
Abstract
A substantial challenge in phenotypic drug discovery is the identification of the molecular targets that govern a phenotypic response of interest. Several experimental strategies are available for this, the so-called target deconvolution process. Most of these approaches exploit the affinity between a small-molecule compound and its putative targets or use large-scale genetic manipulations and profiling. Each of these methods has strengths but also limitations such as bias toward high-affinity interactions or risks from genetic compensation. The use of computational methods for target and mechanism of action identification is a complementary approach that can influence each step of a phenotypic screening campaign. Here, we describe how cheminformatics and bioinformatics are embedded in the process from initial selection of a focused compound library from a large set of historical small-molecule screens through the analysis of screening results. We present a deconvolution method based on enrichment analysis and using known bioactivity data of screened compounds to infer putative targets, pathways, and biological processes that are consistent with the observed phenotypic response. As an example, the approach is applied to a cellular screen aiming at identifying inhibitors of tumor necrosis factor–α production in lipopolysaccharide-stimulated THP-1 cells. In summary, we find that the approach can contribute to solving the often very complex target deconvolution task.
Keywords
Introduction
Despite a focus on target-based approaches for the past decade of drug discovery, most first-in-class new small molecules were discovered via phenotypic screening. 1 Consequently, phenotypic screening programs at the cell or organism level are of strategic importance to the pharmaceutical industry. The molecules identified through these screens can target one or more proteins/pathways, and the observed phenotypic effects often translate into disease. One drawback of these approaches is that the molecular mode of action (MOA) of identified compounds often needs to be characterized to enable efficient further optimization of the screening hit and to enable further research around the new MOA (e.g., safety considerations, functional annotation of target, drug development).1,2 This characterization process, also called the target deconvolution process, is still a substantial challenge in phenotypic drug discovery.
Several types of complementary approaches currently can be used for the target deconvolution process: direct biochemical methods, genetic interaction methods, and computational knowledge-based methods.2,3 A recent review gives a very comprehensive view of several complementary experimental target deconvolution methods. 4 Chemoproteomics is one of the main approaches and is normally used in the absence of hypotheses for which target(s) lead to the phenotypic effect. The molecule of interest is attached to a linker that is used to pull out targets from a cell lysate, which are then profiled using proteomics. The putative target list might be further validated through complementary methods such as small interfering RNA (siRNA). Successful examples of target deconvolution with chemoproteomics are the identification of tankyrase as an essential part of the WNT signaling pathway 5 and the monocarboxylate transporter MCT1 as the target of a novel class of immunosuppressant compounds. 6
Complementarily, computational methods can help infer the targets/MOA of small molecules or can generate hypotheses about key targets involved in the phenotype of interest. Numerous methods are available to predict targets from the molecular structure. 7 They are mainly based on a statistical comparison of chemical features shared with known ligands for a given target. A limitation of these methods, in addition to requiring a known small-molecule ligand, is that structural similarity does not always translate to equivalent bioactivity 8 ; therein, false positives can be generated. Multiparametric phenotypic screens can enable computational methods to analyze/cluster the compounds according to their MOA. The comparison of phenotypic profiles between very well-known tool compounds and other compounds can then lead to the inference of targets and MOA for the latter under the assumption that compounds with similar profiles modulate similar targets or pathways.9,10 A limit to this type of approach is identifying compounds of interest with a new MOA. In this context, interpreting high-dimensional phenotypic profiles from high-content screening is a challenge. To help with this interpretation, and since groups of highly correlated end points are likely to be measuring common underlying traits, Young et al. 11 proposed using standard methods for large data reduction to derive factors that are mechanistically interpretable (e.g., nuclear size, DNA replication levels, chromosome condensation). To further aid the target deconvolution process, these methods can be augmented with additional data sources (e.g., the above-mentioned in silico target prediction from compound structure). 10 Other data sources include side effect profiles, 12 historical bioactivity data available in public databases, 13 and phenotypic profiles available in ChemBank and PubChem.2,14 As many compounds act via the modulation of several targets,15,16 pathway/interactome data sources can also provide valuable insights into the MOA of compounds and support compound optimization strategies as well as the new target identification process. 17 In cases where phenotypic screens only measure a single (i.e., 1D) response, the phenotypic profile for tool compounds is not sufficiently informative on its own, and the incorporation of these data integration strategies becomes critical to enable target deconvolution.
There are two different philosophies regarding selecting screening sets for phenotypic screening. Either a diversity screening approach is taken or a set of tool compounds with a known bioactivity profile is screened. A diversity set consists of compounds that should optimally cover the chemical space in a predefined way. Alternatively, a tool compound set consists of compounds that have been optimized for a specific target and also have been profiled in a selectivity panel. Both screening approaches have pro and cons. The primary advantage with screening a set of tool compounds is that relevant targets for the phenotypic effect can be identified very rapidly. Another advantage is that only a small set of compounds needs to be screened, allowing more complex, costly, and time-consuming assays to be used. A disadvantage is that it is difficult to identify a truly novel target since the tool compounds have been developed with a specific target in mind. An advantage with a diversity screen is the possibility of identifying completely novel targets after optimization of the screening hit and accompanying target deconvolution. Disadvantages with screening a diversity set are that a relatively large number of compounds need to be screened and that there is no guarantee that the optimization and target deconvolution parts will be successful.
