2-MBD protein at 25 nM was added to the same buffer as described above for fluorescence polarization. The FAM-labeled oligonucleotides described above were added in a dilution series, and LanthaScreen Elite Tb-anti-His-Tag terbium-labeled antibody (Life Technologies, Carlsbad, CA) was added to a final concentration of 5 nM. The plate was incubated at 4 °C with gentle shaking for 1 h. The plates were read on a Safire 2 (Tecan) instrument with excitation at 332 nm and emission read at 485 nm (to read FAM emission) and 515 nm (to read terbium FRET emission) after a delay of 50 µs and a total integrated read time of 400 µs. The ratio of the 515 nm and 485 nm readings was used to assess the degree of FRET, which is proportional to the amount of total binding. The ratio from a control sample containing no protein was subtracted from the other data and the binding curves plotted relative to total DNA concentration. Curve fitting was performed using the open-source software R version 2.12.1. The optimal conditions were determined to be 30 nM oligonucleotide and 25 nM MBD2 protein. Positive and negative inhibitor controls consisted of µM of oligonucleotide (either methylated or unmethylated) with the same sequence described above but without a FAM label and were analyzed in 32 replicates. Z′ factors were calculated in the same manner listed above.
LOPAC1280 Screen
The 1280 compound Library of Pharmacologically Active Compounds (LOPAC; Sigma-Aldrich) was screened using the TR-FRET assay described above with optimized conditions in 384-well low-volume plates (Greiner Bio-One) in a reaction volume of 20 µL. A final concentration of 20 µM in 10% DMSO was used for each compound. Four controls were used on each plate with 16 replicates for each control: (1) a 10% DMSO vehicle control; (2) 5 µM of unlabeled, methylated oligonucleotide described above as a competitive inhibitor positive control; (3) unmethylated, FAM-labeled oligonucleotide described above as a technical control; and (4) a second technical control in which no MBD2-MBD protein was added. Plates were incubated for 1 h at 4 °C and read using the Safire 2 (Tecan) and the conditions listed above. The emission ratios were calculated as described above and were adjusted based on the median plate value for each plate. For each compound i on plate j, the Z-score was calculated as shown in eq 1:
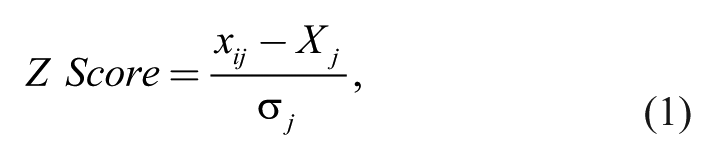
where xi,j is the TR-FRET signal for each compound, Xj is the median plate TR-FRET signal across all compounds on that plate, and σ is the standard deviation across all compounds on plate j. The two-sided p value associated with the Z-score was calculated assuming a normal distribution and was subjected to Bonferroni correction. Compounds with Bonferroni-adjusted p < 0.05 were considered significant hits. The Z′ factor was calculated for each plate separately. In addition, a Z-factor analogous to the Z′ factor was calculated for each plate using all wells on a given plate.
All compounds that significantly inhibited protein binding in the LOPAC screen were tested in a dose-response series (1 nM to 20 µM) using the TR-FRET assay. Also included were a negative vehicle control (DMSO only) and a positive inhibitor control using 5 µM unlabeled methylated oligonucleotide. Curve fitting was performed using the open-source software R version 2.12.1.
Counterscreen with the Transcription Factor SP-1
We adapted the TR-FRET assay for assessment of recombinant Hexa-His-tagged SP1 protein binding to the unmethylated hairpin oligonucleotide described above. The assay was performed using 10 nM of the DNA binding transcription factor SP1 (Abcam, Cambridge, UK), 100 nM FAM-labeled oligonucleotide, and compounds in dose response from 1 nM to 20 µM, with the same plate type and conditions described above.
Results
TR-FRET and Fluorescence Polarization Assay Validation
We developed both TR-FRET and fluorescence polarization–based assays to measure the binding of MBD2-MBD to methylated DNA and evaluated each of these assays for use in HTS for inhibitors capable of disrupting this binding ( Fig. 2 ). Both assays showed strong preferential binding of MBD2-MBD to methylated DNA (EC50 of 86 nM and 42.7 nM, respectively) compared with unmethylated DNA (EC50 >> 1 µM), as expected ( Fig. 2A , B ). 15 Using these experiments, we determined the optimal concentrations of MBD2-MBD, labeled-oligo, and other assay components for each assay (see the Materials and Methods section). Using a series of four controls (n = 32 replicates of each), we evaluated the performance of both assays for use in high-throughput chemical compound screening, as determined by calculating the Z′ factor ( Fig. 2C , D ). The TR-FRET assay showed significantly better signal-to-noise ratios (Z′ factor = 0.59) compared with that of the fluorescence polarization assay (Z′ factor = 0.08). As expected, the negative control (high excess unlabeled unmethylated oligonucleotide) did not significantly disrupt binding of MBD2-MBD in either assay. This suggests that the low degree of binding to unmethylated DNA seen in Figure 2 is due to a nonspecific mode of binding that is different from the specific binding of the protein to methylated DNA. In addition, both assays measured significant DNA binding inhibition by a competitive inhibitor positive control (excess unlabeled methylated oligonucleotide). The TR-FRET assay was therefore selected for further validation in a high-throughput screen.
