
Abstract
eEF-2 kinase is a potential therapeutic target for breast cancer, gliomas, and depression. No potent inhibitors of eEF-2K have been reported, and thus development of high-throughput assay systems may expedite the process. Two high-throughput assays are described for eEF-2K using recombinant, tag-free enzyme purified from bacteria. The first is a fluorescence-based assay that uses the phosphorylation of a Sox-based peptide substrate by eEF-2K, which results in a 5-fold increase in fluorescence emission, allowing for continuous monitoring of the kinase activity. The second is a luminescence-based assay that produces a luminescence signal, which correlates with the amount of adenosine triphosphate remaining in the kinase reaction. Both assays have been optimized and miniaturized for a 384-well plate format and validated in screens. In conclusion, we demonstrated that a traditional radiolabeled assay can be readily transferred to universal spectroscopic assays that are robust and will facilitate high-throughput screening of larger size libraries for the identification of small-molecule inhibitors and significantly contribute to the development of therapies for targeting eEF2K.
Introduction
Eukaryotic elongation factor 2 kinase (eEF-2K), also known as calcium/calmodulin-dependent protein kinase III (CaM kinase-III), shows increased activity in several cancer cell lines and has been identified as a potential therapeutic target.1–3 Most recently, Leprivier et al. 4 provided evidence for an important role for eEF-2K in protecting cells from nutrient deprivation, potentially allowing tumors to exploit eEF-2K to support adaptation to metabolic stress. While small-molecule inhibitors of eEF-2K can facilitate the study of its biology as well as the development of therapeutics, recent attempts to identify small-molecule inhibitors of eEF-2K have had mixed results. For example, NH125 was reported to inhibit eEF-2K activity both in cells 5 and in vivo 3 but was later shown to be a promiscuous aggregator, in which its inhibition activity is prevented by the presence of detergent. 6 In contrast, A-484954, a first-generation inhibitor, exhibits moderate potency in cells and represents a useful agent for studying eEF-2K in a variety of cell lines. 7
Small-molecule inhibitors are often identified through campaigns that begin by screening a large number of compounds against a target enzyme in vitro. Identification and characterization of lead kinase inhibitors through high-throughput screening (HTS) requires robust biochemical or cellular assay systems. A traditional method to determine the catalytic activities of protein kinases is to quantitate the incorporation of radioactive γ-phosphate from [γ-32P] adenosine triphosphate (ATP) into a peptide or protein substrate using a scintillation counter. Although a radiolabeled assay to screen inhibitors against eEF-2K was reported by Ryazanov et al., 8 it is an impracticable approach for a high-throughput application.
Fluorescence or luminescence assays represent better alternatives in high-throughput campaigns, but only a few assays have been reported for identifying inhibitors of eEF-2K, supported by brief experimental information,9,10 and no high-throughput screen (i.e., over 384 well) targeting eEF-2K has been reported in detail. Furthermore, previous assays used a version of an enzyme containing a GST tag, which is dimeric in nature and can interfere with the protein structure and function when fused to the protein of interest (Recombinant Protein Purification Handbook, https://www.gelifesciences.com/gehcls_images/GELS/Related%20Content/Files/1336168762999/litdoc18114275_20130524112435.pdf).
Sox (sulfonamido-oxine amino acid) chromophore-modified peptide substrates (referred here as “Soxtides”) have been shown to be versatile reagents for sensitively reporting peptide phosphorylation via chelation-enhanced fluorescence. 11 Upon phosphorylation of the Sox-containing peptide, the binding affinity of the Sox chromophore for Mg2+ increases significantly, resulting in a large fluorescence increase (2–12-fold), which can be monitored continuously at 485 nm ( Fig. 1A ).11,12 A range of Sox-based fluorescent peptide substrates have been commercialized by Invitrogen (Carlsbad, CA), and this method has been applied to numerous Ser/Thr and Tyr kinases both in vitro 11 and in cell lysates. 12 To date, a Soxtide targeting eEF-2K has not been reported.