In this study, we propose a strategy that combines cheminformatics and bioinformatics to integrate historical bioactivity and pathway data for the analysis of single read-out phenotypic screens. We describe how this data integration can support each step of a phenotypic screening campaign from the compound selection (experiment planning) through to the target deconvolution process. As an illustration and validation of the concept, the strategy is applied to a phenotypic screen measuring tumor necrosis factor (TNF)–α production in lipopolysaccharide (LPS)–stimulated THP-1 cells. An overview of the workflow for the calculations is given in
Materials and Methods
Bioactivity Data Resources
Activity data were extracted from the CHEMBL (
Screening Set Design for Target Enrichment Testing
Since Fisher’s exact test is used to test for enrichment of targets after screening (see the “Enrichment Analysis” section), one can design the library screened in a manner that increases the power of the test for enrichment. By assuming that compound activity on targets in the screen will be identical to their historical target activity, the contingency table will have only nonzero entries on the diagonal, and the significance of the test for target i based on the screening set,
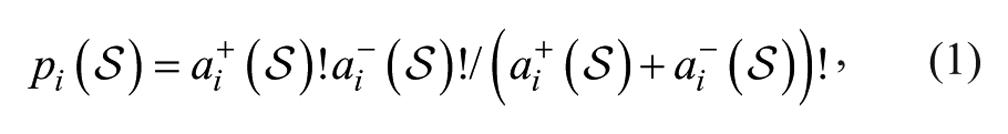
where
In practice, the size of the screening set is fixed and a search is conducted to optimize the composition using the following simulated annealing approach: (1) initially, S is drawn randomly from the compound collection, C; (2) randomly select a candidate compound for exclusion
Selection of Compounds with an Increased Probability of Impact on TNF-α Production
To increase the probability of screening compounds relevant to TNF-α production, a subset of compounds was selected to supplement the above approach according to their activity on proteins putatively related to TNF-α production in pathway databases. The Ingenuity IPA database (Redwood City, CA) 21 was first queried to identify genes whose complete or partial deletion in mouse models leads to a modification of TNF-α levels. GeneGO MetaCore (Philadelphia, PA) 22 was then mined to identify proteins upstream of TNF-α. The linear canonical pathways (noodles) containing TNF-α were extracted from MetaBase. In these noodles, TNF-α was affected through five different mechanisms: “influence on expression,” “transcription regulation,” “co-regulation of transcription,” “cleavage,” and “unspecified (but related to TNF-α production).” Consequently, all upstream proteins in these canonical noodles can affect TNF-α production. The distance to TNF-α was limited to five to select proteins most likely to be causally linked to effects on TNF-α production (as the distance increases, this causal link is expected to reduce due to feedback loops and redundancies).
Targets in the above lists not already well covered by the designed screening set were identified. For each target i identified, available tool compounds were found and included in the screening set S if, upon inclusion,
Assay
The assay employed a phenotypic, functional readout for the identification of inhibitors of LPS-induced TNF-α production using a surrogate monocytic cell line, THP-1 (TIB-202; ATCC, Manassas, VA). TNF-α production was measured by fluorescent-linked immunosorbant assay (FLISA).
The FLISA was constructed using goat anti–mouse (GAM) beads coated with a mouse anti–human TNF-α antibody. TNF-α was captured by the antibody and detected using a biotinylated anti–human TNF-α antibody coupled to streptavidin Cy5. Fluorescence was measured using the Applied Biosystems 8200 Cellular Detection System, which employs the Fluorometric Microvolume Assay Technology (FMAT; Applied Biosystems, Life Technologies Corp., Paisley, UK). All materials were purchased from Sigma (Sigma-Aldrich, St Louis, MO) unless otherwise stated.
Cell culture and preparation
THP-1 cells were maintained in THP-1 cell media (Roswell Park Memorial Institute media [RPMI-1640, R0883] containing 10% [v/v] fetal calf serum [FCS; CR1207] and 2 mM L-glutamine [G7513]) at a density between 2 × 105 and 1 × 106 cells/mL under typical cell culture conditions (37—°C, 5% CO2, 95% humidity). For the assay, cells were centrifuged (300 g for 5 min), the supernatant discarded, and the cell pellet resuspended in assay media (Dulbecco’s modified Eagle’s medium [DMEM; D5671] containing 0.1% [v/v] FCS, 2 mM L-glutamine, and 100 U/mL penicillin/0.1 mg/mL streptomycin [P4333]). This was repeated, finally resuspending the cells at 5 × 105 cells/mL in assay media in the presence of 3 nM phorbol 12-myristate 13-acetate (PMA; P1585, 100-µM stock in 100% [w/v] DMSO [Fisher Scientific, Loughborough UK SP/1044/99SS]).
Compound preparation
Compounds were supplied solubilized in DMSO at a stock concentration of 10 mM and were diluted 100-fold in assay media to give a 10× final assay concentration in 1% (v/v) DMSO.
Preparation of anti–TNF-α monoclonal antibody-coated GAM beads
GAM beads (Saxon, Spherotech, Lake Forest, IL; MPFc-60-5; 6 mm, 0.5% [w/v]) were washed twice with bead-coating buffer (phosphate-buffered saline [PBS; D8537] containing 0.01% [w/v] sodium azide [S-2002]). Then, 40 µL stock anti-TNF monoclonal antibody (R&D Systems, Minneapolis, MN; MAB610; 500 µg/mL) was added to 1 mL beads and incubated, mixing at 4—°C overnight. Beads were washed twice, centrifuged at 13,000 rpm for 1 min, and resuspended in bead-coating buffer, finally adding 2.5 mL to give a final 0.2% (w/v) beads concentration.
Preparation of FLISA bead mix (detection mix)
The detection mix consisted of PBS, 0.5% (v/v) FCS, 2% (v/v) penicillin/streptomycin, 0.002% (w/v) anti–TNF-α mAb-coated GAM beads, 60 ng/mL Biotin anti-TNF antibody (BAF210; R&D Systems), and 1 µg/mL Streptadvidin-Cy5 label (43-8316; Cambridge Biosciences, Cambridge, UK).
Preparation of recombinant human TNF-α (rhTNF-α) standard curve
A 500 µg/mL stock rhTNF-α (210-TA-010; R&D Systems) in PBS was used to produce a 10-point, double-diluted standard curve in assay media (8 ng/mL to 7.8 pg/mL rhTNF-α). Assay media was used as a zero standard control.