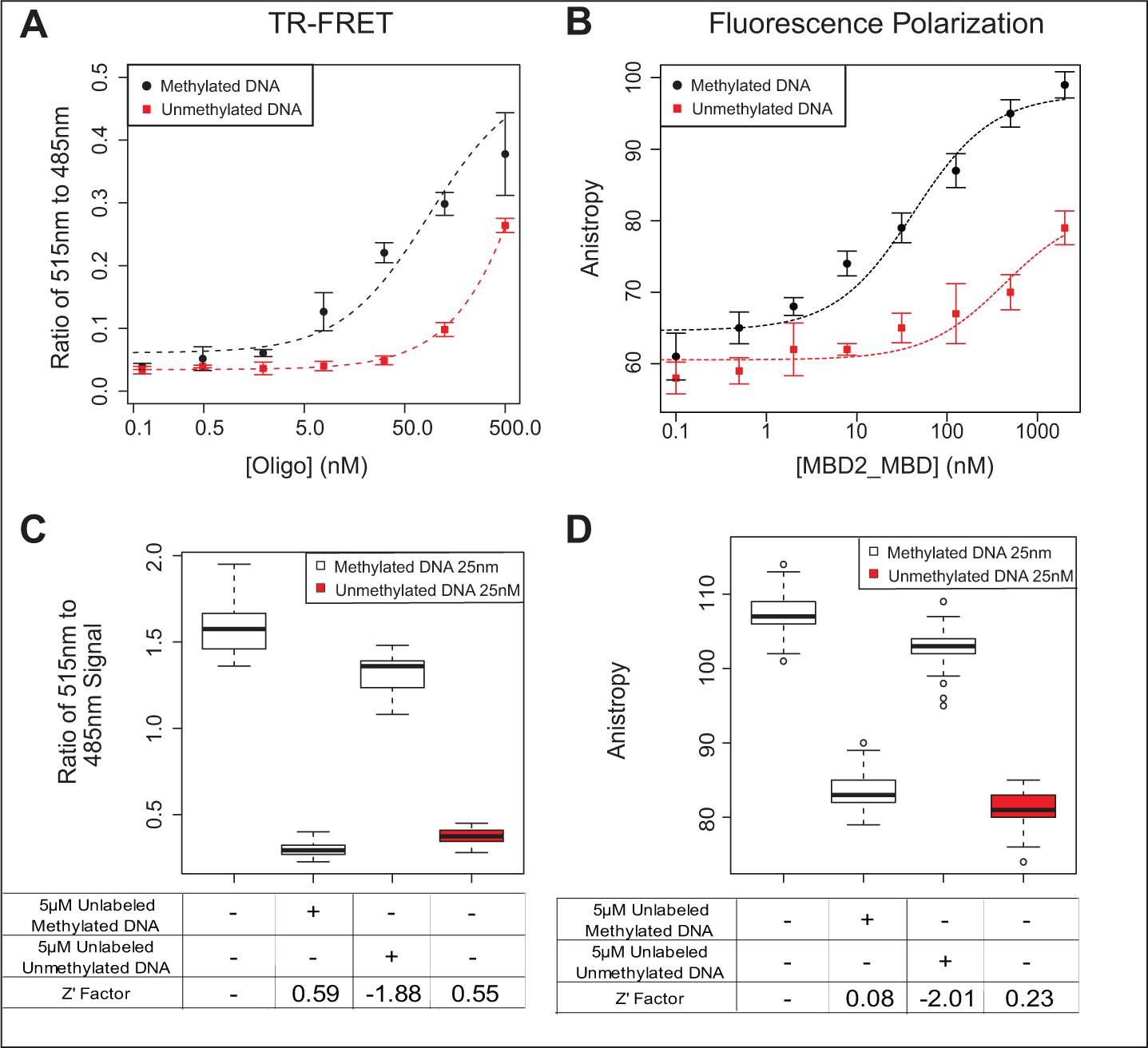
Assay performace of time-resolved fluorescence resonance energy transfer (TR-FRET) and fluorescence polarization (FP) MBD2-MBD DNA binding assays. (
Given the ability of MBD2-MBD to bind selectively to methylated versus unmethylated cytosine in the context of double-stranded DNA, we assessed whether methylcytosine and cytosine, either as free bases or as nucleoside monophosphate derivatives, were capable of inhibiting the interaction between MBD2-MBD and methylated DNA. Interestingly, cytosine, 5-methylcytosine, 5-methyl 2′-deoxycytidine monophosphate, and 2′-deoxycytidine monophosphate all failed to show any appreciable inhibition of the MBD2-MBD interaction with methylated DNA in our TR-FRET assay (
Pilot Screen of LOPAC1280 Compounds for Inhibition of MBD2-MBD Binding to Methylated DNA
To assay inhibitors of MBD2-MB2 binding a methylated oligonucleotide, we used the 1280-compound LOPAC (Sigma Aldrich). This library consists of a variety of drug and druglike compounds with well-studied mechanisms, as well as molecules from larger chemical compound libraries. Test compounds from the LOPAC were added to our optimized TR-FRET assay at a final concentration of 20 µM. The compounds were assayed across four 384-well plates, with each plate containing positive and negative controls (