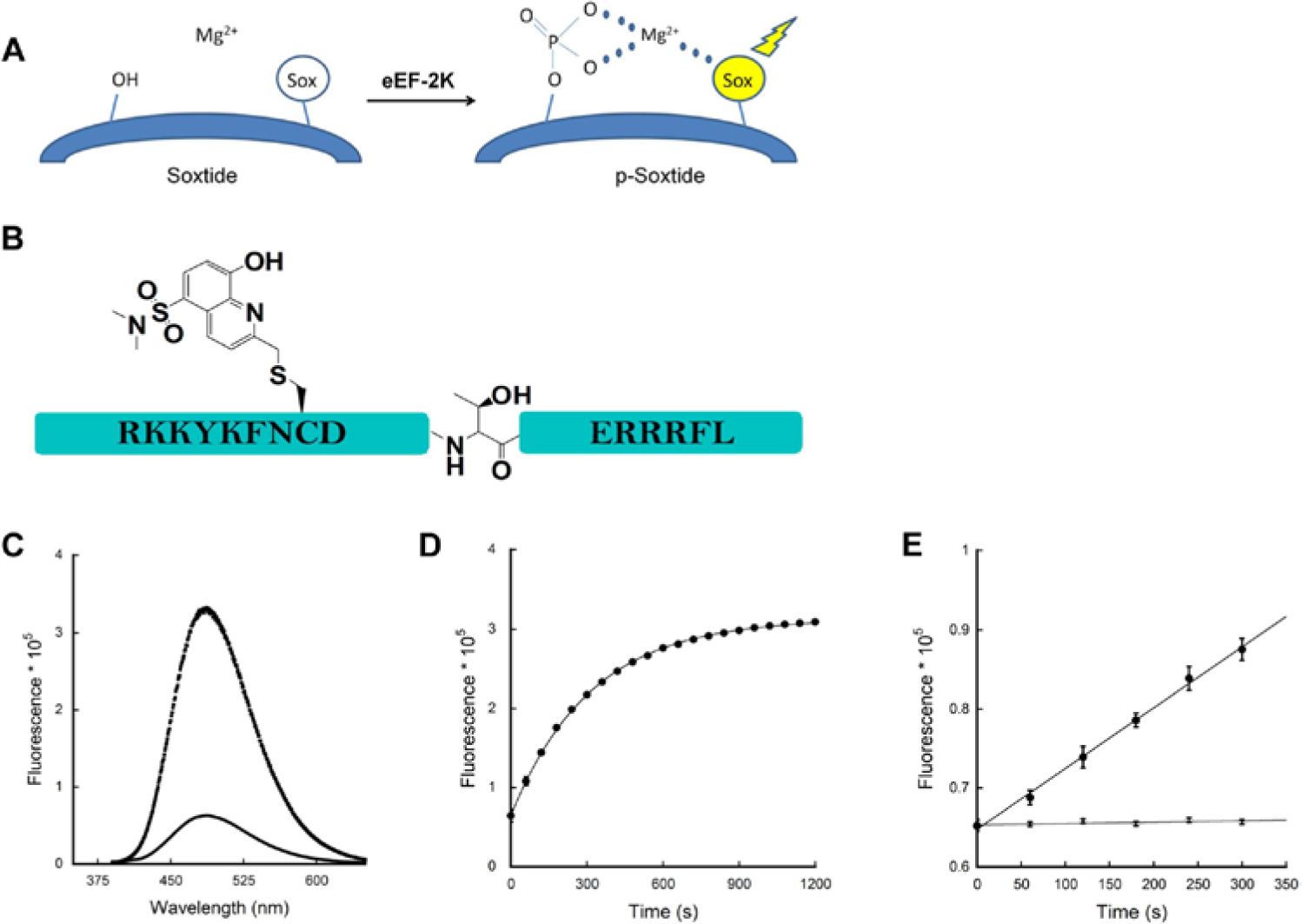
Sox-assay development. The fluorescence was monitored using a Fluorolog model FL3-11 fluorometer (Jobin Yvon, Edison, NJ) equipped with excitation (360 nm) and emission (485 nm) filters and three-window fluorescence grade quartz cuvettes (path length of 10 mm; Helma, Plainview, NY). (
We previously reported a protocol for fluorescence-based assays to determine the activity of kinases. 13 In the past, we also purified a tag-free version of eEF-2K expressed in Escherichia coli and developed an assay that can sensitively monitor the enzymatic activity against a peptide substrate using radiolabeled ATP. 14 The tag-free eEF-2K showed almost 350-fold higher activity toward a peptide than the GST-tagged eEF-2K that was earlier reported by Pavur et al. 15
Herein we report the development of the Sox-based florescence assay toward eEF-2K to a 384-well format and demonstrate its potential for high-throughput applications. We also report the miniaturization of a luminescence-based assay against tagless eEF-2K using the Kinase-Glo Luminescent Kinase assay kit (Promega, Madison, WI). The luminescent signal represents an orthogonal assay format allowing the convenient elimination of false positives from other assay platforms. The feasibility of the luminescence-based assay for high-throughput application was demonstrated in a 384-well plate format. Together, the Sox-based fluorescence- and luminescence-based assays are demonstrated to be robust for the identification of small-molecule inhibitors targeting eEF-2K.
Materials and Methods
The expression and purification of eEF-2K were followed accordingly as explained elsewhere.