Assay build
In total, 40 µL of cells (20,000 cells/well) was added to black 384-well clear-bottom plates (assay plate; 781091; Greiner, Stonehouse, UK). Plates were incubated under typical cell culture conditions overnight. Then, 5 µL of each compound was transferred to the assay plates and incubated as above for 30 min. Next, 5 µL of 10 µg/mL LPS (Escherichia coli 0111:B4; L4130, diluted in assay media from a 3-mg/mL stock in RPMI-1640) was added to the assay wells, and 5 µL of assay media was added to unstimulated control wells. Plates were incubated as above for 4 h. Then, 25 µL of supernatant was removed and transferred to black clear-bottom 384-well plates (FMAT assay plate; Greiner 781091).
TNF-α determination by FLISA
In total, 25 µL of detection mix was added per well, to the FMAT assay plates containing the supernatants. Plates were incubated at ambient temperature overnight and read on the FMAT (mean fluorescence count, factory settings for the photomultiplier tubes and event cutoff = 20).
Data analysis
Data conversion of fluorescence units (FLU) readings to TNF-α concentrations was based on the standard curve and used for subsequent calculation of % inhibition. The effect of each compound was calculated as
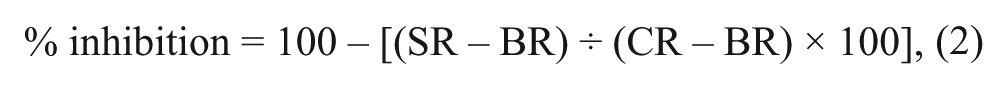
where control reading (CR) denotes LPS-stimulated cells and compound solvent, sample reading (SR) denotes LPS-stimulated cells and compound, and background reading (BR) denotes cells, no LPS, and compound solvent. Active compounds were considered those showing % inhibition ≥40% at the 10-µM test concentration. This cutoff was applied as it was the point at which it was considered that compounds showed activity beyond the noise of the assay, being approximately 1.5 interquartile ranges from the third quartile, and hence defined as an outlier. Compounds were routinely tested in duplicate in a single experiment and the maximum % inhibition achieved reported.
A selection of actives was taken into concentration-response confirmation; this was performed under the conditions described above for the single-concentration screening, with a maximum concentration of 30 µM.
Enrichment Analysis
Screening results were analyzed at the target, pathway, and biological process levels. Let S+ denote the subset of compounds found active in the screen and
To assess the null hypothesis of no association between historical bioactivity data on target i and the screening results, Fisher’s exact test was applied to Table 1 .
Contingency Table.
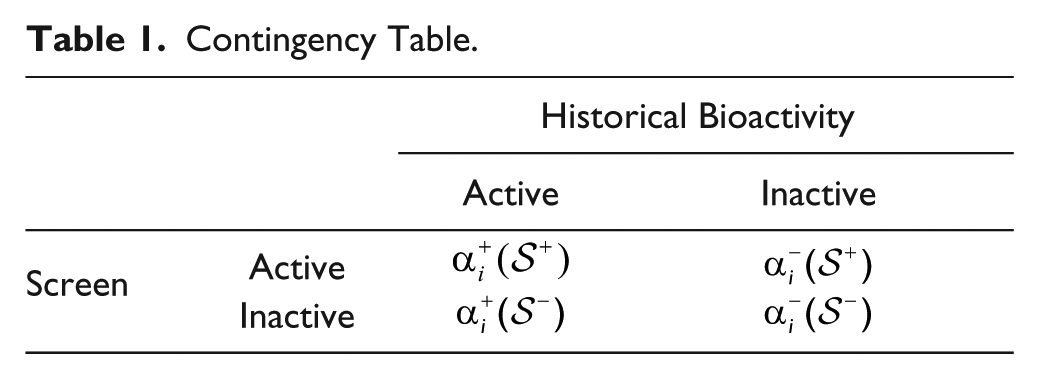
To correct for multiple hypothesis testing, a Bonferroni correction was applied and significance was called at the
Two types of pathway enrichment were assessed.
First, all canonical maps from GeneGO MetaBase were considered. For each map, all compounds active on any target belonging to this map were considered active, and compounds inactive on all targets from this map were considered inactive. The contingency table can thus be populated and the null of independence assessed as above for each pathway.
Second, a subnetwork enrichment considering the whole interactome available in GeneGO MetaBase was assessed. All targets were clustered using hierarchical clustering (Ward’s method 23 ) with a similarity measure, which was the number of common neighbors in the graph weighted by the inverse logarithm of their degrees. 24 The null of no association between a cluster and the screening results was assessed as above with the contingency table formed as the sum of contingency tables of all targets in the cluster.
Similarly, enrichment of GO biological processes 25 was determined by testing GO terms (including ancestors) for all targets in the design under the null of independence with the targets found to be enriched.
Results
Selection of the Compound Set for Screening
A library of approximately 650,000 compounds was used as a starting point to select the compound set for screening. For those 650,000 compounds, a total of 3,050,580 compound-target pairs were extracted from CHEMBL, GoStar, BioPrint, and in-house activity data across 3060 proteins.
Using the optimization method described in the Materials and Methods, a subset of 10,000 compounds maximizing the number of proteins for which enough data were available for the statistical analysis was selected using α = 10–6. Using this value of α, chosen conservatively to allow some discrepancy with historical bioactivity data (see
To enrich this screening set with compounds active on targets potentially affecting TNF-α production, the pathway databases IPA and GeneGO were queried. Sixty-nine genes whose complete or partial deletion in mouse models leads to a modification of TNF-α levels were extracted from IPA, and 180 proteins upstream of TNF-α in canonical linear pathways were extracted from GeneGO MetaBase (from 4850 linear pathways containing TNF-α). The two lists of targets were complementary to each other, with only nine targets in both. Of these 240 targets, 178 were not in the 665 proteins that can be identified as enriched. An analysis of the historical bioactivity data available (see Materials and Methods) identified a few more targets where the coverage could be improved, and in total, 165 compounds were added to the screening set. This increased the number of targets that could potentially be identified as enriched to 672 and provided a final screening set of 10,165 compounds.