Fig. 3A
). To confirm that the 10% DMSO used to dissolve the compounds was not interfering or causing bias with the binding assay, we ran a set of negative controls on each plate with water and a separate set with 10% DMSO (n = 8 wells on each plate for a total of 32 replicates each). There was no significant difference between these controls (
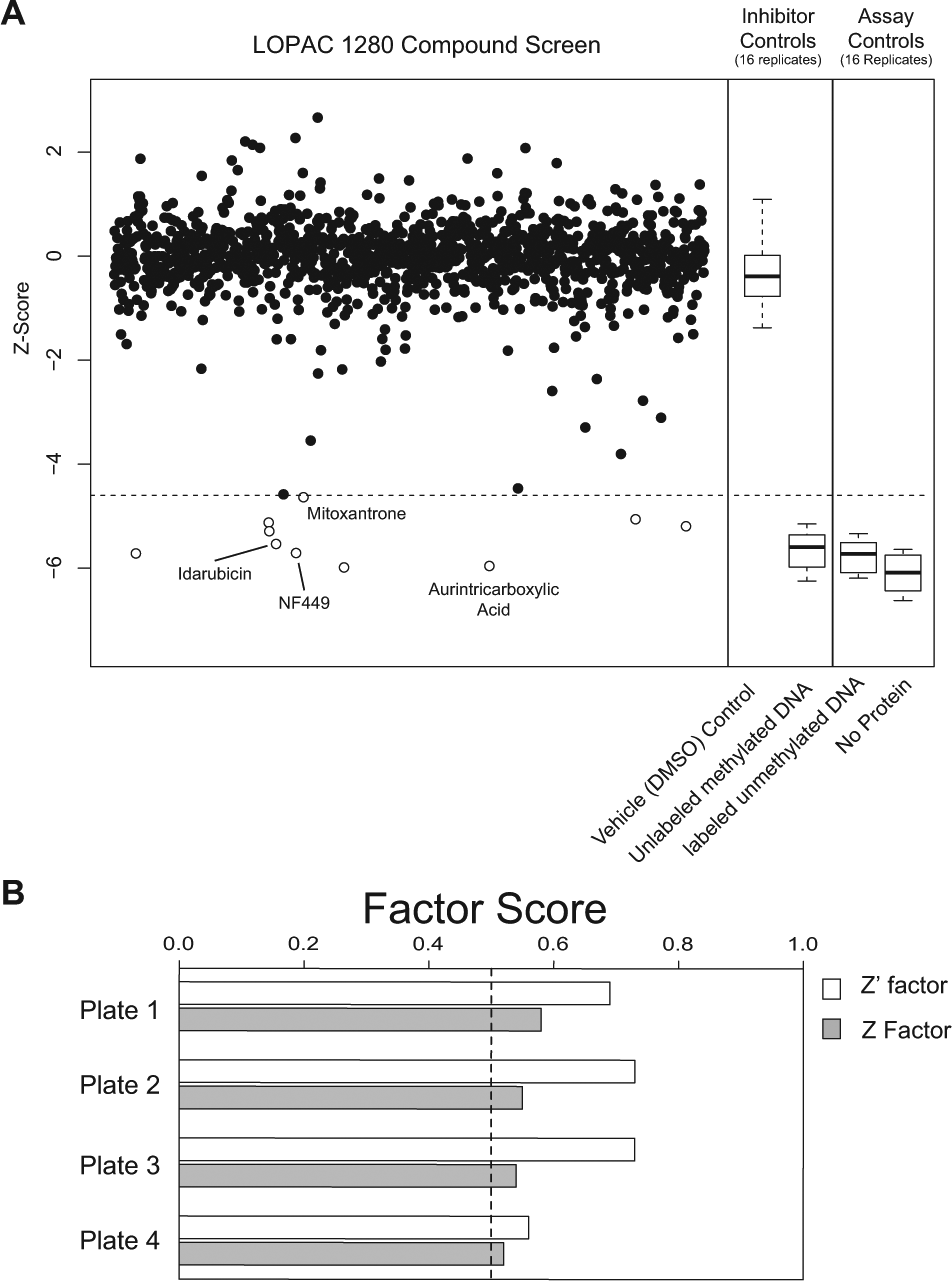
Screening of 1280-compound LOPAC library with MBD2-MBD time-resolved fluorescence resonance energy transfer (TR-FRET) assay. (
The TR-FRET signal from most compounds followed a normal distribution centered on the median of the negative control. Z and Z′ factors were calculated for each plate ( Fig. 3B ); Z′ factors for each plate were 0.69, 0.73, 0.73, and 0.55, respectively. As expected, Z factors for each plate were lower than their respective Z′ scores, but all were greater than 0.5. The LOPAC screen replicated the sensitivity of the initial validation and also identified 10 compounds that significantly inhibited MBD2-MBD binding to methylated DNA after correction for multiple hypothesis testing ( Fig. 3A , open circles; Table 1 ).