14
Details on buffer components used in the enzyme purification and assays are provided in
Sox-Based Fluorescence Assay Development
Fluorescence assays were performed with 10 nM eEF-2K, 100 µM ATP, and 10 µM Soxtide in 100 µL volume at room temperature. For estimating the background activity, the same assays were performed in the absence of eEF-2K. Assays were initiated and monitored at 1-min intervals. For determining maximum signal change upon phosphorylation, assays were allowed to proceed to the end point using 100 nM (10×) eEF-2K before the emission spectrum was recorded. The time required for the reaction to reach the end point was estimated by incubating 100 nM (10×) enzyme with 10 µM Soxtide and monitoring activity for up to 5 half-lives. The effect of magnesium concentration was examined by varying MgCl2 concentration.
Assay tolerance in the presence of DMSO and Brij-35 was tested by varying DMSO and Brij-35 concentrations in the assay. To test the effect of incubation time on assay stability, assay mixture was incubated at room temperature for different amounts of time before initiating the assay. Assay validation was performed by filling half of the plate with assay mixture, while the other half of the plate was filled with assay mixture excluding enzyme. In all cases, assay mixtures containing enzyme and Soxtide were prepared initially, and the fluorescence assays were initiated with the addition of ATP.
Application of Sox-Based Fluorescence Assay for HTS
Of the assay mixture, 7 µL was first dispensed to an assay plate. Compounds dissolved in 100% DMSO at a 10-mM concentration were first diluted in water and then transferred to assay plates (two-step dilution). In detail, for compounds screened at a final concentration of 25 µM, first 0.5 µL of the compound (10 mM stock in 100% DMSO) was dissolved in 19.5 µL of water to achieve a compound concentration of 250 µM; then, 1 µL of the dilution was added in a 10-µL assay to achieve a final concentration of 25 µM. For compounds screened at a final concentration of 100 µM, first 2 µL of the compound was dissolved in 18 µL of water to achieve a compound concentration of 1 mM; then, 1 µL of the dilution was added in a 10-µL assay to achieve a final concentration of 100 µM. Enzyme was allowed to incorporate with compounds for 30 min at room temperature prior to being initiated with the ATP. Following the addition of 2 µL of ATP, fluorescence was monitored for 5 min with 1-min intervals.
Luminescence-Based Assay Development
Initially, assays were performed manually in white 96-well plates by varying reagent (enzyme, ATP, or unlabeled peptide) concentrations or reaction time to determine the optimum condition. In detail, 60 µL of the assay mixture (enzyme and peptide diluted in assay buffer) was pipetted into a plate, followed by the addition of 40 µL ATP to initiate the enzymatic reaction. After incubating at room temperature for various times, 100 µL of the Kinase-Glo Luminescent Kinase assay kit prepared according to the manufacturer’s protocol was added and the luminescence was measured 10 min later. Assay tolerance, miniaturization, and validation were performed according to the methods applied to Sox-based fluorescence assay described above.
Application of Luminescence-Based Assay for HTS
Final concentrations of eEF-2K, ATP, and peptide were set to 5 nM, 1 µM, and 30 µM, respectively. In detail, 5 µL of assay mixture (eEF-2K and peptide diluted in assay buffer) was first dispensed to an assay plate. Compounds were diluted and transferred to an assay plate as described above. Enzyme was first allowed to incorporate with compounds for 30 min at room temperature and then incubated an additional 80 min in the presence of 4 µL ATP. Following the addition of 10 µL of the Kinase-Glo Luminescent Kinase assay kit and incubation for 10 min at room temperature, luminescence was measured.
Confirmation Screen
First, compounds showing greater than 50% activities from the Sox-based fluorescence screen were cherry-picked and rescreened using the same assay in duplicates. Then the compounds showing reproducible activities (>30%) in the rescreen were further confirmed using an orthogonal radioactive assay (details are described in
Data Analysis
Data calculations were performed using Excel (Microsoft, Redmond, WA), whereas all data plots and fittings were performed using Kaliedagraph 4.0 (Synergy Software, Reading, PA). Each data point represents mean ± standard deviation. Data acquired from screening were mined for hit identification using the CDD database (Collaborative Drug Discovery, Burlingame, CA).
Results and Discussion
Sox-Based Fluorescence Assay Development
Recently, we reported a peptide substrate for eEF-2K, Ac-RKKYKFNED
We then asked whether Soxtide supports a fluorescence assay and whether its signal enhancement is sufficient to support a high-throughput campaign. The same assay protocols for buffer, peptide substrate, eEF-2K, and ATP were directly employed based on earlier studies from the radiolabeled assay,6,14 without further optimization. In the same way as the radiolabeled assay, higher concentration of ATP (100 µM) than its Km (40 µM) was employed to search ATP noncompetitive inhibitors. Effect of magnesium concentration on the assay performance was examined. As the enzyme activity was consistently above 10 mM of MgCl2 (data are shown in
Unlike the end-point–based assays, kinetic assays monitoring the reaction continuously over time ( Fig. 1D ) and measuring signal intensity in terms of the initial velocity (v) allow us to exclude false hits due to any signal interference. As v is usually measured under assay conditions where the substrate conversion to product is less than 10%, it is necessary to make sure that the assay is sensitive enough to be monitored under such conditions. Initial velocity was calculated and the signal enhancement was linear for the first 5 min, resulting in a v of 74 relative fluorescent units (RFU)/s ( Fig. 1E ). On the other hand, no significant signal change occurred in the absence of eEF-2K, as expected. These results demonstrated that Soxtide is a sensitive fluorescent reporter for the monitoring of the enzymatic activity of eEF-2K. Measurements over 5 min with 1-min intervals per assay were employed for further optimization and HTS.