Screening and Enrichment Analysis
The assay performed as expected and within the performance expectations of a robust assay,
26
with a Z′ of 0.5 or greater and at least a 10-fold assay window. The detection range for rhTNF-α was approximately 60 to 2000 pg/mL, with a midpoint of 1 ng/mL. In total, 1045 of the 10,165 compounds selected were classified as active by the criteria described (see
Integrating both the TNF-α production screen results and the historical bioactivity data from CHEMBL, GoStar, BioPrint, and AstraZeneca, an enrichment analysis was run across all targets for which activity data were available (2750 proteins for the 10,165 compounds selected). Figure 1 presents the p values and odds ratios obtained for each target. Sixty-four protein targets were highlighted as enriched (targets above the significance threshold—indicated by the green line—in Figure 1 and Table 2 ). Half were kinases (32 proteins). The five top-ranking targets were MET, GSK3B, JAK3, PAK1, and PTK2.
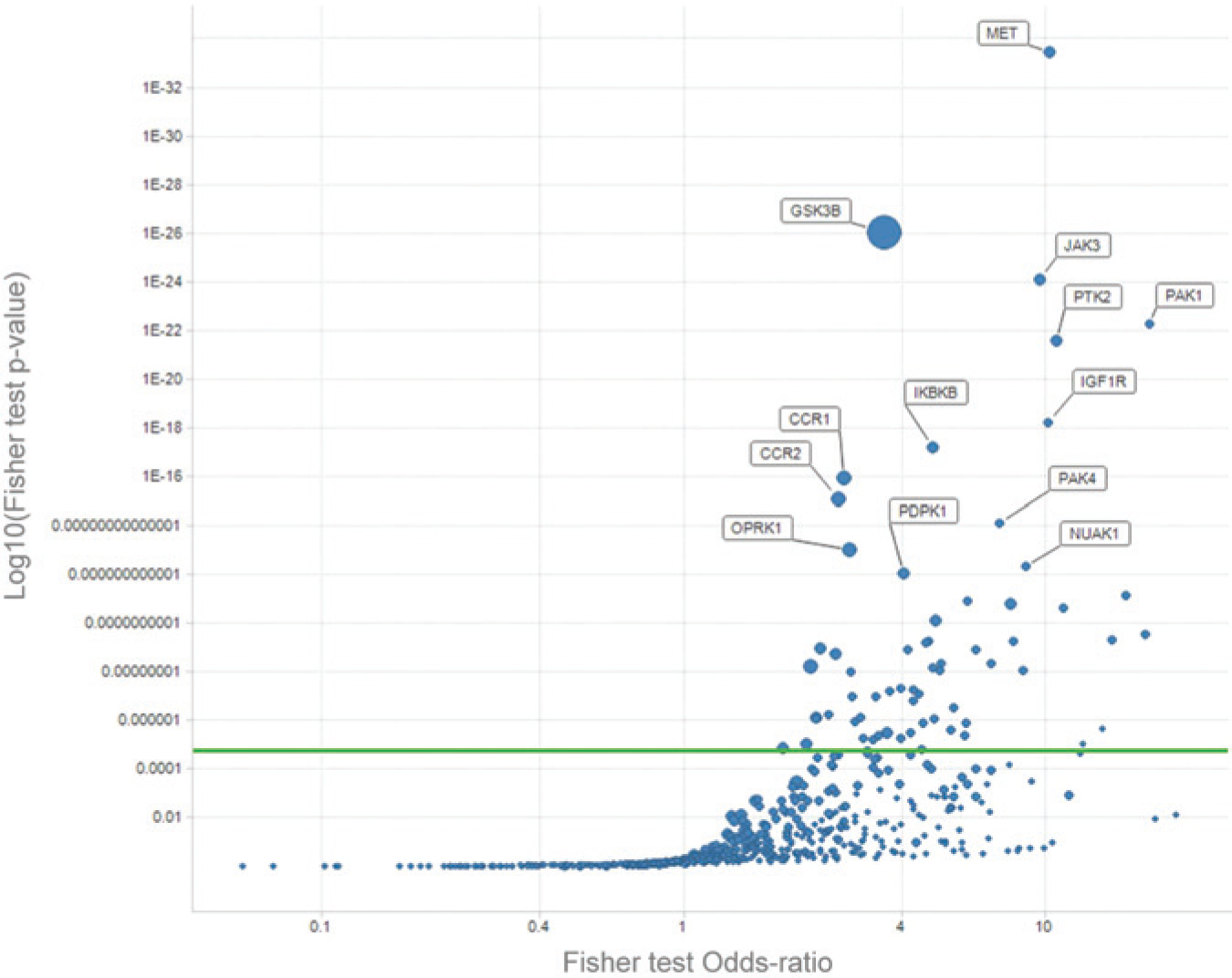
Target enrichment results for tumor necrosis factor (TNF)–α production. Each dot represents a protein target. The green horizontal line is the Bonferroni threshold for the enrichment analysis. All targets above the green line are statistically enriched with compounds active on them and on TNF-α production. The higher and the further right a target is, the more it is associated with TNF-α production. The size of the dots corresponds to the number of compounds active on the target and on TNF-α production. The most significantly enriched targets have been tagged with their short names.
Targets Enriched in Compounds Both Active on Them and on TNF-α Production.