Significant Hits from the MBD2-MBD Time-Resolved Fluorescence Resonance Energy Transfer Binding Assay Applied to the Sigma LOPAC Screen.
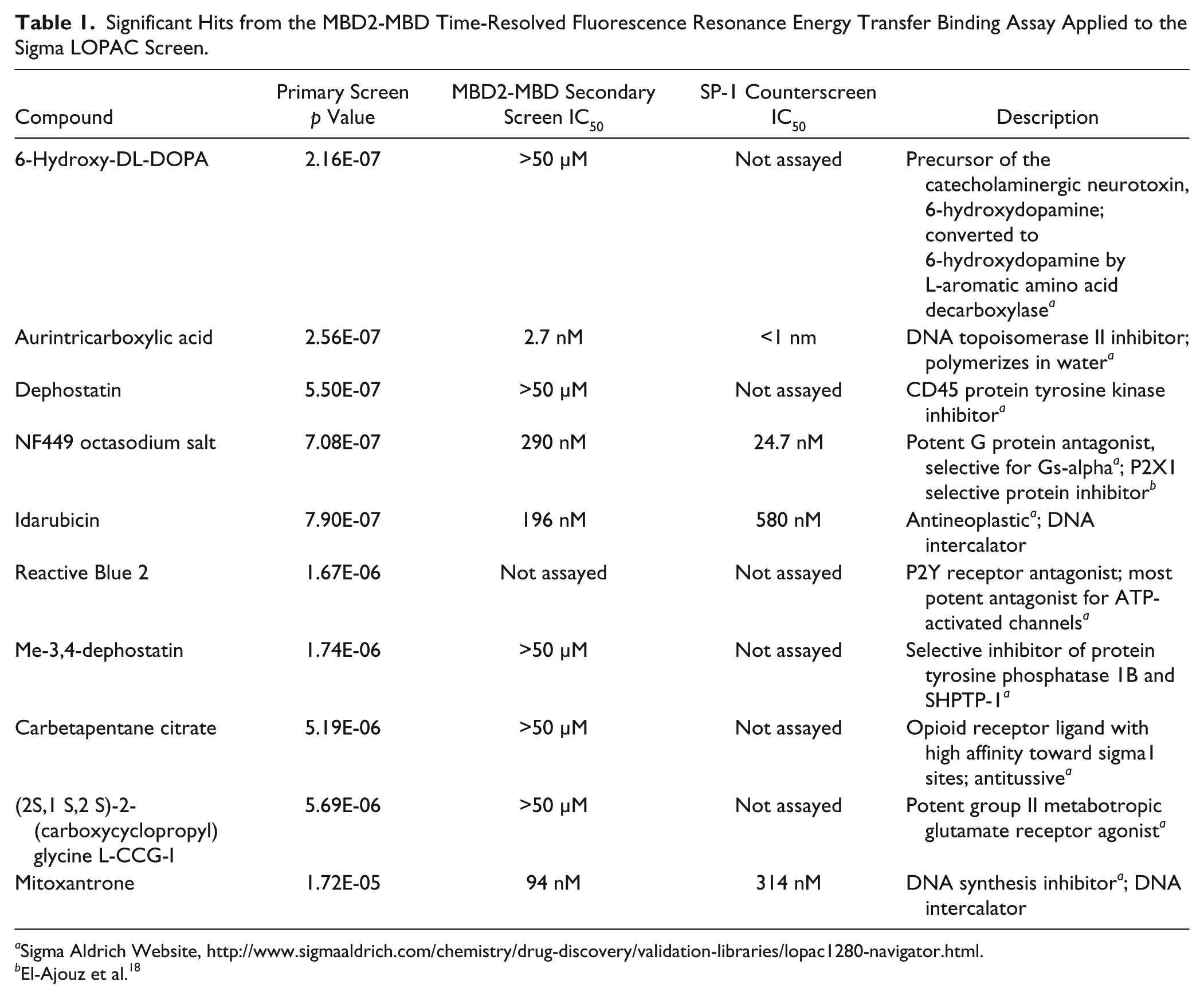
Sigma Aldrich Website, http://www.sigmaaldrich.com/chemistry/drug-discovery/validation-libraries/lopac1280-navigator.html.