The next step was to implement the assay for HTS, where it should be subjected to automation in a plate format, preferably miniaturizing the assay volume but maintaining high sensitivity. The assay was transferred to 384-well plates by reducing the assay volume to 10 µL while retaining the same assay compositions as described above. As high-throughput screens are usually conducted at room temperature where reagents may need to be stored for a long time, the assay should be tolerable for extended operation at room temperature. Incubation for up to 3 h at room temperature did not reduce the enzyme activity significantly, while only a slight reduction in activity was observed after 5 h (data not shown).
Assessing the tolerance under DMSO is another critical step in high-throughput assay development, because standard chemical libraries are typically dissolved in 100% DMSO and the degree of DMSO tolerance limits the concentration of compounds that can be tested. Up to 2.5% DMSO had no significant effect on the enzymatic activity (data not shown).
Usually, the high-throughput assays contain a small amount of additives to improve assay quality by avoiding nonspecific adherence of reagents to assay plastics (robotic tips, assay plates, and reagent reservoirs) and by eliminating the false hits due to promiscuous aggregation of small molecules. Brij-35, a nonionic detergent, is one of the common additives whose use has been reported in several protein kinase assays and high-throughput screens. As this was also our choice of detergent, its effect on eEF-2K stability was tested and no significant effects on the enzyme activity were observed within the range of concentrations tested (data not shown). Therefore, a final concentration of 0.03% Brij-30 (w/v) was chosen for further assay optimization.
Last, assay validation produced a reproducible Z′ factor (0.75 ± 0.1) from three consecutive experiments, supporting the robustness of the assay for high-throughput application. Although the absolute fluorescence intensity after 5 min was only around 1.5-fold higher than the background, the signal-to-background ratio (S/B) and the signal-to-noise ratio (S/N) were almost 70:1 and 21:1 when measured as an initial velocity, respectively. These results also warrant the benefit of kinetic measurement over absolute intensity assessment.
Sox-Based Fluorescence Assay Applied for HTS
Compound libraries in a 384-well plate were typically formatted by locating compounds dissolved in 100% DMSO through columns 3 to 22, while columns 1, 2, 23, and 24 were filled with DMSO only. Therefore, a maximum 320 compounds are plated in a 384 well-plate format. This standard format allows the use of columns filled with DMSO (without chemicals) as controls to validate assay plates individually—for example, columns 1 and 2 as positive controls and columns 23 and 24 as negative controls, or vice versa. To correspond with the standard library plate format, assay plates were prepared by dispensing positive control into columns 1 through 22 and dispensing negative control (assay mixture excluded with eEF-2K) into columns 23 and 24.
Initially, the pilot screen was performed using an NIH Clinical collection (NCC collection) (Evotec Inc., South Francisco, CA) containing a small library of 446 compounds with known toxicity profiles and a Microsource Spectrum collection (Discovery Systems Inc, Gaylordsville, CT) (2000 compounds) at a concentration of 100 µM without duplicates. A pilot screen is often useful to get an estimation of hit rate and to adjust the compound concentration accordingly, if needed, before progressing into the larger sized libraries. The average Z′ and S/N values calculated from our pilot screen for both the NCC and Spectrum collections were 0.76 ± 0.8 and 11:1, respectively. Twenty-two (4.9% hit rate) from the NCC collection and 131 (6.6% hit rate) compounds from the Spectrum collection showing over 50% activity were identified as primary hits. The resulting hit rates were higher than the commonly manageable range of 1%, and several of these hits also exhibited very high levels of intrinsic fluorescence that interfered with assay responses. As a result, the compound concentrations were reduced to 25 µM for the Chembridge Kinase set (ChemBridge Corporation, San Diego, CA) (11,250 compounds) and Maybridge Hitfinder v9 (Fisher Scientific, Pittsburgh, PA) (14,400 compounds) for following screens. Unlike typical libraries that follow Lipinski’s “rule of 5,” Fragment collection is usually designed by “rule of 3,” consisting of lower molecular weight and complexity. The small molecular size makes the Fragment set beneficial for lead optimization specially. Because smaller molecules tend to show weaker activity than bigger compounds, the Chembridge Fragment library (4000 compounds) was screened at 100 µM to obtain a comparable hit rate.