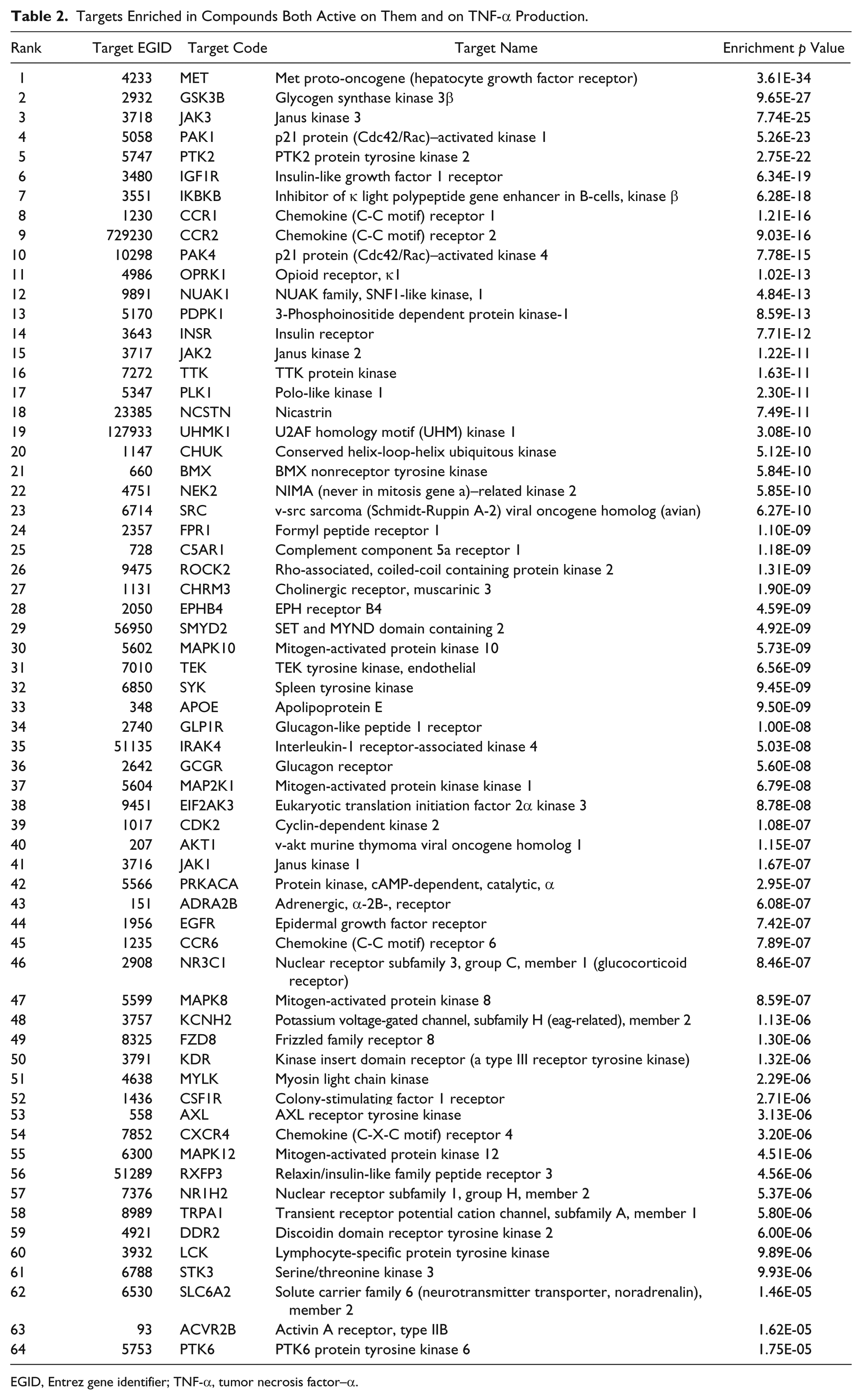
EGID, Entrez gene identifier; TNF-α, tumor necrosis factor–α.
To capture targets for which compounds were not available or for which historical data were insufficient for the target enrichment analysis, a canonical pathway map enrichment was carried out as described in the Materials and Methods based on canonical maps from GeneGO. Of the 862 maps available, 504 maps were found enriched. This high number of enriched pathways was likely due to the high number of screened compounds that were active on various kinases and on the high number of kinases in the signaling cascades presented in the GeneGO canonical pathways. Nevertheless, among the enriched maps, 47 included TNF-α, and 83 were related to the immune response or to inflammation. Three of the five top-ranking maps (see Table 3 : “Activation of PKC via G-protein coupled receptor,” “cAMP signaling,” and “VEGF signaling via VEGFR2—generic cascades”) represented generic signal transduction cascades involving mainly the protein kinase C (PKC) family and various MAP kinases (including p38). They also included GSK3B, SRC, and PAK1, for instance, all identified during the previous enrichment analysis. Finally, they involved CREB1, NF-κB, c-Jun, and Elk-1, with all four being recorded as potential regulators of TNF-α production in GeneGO Metacore. 27 The two remaining top-ranking canonical maps concerned the immune response and embryonal myogenesis. They also included relevant targets for TNF production such as CREB1, the PKC family, p38, NF-κB, and PAK1 (the latter also identified in the previous target enrichment analysis).
Top-Ranking Canonical Pathways from the Enrichment Analyses.

TNF-α, tumor necrosis factor–α.
The five top-scoring canonical maps including TNF-α, respectively, ranked 17, 35, 49, 53, and 60 ( Table 3 ). Those maps contained most of the general signaling cascades and targets highlighted above. The map with GeneGO map ID 6241 (“IgE- and MGF-induced Lyn-mediated production of cytokines and arachidonic acid metabolites”), for instance, presents several paths that were affected by the compounds screened and that were observed to have an effect on TNF-α production, providing a mechanistic rationale with regard to how TNF-α and inflammation can be affected in mast cells in asthma.