El-Ajouz et al. 18
Dose Response of Hits for Inhibition of MBD2-MBD Binding to Methylated DNA
To validate the hits observed on the LOPAC screen, we assayed each compound in a dose response ( Figure 4 ; Table 1 ). A suramin analog, Reactive Blue 2, emerged as a hit from the primary screen but was excluded from the secondary analysis because its coloration and light absorption could interfere with the assay. Of the remaining nine compounds, four compounds (mitoxantrone, idarubicin, aurintricarboxylic acid, and NF449) showed a sigmoidal dose response for inhibition of MBD2-MBD binding to methylated DNA, whereas five compounds (6-hydroxy-DL-DOPA, carbetapentane citrate, dephostatin, Me-3,4-dephostatin, and L-CCG-I) failed to show appreciable inhibition in the dose range tested ( Figure 4 ; Table 1 ). Mitoxantrone and idarubicin are known DNA intercalators,19,20 and aurintricarboxylic acid is known to polymerize in water and can inhibit many macromolecular protein-nucleic acid interactions.18,21 These three compounds would be expected to nonspecifically inhibit DNA-protein interactions. Interestingly, a suramin analog, NF449, known to inhibit P2X1 receptors with picomolar potency and high selectivity compared with P2X2 receptors,22,23 also emerged as a dose-response validated inhibitor of the MBD2-MBD interaction with methylated DNA, showing an IC50 of 290 nM.
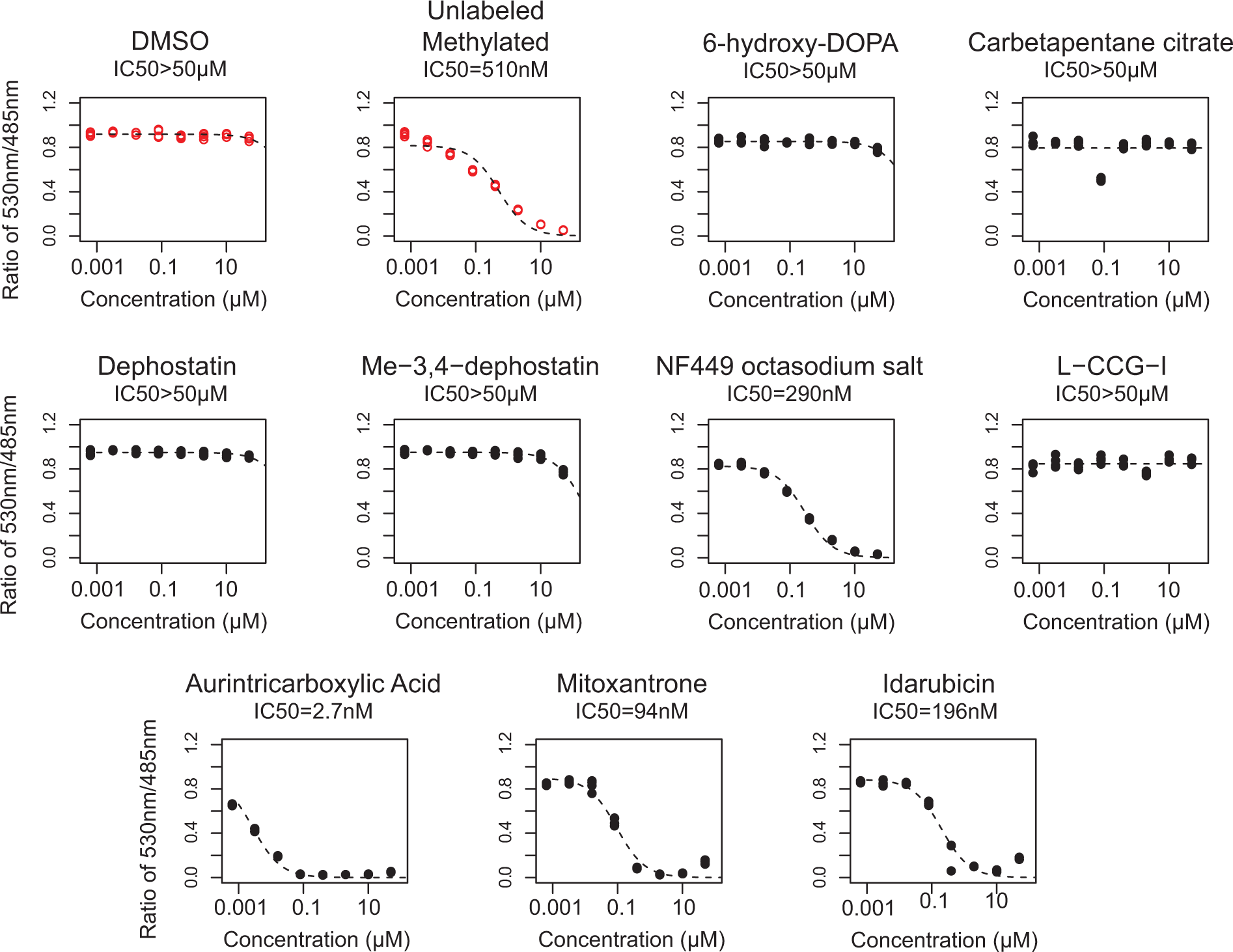
Dose-response curves of significant hits. The nine hit compounds from the primary screen were tested with the time-resolved fluorescence resonance energy transfer assay using a fourfold dose-response series from 1.2 nM to 20 µM (four replicates each). Controls are shown as red open circles. Four of the nine compounds showed a potency for inhibition of MBD2-MBD binding to DNA within the dose range tested. IC50 values for each compound are listed below the title for each dose response.
Counterscreen with the DNA Transcription Factor SP-1