The quality of more than 100 assay plates (
The most common drawback of a fluorescence assay is that compounds may absorb light, resulting in signal amplification and an increase in false positives. In our Sox assay, almost 9% of the total compounds had some level of intrinsic fluorescence. Among them, 7% of the total compounds had a weak autofluorescence (signal intensity within 5-fold of control), whereas the rest, 2%, had high levels of autofluorescence (signal intensity was five times greater than that of control). The kinetic measurement approach easily eliminated the effect of the higher background observed for the low autofluorescent compounds. However, the data analysis was more complicated for the highly autofluorescent compounds, as determination of an accurate initial velocity was affected by the high background from these compounds. For that reason, a fluorescence assay that does not involve light absorption, such as a luminescence-based platform, is considered a useful alternative if compounds’ autofluorescence is a limiting factor.
Luminescence-Based Assay Development
An enzyme-coupled luciferase-kinase assay has a universal mechanism of signal development for any kinases and has been demonstrated with tagged eEF-2K. 10 In this regard, we investigated if a Sox-based fluorescence assay can be imported to a luminescence-based assay for targeting tagless eEF-2K in a high-throughput format by replacing the Soxtide with a nonlabeled peptide. First, the assay was optimized by varying concentrations of reagents (enzyme, ATP, and nonlabeled peptide) and at various assay times (data not shown). A luminescence intensity profile ( Fig. 2A ) with an 80-min incubation time resulted in around a 14-fold signal decrease compared with background signal. We considered that this signal change is sensitive enough for evaluating compound activities and thus used this assay condition for further optimization. Assessment of DMSO and detergent effect was conducted as described in the Sox-based fluorescence assay. Final concentrations of 0.01% (w/v) and 0.25% (v/v) were chosen for Triton X-100 and DMSO, respectively, for further assay optimization.
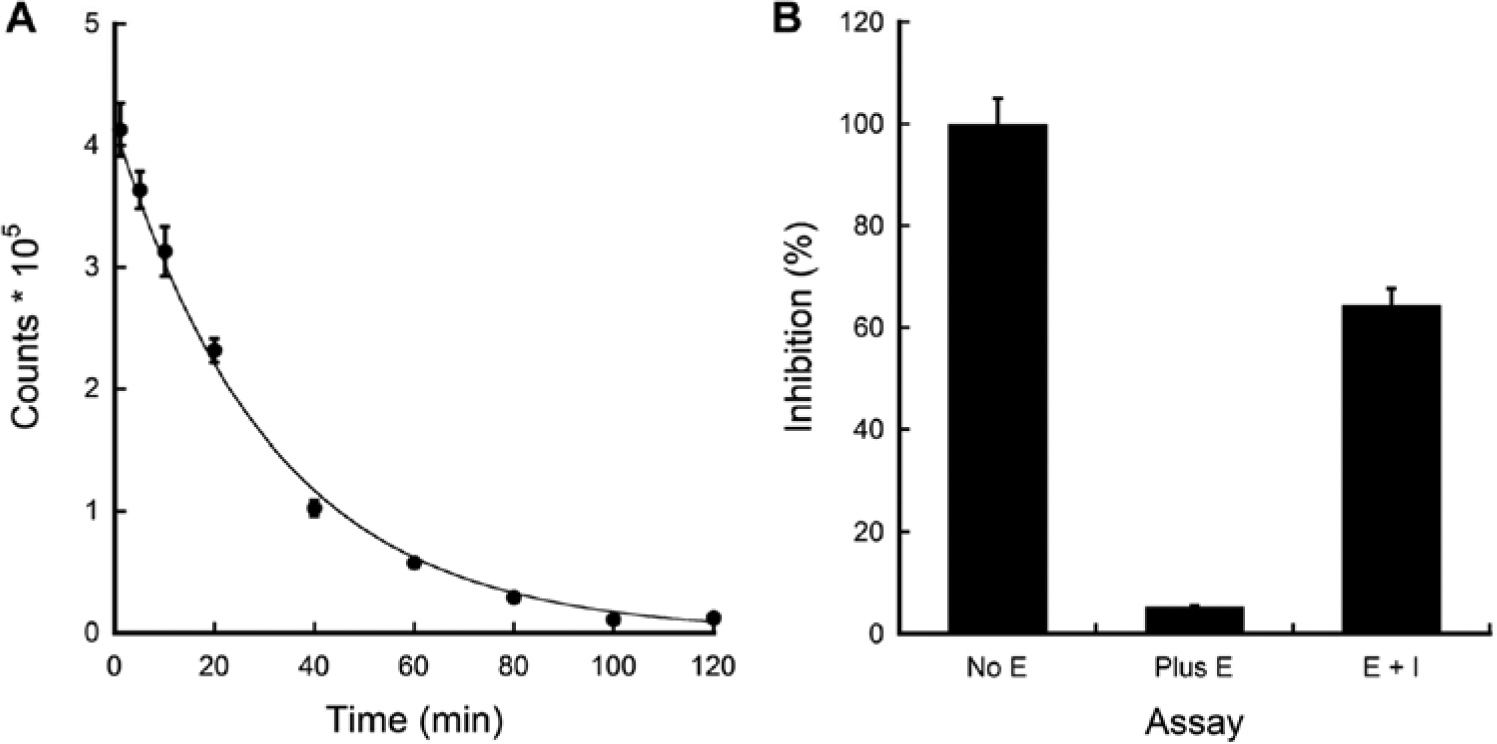
Luminescence assay development and validation. (