The network enrichment based on interactome clustering is summarized in Table 4 . To limit the size of the networks considered for enrichment, only clusters with a merging cost less than 0.1, equivalent to approximately the first 1/10th mergers and for which agglomerations led to improved p values, were included. A total of 15 subnetworks were identified, with JAK1-3, PTPN6/11, and STAT5B being most significant.
Subnetworks Enriched in Compounds Both Active on Them and TNF-α Production.
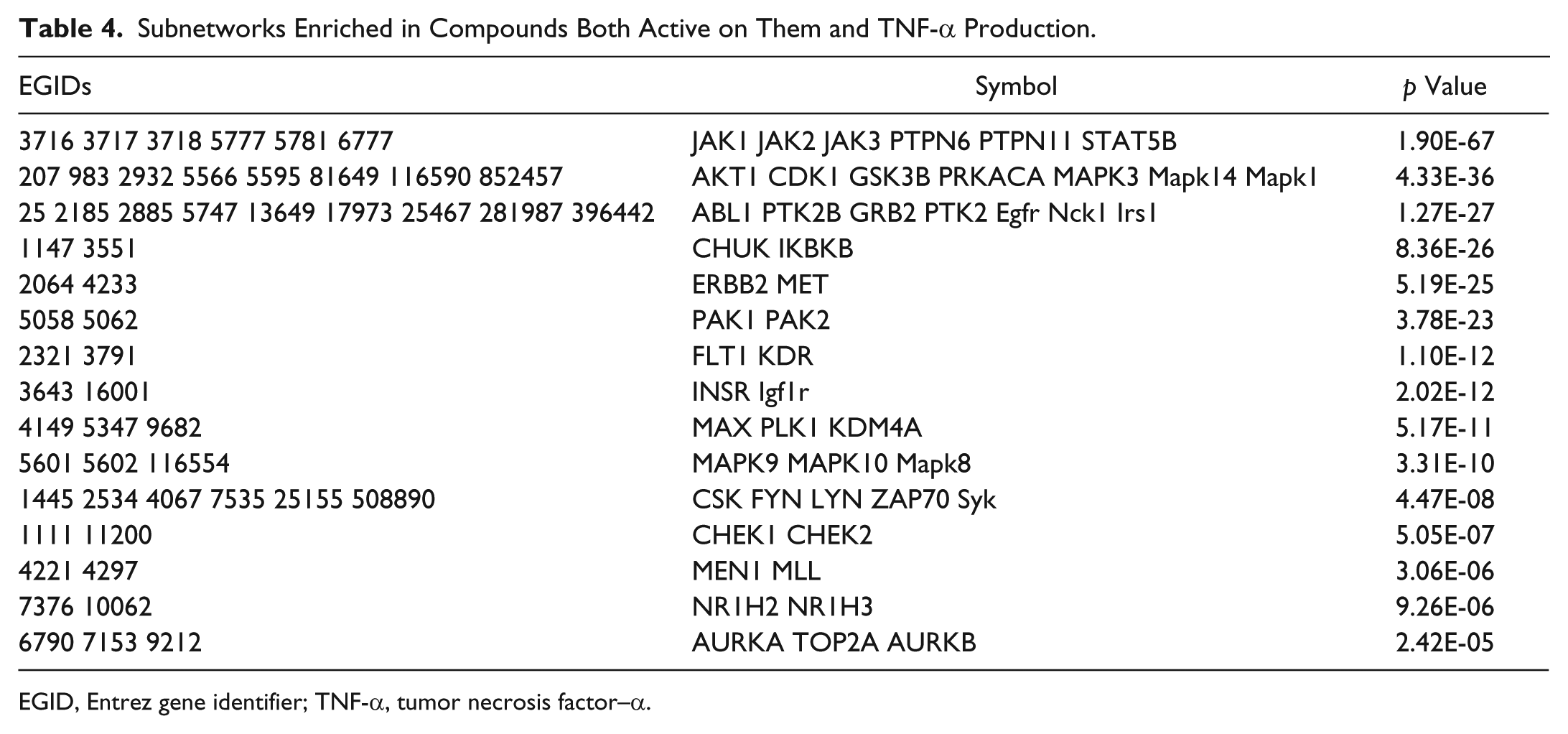
EGID, Entrez gene identifier; TNF-α, tumor necrosis factor–α.
The biological process analysis identified 71 enriched GO terms from a total of 4358 terms containing the targets in the design (see
Discussion
The LPS-stimulated TNF-α assay selected for this screen was initiated to identify compounds that could be optimized for treatment of respiratory disease as bacterial infections have been implicated in exacerbations of both asthma and chronic obstructive pulmonary disease. 28 The pathway by which LPS binds to TLR4 and stimulates production of proinflammatory cytokines such as TNF-α and interleukin (IL)–6 via a MyD88-dependent mechanism has been well characterized. 29 Several of the 64 protein targets that were identified by the enrichment analysis are known to be involved in the pathway of LPS-stimulated TNF-α production ( Table 2 and Fig. 1 ). These include IRAK4, which interacts with MyD88, CHUK, and IKBKB (IKKα and β), which are upstream of NF-κB transcription, and MAPK8 and MAPK10 (JNK1 and 3), which are involved in the activation of the transcription factor AP-1. The transcription factors NF-κB and AP-1 are known to be required for expression of inflammatory cytokines, including TNF-α. In addition, the target enrichment approach highlighted MAP2K1 (MEK1), which activates the MAP kinases ERK1 and ERK2, leading to enhanced TNF-α transcription via Egr-1 binding to the TNF-α promoter. LPS has also been shown to stimulate other pathways leading to TNF-α production, and members of these pathways, such as AKT1, which is downstream of PI3-kinase, and SYK, which can activate MAP2K1 and JNK, were also identified. Interestingly, a number of proteins that have been implicated in the negative regulation of the pathway were also identified, such as GSK3B and the JAK kinases JAK1, 2, and 3. This may be because the enrichment analysis only considered association of the target with the pathway and not the direction of effect, so that both agonists and antagonists of a target would be included. Cross-talk between pathways may also account for the inclusion of some proteins on the target list such as the focal adhesion kinase FAK (PTK2), which has been shown to interact with the MyD88 pathway, and the insulin receptor INSR and insulin-like growth factor receptor IGF1R, which activate the PI3K/Akt and ERK/MAPK pathways. Further targets such as PAK1 and the hepatocyte growth factor receptor MET are known to be activated by LPS signaling. It is perhaps surprising that p38 MAP kinase (MAPK14) was not identified as a target from the enrichment analysis as p38 is involved in the transcriptional and posttranscriptional control of TNF-α production, and p38 inhibitors block LPS-stimulated TNF-α production in human monocytes.