To assess the specificity of the four verified hit compounds for inhibiting binding of MBD2-MBD to methylated DNA, we developed a counterscreen ( Fig. 5A ) using the transcription factor SP-1 to identify those hits that would also inhibit binding of an unrelated protein to DNA or some other components of the assay. The transcription factor SP1 has been shown to localize to GC-rich regions in the human genome, including promoter CpG islands where MBD2 can be found. 7 However, SP-1 has no known specificity for methylated DNA, which we verified (EC50 = 22 nM for both methylated and unmethylated DNA; Fig. 5B ). 24 We then tested MBD2-MBD inhibitors from the LOPAC for cross-inhibition of SP-1. All four hit compounds inhibited SP-1 in a dose-response manner ( Fig. 5C ), suggesting that these compounds are unlikely to be selective inhibitors of MBD2-MBD binding to DNA but may have activity in inhibiting DNA-protein interactions more generally.
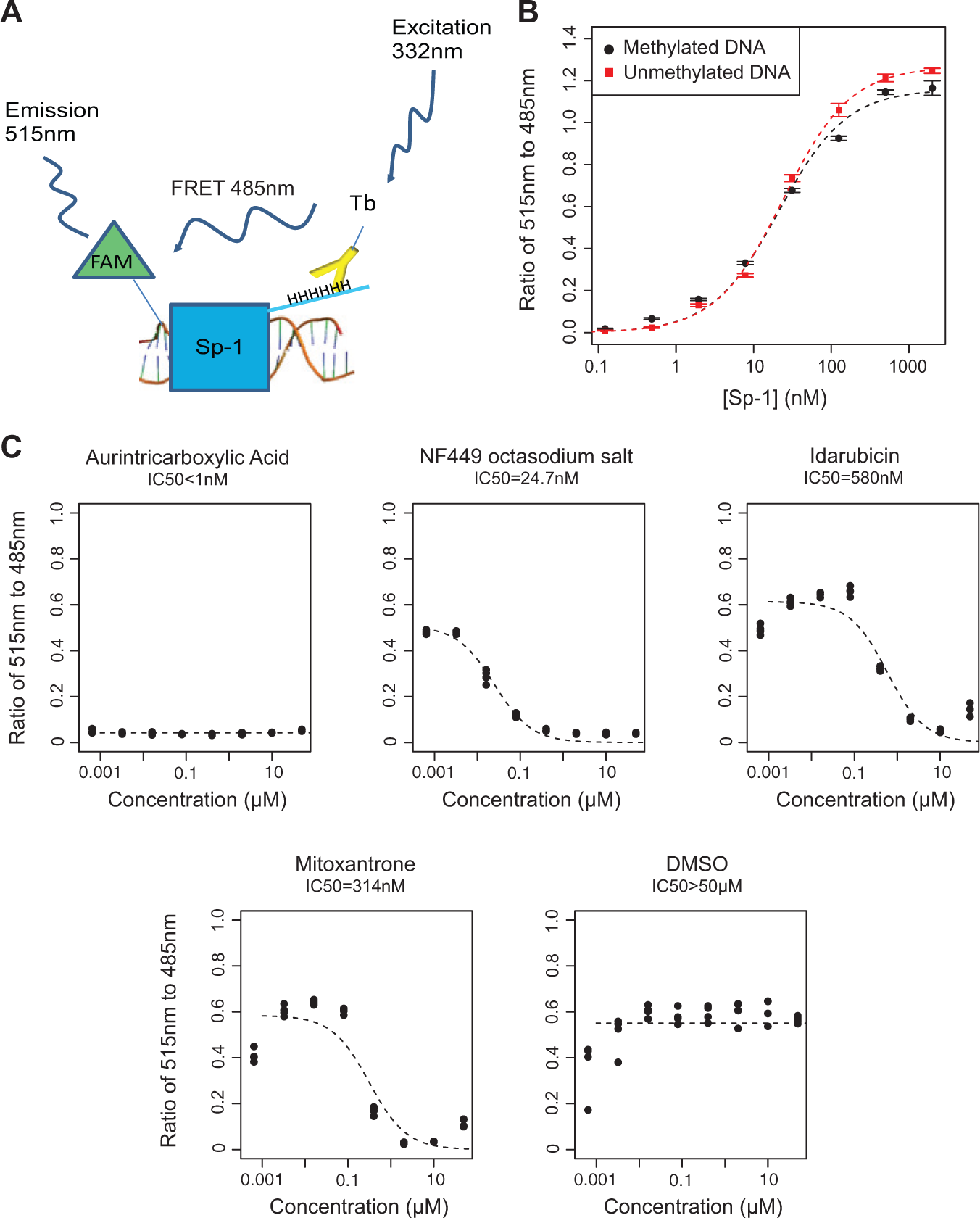
Counterscreen with DNA transcription factor SP-1 to assess specificity of identified hit compounds. (
Discussion
Our overall goal is to develop small-molecule inhibitors capable of disrupting the interaction between methylated DNA and MBD2, a key transcriptional repressor involved in epigenetic repression of methylated DNA in cancer cells. On the surface, this goal appears to be somewhat impractical given that numerous previous attempts at inhibiting protein-DNA interactions have had limited success, with some categorizing such protein-DNA interactions as “undruggable” targets. However, unlike most DNA protein interactions, which involve broad electrostatic and nonelectrostatic interaction surfaces, the interaction of methyl binding domains with methylated DNA appears to involve relatively few critical contacts, as illustrated by the structure of the methyl binding domain from MECP2 bound to methylated DNA, 25 and might be targetable by small molecules. In addition, given the exquisite selectivity of the MBD2-MBD for symmetrically methylated DNA compared with unmethylated or even hemi-methylated DNA, 15 we reasoned that it might be possible to identify small molecules capable of disrupting this interaction.