Next, assay miniaturization was begun with an inhibitor identified from our previous fluorescence screen and confirmed in a radiolabeled assay. There was a 19-fold signal difference between the negative control (no enzyme, 100% signal) and the positive control (enzyme but no inhibitor). In the presence of compound, the signal was inhibited by 60% ( Fig. 2B ). The ability to use this luminescence-based assay in a high-throughput campaign was validated as described above. A reproducible Z′ (0.80 ± 0.7) was obtained and the S/N ratio was calculated to be 11:1. These results supported the notion that the luminescence-based assay could be employed for small-molecule screening targeting tagless eEF-2K.
Luminescence-Based Assay Applied for HTS
The practicality of the luminescence-based assay for HTS was demonstrated by examining the inhibition activities of a Kinase Focused library, which was custom-selected by Texas Screening Alliance for Cancer Therapeutics (TxSACT, Austin, TX). This unique collection is composed of more than 600 small molecules with known activities against around 100 kinases and represents a useful panel of compounds for developing leads against novel ATP-dependent proteins based on their off-target activities. Again, reproducible Z′ values (0.80–0.85) were observed, and this result indicated the assay was in high quality. These studies confirm that the luminescence-based assay is a good alternative to the Sox-based fluorescence assay.
Confirmation Screen
Overall workflows for the inhibitor screening and confirmation are summarized in Figure 3A , B . Compounds demonstrating positive activity in the primary screen were first rescreened using the same screening conditions in duplicates to eliminate any false-positive hits caused by experimental aberration such as liquid-handling errors. The overall purpose of the screen was to identify the maximum number of hits that could lead characterization of common subscaffolds for lead development. In this regard, hit cutoff for the rescreen was lowered to 30% to increase the number of hits. In total, 123 compounds from both Sox-based fluorescence and luminescence assays reproduced inhibition activities, and this high dropout rate is suspected because the primary screens were performed in singleton, which could cause higher experimental deviation as mentioned above.
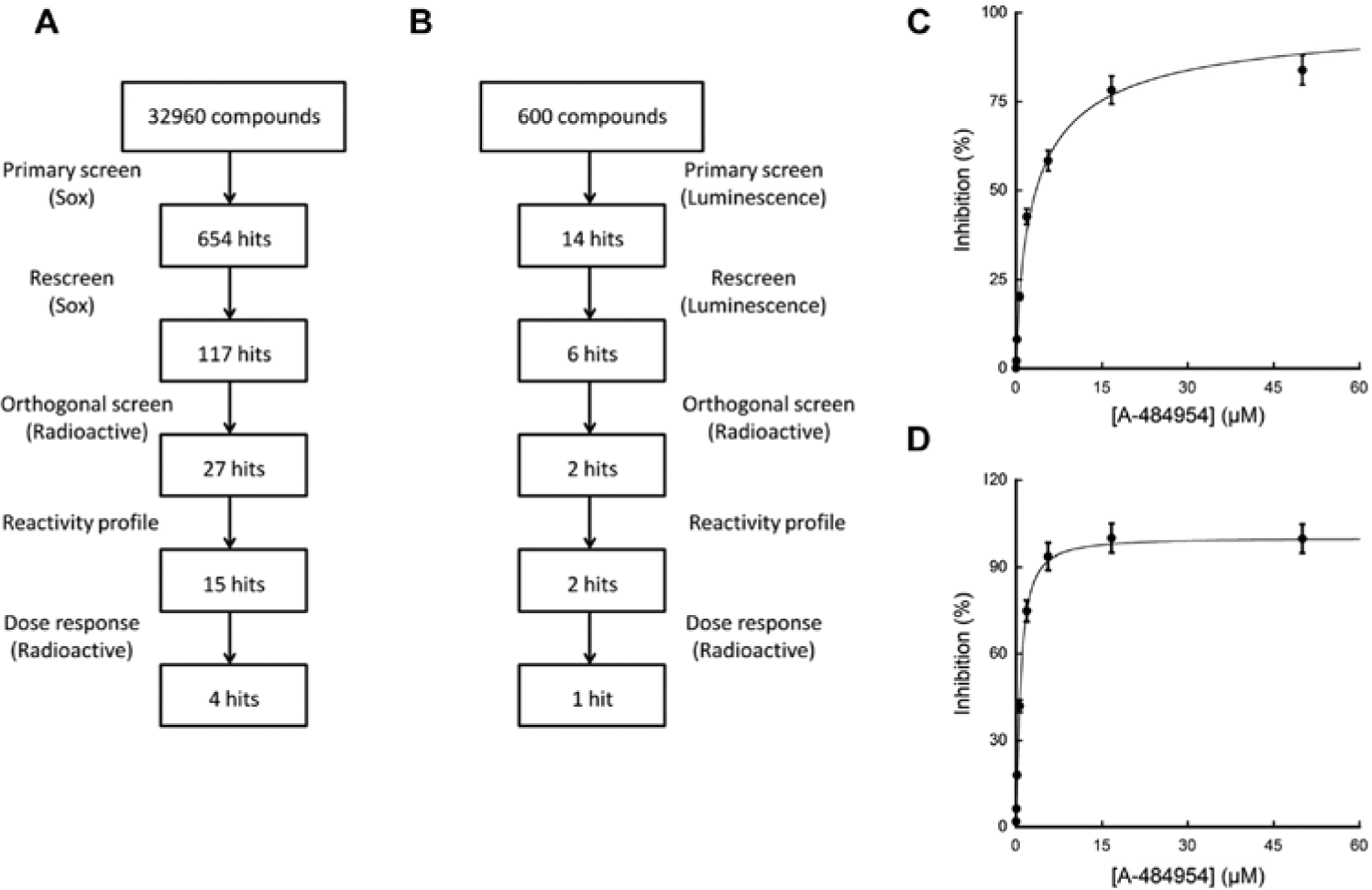
Diagrams of workflow for inhibitor discovery and confirmation for Sox-based (