The information obtained from pathway map enrichment is broadly supportive of the results of the target enrichment analysis ( Table 3 and Fig. 2 ). The top-ranking canonical pathways not including TNF-α comprise multiple signaling molecules known to be involved in LPS-stimulated TNF-α production such as GSK3B, ERK, JNK, and p38 MAPK ( Table 3 ). Although the target enrichment did not highlight the cAMP signaling pathway as important, this was identified by the pathway analysis as the third ranking pathway, which is consistent with literature showing that agents that elevate cAMP suppress TNF-α production. This also raises the question of why the β2 adrenergic receptor, as a G-protein–coupled receptor (GPCR) coupled to intracellular elevation of cAMP, was not identified on the target list. A possible explanation is that in the target enrichment analysis, the statistical significance of an inhibitory effect of an agonist for a given target in the assay would be reduced by lack of effect of an antagonist for the same target. An enhancement of the approach taken in the current study would be to separate the analysis on the basis of whether compounds are agonists or antagonists of the target.
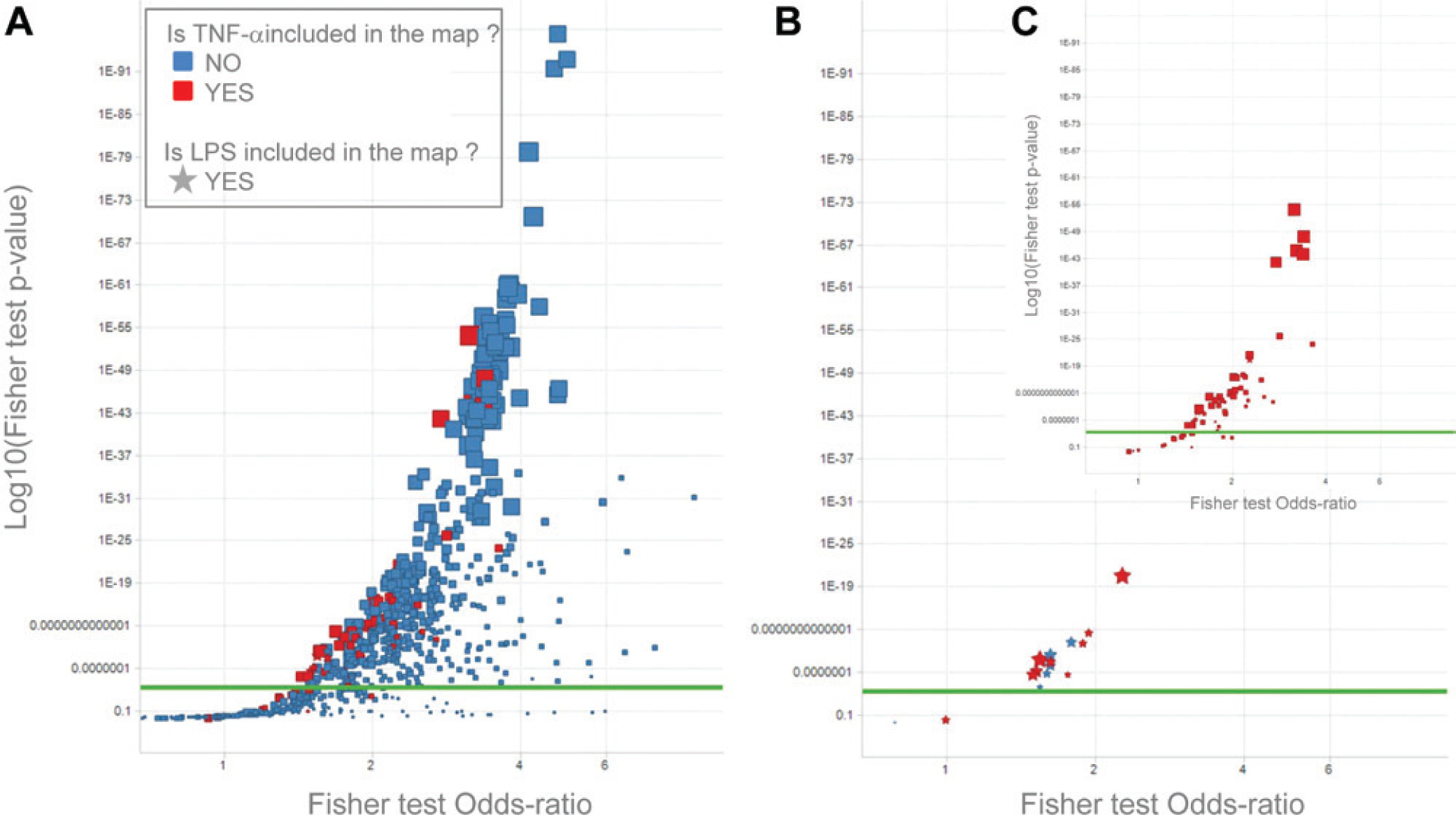
Canonical pathway map enrichment results for tumor necrosis factor (TNF)–α production. (
The pathways identified that include TNF-α also incorporate many signaling molecules involved in LPS-stimulated TNF-α production such as AKT, GSK3B, and ERK ( Table 3 and Fig. 2C ). Interestingly the top-ranking pathways including TNF-α have a strong linkage to respiratory disease and include pathways involved in hypertrophy of airway smooth muscle cells and mast cell mediator production and survival. The overlap between the pathways gives the potential to identify targets and compounds from the analysis of the LPS-stimulated TNF-α production assay in monocytes that might have activity against multiple cell types implicated in the asthma phenotype. A further analysis to show pathways that include LPS ( Fig. 2B ) identified pathways that are predominantly involved in the innate immune response to bacterial infection. Several of the pathways are linked to cystic fibrosis (CF), suggesting that there may be common mechanisms and targets between asthma and CF that are worth further investigation. A high proportion of the pathways involving LPS also include TNF-α (8 of 13), which is due to the strong linkage of LPS signaling to TNF-α production in addition to the similarity of the signaling pathways by which TNF-α binding to the TNF receptors and LPS signaling via TLR4/MyD88 stimulate NF-κB activation.