We have developed a robust, modular, and high-throughput TR-FRET–based assay for screening small-molecule libraries for inhibitors of MBD2-MBD protein binding methylated DNA. The modularity of the assay is attributable to the fact that we can potentially substitute the MBD2-MBD and DNA oligo in the assay for any His-tagged protein and/or FAM-labeled DNA binding partner. Indeed, we were able to capitalize on this modularity to use the same assay format to counterscreen our hits by assessing whether they could also inhibit the interaction of the transcription factor SP-1 with its DNA substrate.
Compared with a fluorescence polarization–based approach, our TR-FRET assay provided a better signal-to-noise ratio, resulting in improved Z′ factors under optimized screening conditions. The better performance of the TR-FRET assay compared with the fluorescence polarization assay in the context of measuring the MBD2-MBD polypeptide to methylated DNA is likely due to the biophysical characteristics of the protein DNA complex. Whereas fluorescence polarization assays achieve optimal dynamic range and signal-to-noise when the size of the fluorescently labeled molecule is much smaller than the size of the bound complex, TR-FRET assays often favor bound complexes in which both binding partners are relatively small so that the distance between the two labels can facilitate FRET. 11 Therefore, the relatively small and equal size of the MBD2-MBD polypeptide and the methylated duplex DNA in our assay system likely favored the use of the TR-FRET assay. Indeed, although the fluorescence polarization assay exhibited reasonable performance in 96-well plate format (with Z′ factor ~0.6), allowing robust measurement of the MBD2-MBD binding to methylated DNA in Figure 2 , miniaturization of the assay to 384-well plate format resulted in considerable decay in signal-to-noise. The TR-FRET assay maintained performance in both 96-well and 384-well plate formats and was therefore better suited for translation to the high-throughput setting.