Compounds possessing inherent fluorescence or directly inhibiting luciferase can often be included as primary hits in either fluorescence- or luminescence-based assays. A radiolabeled assay that was previously developed in our laboratory6,14 was used to discriminate these false hits. Of the 123 compounds, 29 showed reproducible inhibition activity in a radiolabeled assay. This high attrition is suspected since the libraries we tested had a significant number of compounds possessing high intrinsic fluorescence, which made the accurate kinetic measurement difficult as mentioned earlier. In addition, 12 compounds from the Sox-based screen were classified as false-positive hits, which possessed reactive chemical groups or were likely to interact nonspecifically, and thus eliminated from further consideration.
Next, we examined the dose-dependent activities of the remaining 17 compounds in the same radioactive assay. Two compounds, 2,6-diamino-4-(2-fluorophenyl)-4H-thiopyran-3,5-dicarbonitrile and 2-((3-cyano-4-(4-methoxyphenyl)pyridine-2-ylthio)-2-phenylacetic) acid, identified originally from the Sox-based fluorescence assay, showed the most potent inhibition. The IC50 values and Hill coefficients for these two compounds were 60 µM, 77 µM, 1.0, and 2.9, respectively. Particularly, compound solubility and mechanism of inhibition are now under investigation to elucidate the higher Hill coefficient of 2-((3-cyano-4-(4-methoxyphenyl)pyridine-2-ylthio)-2-phenylacetic acid.
As these hits were primarily identified from the Sox-based fluorescence screen and confirmed using a radioactive assay, we further examined the inhibition activities of these two compounds in both Sox- and luminescence-based assays. Upon performing a dose response using these two assays, we found that the IC50 values for these hits were in a similar range to those obtained from the radioactive assay ( Table 1 ) even though 100-fold lower ATP concentration was employed in the luminescence assay. It was predicted that these hits could be ATP noncompetitive, and further investigation on the mechanism of inhibition is under way. Nonetheless, this suggested that both Sox- and luminescence-based assays are reproducible in terms of identifying similar hits.
Top Two Hits Identified Primarily from Sox-Based Fluorescence Assay and Confirmed in Radioactive, Sox, and Luminescence Assays.