The network enrichment builds on the target and pathway enrichment by clustering targets into molecules with similar neighborhoods in the interactome and, thus, potentially with similar functions. The results of this analysis are also consistent with the known signaling pathways for LPS-stimulated TNF-α production and highlight the JAKs, MAP kinases, IKKs, and PAKs as families of key signaling molecules. Similarly, the biological process analysis is very strongly enriched for processes related to protein phosphorylation and regulation of protein kinase activity (see
The approach described in this article has the advantage of combining historical bioactivity data for multiple compounds to strengthen the evidence that a particular pathway is involved in the biological effect under investigation. The relatively high active rate of about 10% in the screen was expected since the screening set was specifically designed to enable investigation of activity against TNF-α, not only because of including compounds with activity on targets upstream of TNF-α but, more important, because of selecting compounds with activity profiles allowing coverage for enrichment analysis of almost 700 targets and with recorded activity on 2111 proteins.
In addition, since many of the compounds in the set exhibit polypharmacology, the enrichment analysis can help to identify compounds that show efficacy through inhibition of multiple pathways. These compounds could be used as starting points for lead optimization and combined with a more traditional chemoproteomics approach to identify the interacting proteins to fully deconvolute the target(s). Understanding the mechanism of action of a compound is important in the drug discovery process to assess potential target-related side effects and selectivity to related targets, as well as facilitate rational drug design. However, there can be limitations of the chemoproteomics approaches due to low abundance of the target protein, low potency of the interaction between compound and relevant target, and identification of a large number of interacting proteins. In this situation, combining the output of the chemoproteomics with the target and pathway enrichment analysis can aid the identification of the relevant target protein(s).
The enrichment analysis reported was based on data from an assay in which compounds were screened at a single concentration and activity determined based on a fixed threshold for inhibition (40% at 10 µM). In the same way, a threshold was applied to historical bioactivity data: a compound is considered active on a target if 10 µM or less is sufficient for a minimum percentage effect of 50%. Fifty percent activity was selected for the historical data as this was considered an appropriate cutoff to apply across a range of targets to ensure the result was outside of the noise of the assays. This binarization of the data may not be suitable for all compounds or targets (e.g., for some targets, a threshold of 1 µM might make more sense). An improvement of this strategy might be to take into account compound-target affinity to perform correlation analyses linking compound effects on TNF-α and compound affinity for specific targets. This could provide a more accurate view of the targets affecting TNF-α. This could also decrease the number of false-positive targets in the enrichment analyses due to compound polypharmacology. If two targets have a similar binding site (therein a similar set of active compounds) and if one of the targets is affecting TNF-α production, the second protein will also be identified as an enriched target with the approach described herein. The use of experimentally determined potency values could help capture more subtle differences between targets and thus improve this limitation of the method. A further refinement would be to perform secondary screens such as a cytotoxicity assay and an LPS-stimulated TNF-α production assay in primary human monocytes to increase confidence in the relevance of the compound activity to the biological effect of interest.
One disadvantage of the approach described herein is that it is limited by the availability of historical data for compounds and targets. A target without enough bioactivity data cannot be identified as enriched. The amount of data available per target could be increased by including activity data from orthologs as well as from predicted target activity based on chemical structure. This might also help reduce the bias due to known “druggable” targets such as kinases and GPCRs as compounds with activities against these target classes are overrepresented in the compound collection. A phenotypic screen on a broader set of compounds that was either not subject to bioactivity-based preselection criteria or selected on the basis of chemical diversity would increase the potential to identify a compound with a completely novel mechanism of action. However, in this case, a relatively large number of compounds would need to be screened, thus demanding an assay format that is amenable to high throughput. In addition, the identified hit might be only weakly active.
As already highlighted, computational target deconvolution is a first step in the process of phenotypic drug discovery. The identified targets can be validated by siRNA techniques or sometimes pharmacologically by adding a compound that would antagonize the effect of the screening hit. In conclusion, we have described a novel screening set design and target deconvolution method using historical bioactivity data to identify targets and pathways involved in a phenotypic response. We also illustrated that these methods can provide valuable targets in the TNF-α pathway that could be progressed for further target validation.
Footnotes
Acknowledgements
The authors acknowledge Dr. John Unitt for the phenotypic screening design concept and implementation and Philip Rawlins for his support and input into the experimental methodology. The authors also thank Dr. Mike Firth and Dr. Péter Várkonyi for their support during the extraction of historical activity data. Dr. Firth also supported the extraction of linear pathways from MetaBase.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.