We used this TR-FRET assay to analyze the 1280 compound LOPAC library to identify small molecules capable of inhibiting the interaction of MBD2-MBD with methylated DNA. Of the initial nine hits, four showed a robust sigmoidal inhibitory dose response. Interestingly, all of the hit compounds that failed to confirm in the dose-response analysis exhibited extremely low raw donor counts (emission at the 485 nm wavelength less than two standard deviations below the mean, suggesting significant optical interference of the assay), whereas only one of the four compounds that did verify (mitoxantrone) exhibited such low raw donor counts. We may therefore be able to use the raw donor counts as a way to screen out potential false-positive hits and reduce the overall false discovery rate in larger-compound screening efforts. Three of the four compounds that did confirm, mitoxantrone, idarubicin, and aurintricarboxylic acid, would be expected to inhibit MBD2-MBD binding to methylated DNA given that each are known to inhibit multiple DNA-protein interactions. This was confirmed by our counterscreen, which showed that these compounds also prevented binding of SP1 to DNA with similar potency. However, the ability of NF449 to inhibit MBD2-MBD binding to methylated DNA was not anticipated.
NF449 is a suramin analog that can inhibit the P2X1 receptor with subnanomolar potency.23,26 The P2X family forms homo- or heterotrimeric complexes on the surface of many cell types, forming ATP-gated cation channels 27 that have been implicated in multiple cellular processes. 28 The mechanism of inhibition features ionic interactions between the eight sulfonic acid groups on NF449 with positively charged basic amino acids at the base of a cysteine-rich loop on P2X family members. 26 The selectivity of NF449 to P2X1 compared with other P2X and P2Y members is thought to arise through a second favorable contact between the compound and lysine 138 near the ATP binding pocket. 26 NF449 has also been shown to inhibit other proteins, including the G protein alpha subunit 29 and FGFR3. 30 In each case, the number and orientation of the negatively charged sulfonic acid groups on NF449 were thought to be critical for inhibition. A query for NF449 in the PubChem database (http://pubchem.ncbi.nlm.nih.gov/summary/summary.cgi?cid=6093161; searched on December 27, 2013) showed that it was identified as an active hit in screens of inhibitors of multiple DNA binding proteins, including a screen for inhibitors of the interaction of the MLL CXXC domain with DNA. Based on all of these previous studies, we speculate that NF449 may similarly inhibit MBD2: the negatively charged sulfonic acids on NF449 may disrupt the binding of positively charged basic residues on the MBD2 surface with the negatively charged phosphate backbone of DNA. 31 A similar mechanism may also be responsible for the observed disruption of SP-1 binding to DNA by NF449 in our counterscreen. In addition, the previous work examining the inhibition of FGFR3 by NF449 suggested that the compound may be capable of interacting with the intracellular tyrosine kinase domain of FGFR3. 30 NF449 may therefore be available in the intracellular space for activity. If this proposed intracellular bioavailability is confirmed, then NF449, or its derivatives, may have promise as pharmacologic probes for studying MBD2 function in cells. In addition, in future work, it may be possible to find NF449 derivatives that are capable of selectively inhibiting MBD2 and not other DNA binding proteins, analogous to the selectivity of NF449 for P2X1 compared with other P2X receptors. 26
This study demonstrates the utility of our highly modular TR-FRET–based MBD2 methylated DNA binding assay and coupled counterscreen to identify and test the specificity and activity of small molecules. This TR-FRET–based primary screening and counterscreening assay should be easily adaptable to more extensive small-molecule libraries to discover novel, specific inhibitors of the MBD2 methylated DNA interaction. One potential problem commonly encountered by target-based, as opposed to cell-based, screening strategies such as the one used here is that the compounds that are identified in large-scale compound screening may not be bioavailable inside cells, where the target is active. Maneuvers such as lowering the DMSO concentration in the screening assay and using medicinal chemistry approaches to improve the cellular bioavailability of identified hits can help to identify compounds or develop derivatives that can exert their inhibition of the MBD2-DNA binding interaction in cells. In addition, the modular nature of the TR-FRET assay developed here could facilitate screening with full-length MBD2, potentially yielding allosterically acting inhibitors, and screening against other methyl-binding domain proteins, yielding inhibitors that selectively inhibit MBD2 and not other MBD family members such as MECP2. This assay format might even be readily extendable to facilitate the study of any DNA-protein interaction.
Footnotes
Acknowledgements
The authors would like to thank the laboratory of Dr. Dan Leahy, Dr. Ping Liu, and Jackie McCabe for expertise with protein expression and use of their gel filtration system. The authors would also like to thank the Johns Hopkins School of Medicine High Throughput Biology (HiT) center, in particular Dr. Min Li, Alan Long, and Melissa Miller, for assistance with the LOPAC screen and for helpful discussion.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by funding from the Prostate Cancer Foundation, the Patrick C. Walsh Prostate Cancer Research Fund, Department of Defense Prostate Cancer Research Program predoctoral training grant (W81XWH-11-1-0618 to N.W.), and NIH/NCI grants CA70196 and CA58236.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.