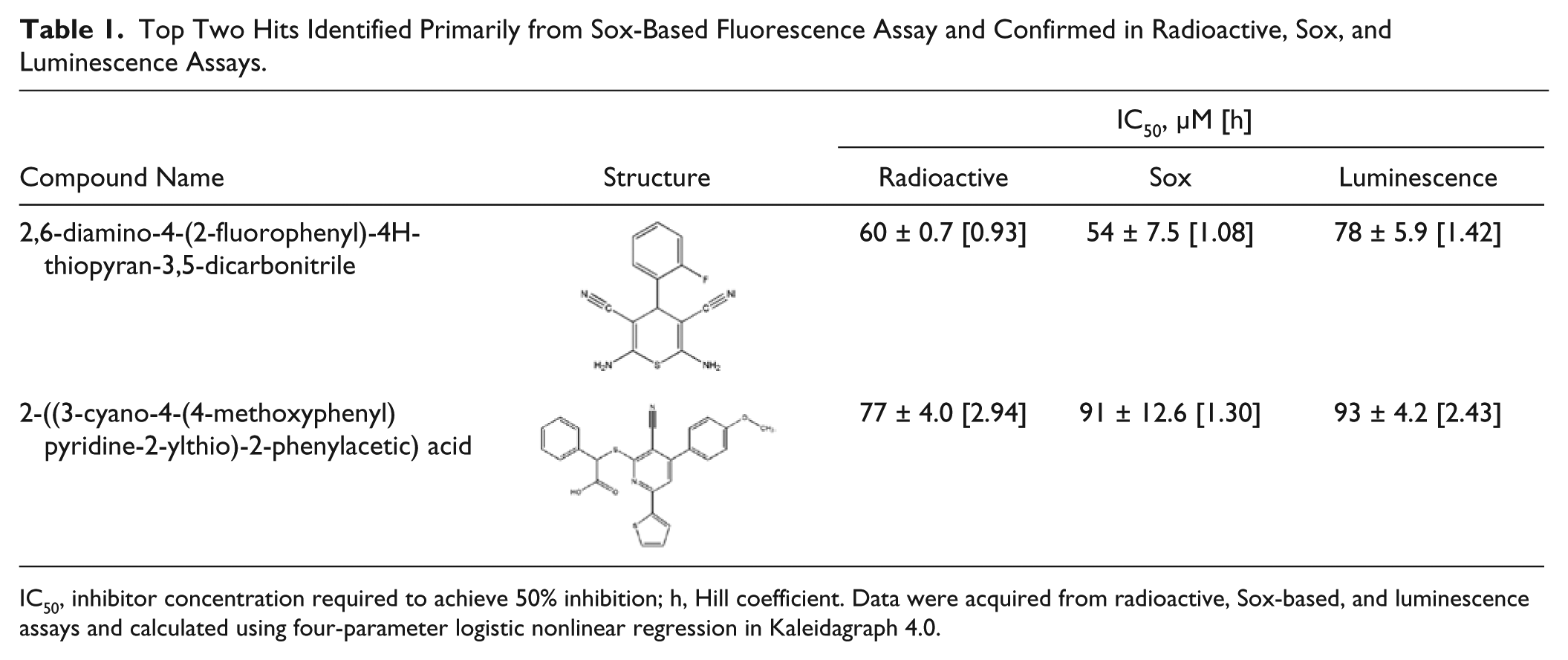
IC50, inhibitor concentration required to achieve 50% inhibition; h, Hill coefficient. Data were acquired from radioactive, Sox-based, and luminescence assays and calculated using four-parameter logistic nonlinear regression in Kaleidagraph 4.0.
Neither the Sox- nor the luminescence-based screens identified potent hits ( Table 1 ). eEF-2K belongs to an atypical class of protein kinase whose catalytic domain is distinct from a number of typical kinases. Thus, it is perhaps not surprising that a library derived from inhibitors of conventional kinases may yield significant off-target activity toward eEF-2K. To eliminate any possibility that this was associated with assay limitations, we tested an authentic compound, A-4849547 (a specific inhibitor of eEF-2K, which was released after completion of our primary screens), in dose-response experiments against tagless eEF-2K. IC50 values of 3.4 µM and 0.76 µM were obtained from Sox- and luminescence-based assays, respectively ( Fig. 3C , D ), which are consistent with the previous report (0.28 µM), thereby verifying the suitability of our assays for screening potent hits. A slightly higher IC50 value was observed in the Sox-based assay, consistent with the higher concentration of ATP (100 µM) used when compared with the luminescence assay (1 µM).
In conclusion, we developed, miniaturized, and validated fluorescence (Sox)– and luminescence-based high-throughput assays for a tagless eEF-2K using a 384-well plate format. Unlike many other fluorescence assays, Sox-based assays are simple to develop and are highly sensitive.11,12 Sox-based kinetic assay demonstrated a 70-fold higher initial velocity compared with that of a negative control. This represents the first time a Sox-based assay has been reported for eEF-2K. Both assays were proven to be robust, automation friendly, and time and cost-effective. Therefore, by developing two simultaneous assay platforms for eEF-2K, we provide researchers with a choice of using a Sox-based assay for primary screening and a luminescence-based assay for a confirmation screen (or vice versa).
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was supported in part by the grants from the Welch Foundation (F-1390), CPRIT (RP110532-P1), and National Institutes of Health (GM059802 and CA167505).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.