
Abstract
Phenotypic screening seeks to identify substances that modulate phenotypes in a desired manner with the aim of progressing first-in-class agents. Successful campaigns require physiological relevance, robust screening, and an ability to deconvolute perturbed pathways. High-content analysis (HCA) is increasingly used in cell biology and offers one approach to prosecution of phenotypic screens, but challenges exist in exploitation where data generated are high volume and complex. We combine development of an organotypic model with novel HCA tools to map phenotypic responses to pharmacological perturbations. We describe implementation for angiogenesis, a process that has long been a focus for therapeutic intervention but has lacked robust models that recapitulate more completely mechanisms involved. The study used human primary endothelial cells in co-culture with stromal fibroblasts to model multiple aspects of angiogenic signaling: cell interactions, proliferation, migration, and differentiation. Multiple quantitative descriptors were derived from automated microscopy using custom-designed algorithms. Data were extracted using a bespoke informatics platform that integrates processing, statistics, and feature display into a streamlined workflow for building and interrogating fingerprints. Ninety compounds were characterized, defining mode of action by phenotype. Our approach for assessing phenotypic outcomes in complex assay models is robust and capable of supporting a range of phenotypic screens at scale.
Introduction
The standard application of phenotypic cellular assays in drug discovery in the recent past has typically been in lead optimization to prioritize and refine targeted compounds selected by high-throughput screening (HTS) campaigns. An alternative starting point for such assays is unbiased phenotypic profiling to identify substances that modulate phenotypes in a desired manner with the aim of either identifying novel targets for HTS campaigns or to progress compounds rapidly to candidate selection. Successful phenotypic screening campaigns are not without challenges. These include both the requirement for robust screening in an assay with appropriate physiological relevance and an ability to deconvolute perturbed pathways to de-risk future drug development.
Angiogenesis, the formation of new blood vessels from preexisting vasculature, is a complex process that plays a significant role in the progression of a number of pathological conditions, including tumor growth, macular degeneration, and psoriasis. 1 The angiogenic mechanism is a multistep, multicellular process that involves endothelial cell migration, proliferation, and differentiation and is regulated by autocrine and paracrine cell signaling interactions with stroma, tumor, and bone marrow–derived progenitor cell types. 2 Key mediators of the angiogenic process include the cytokines, vascular endothelial growth factor (VEGF), and several secondary mediators, including basic fibroblast growth factor (bFGF), epidermal growth factor (EGF), and platelet-derived growth factor (PDGF). 3 Agents targeting VEGF signaling, including monoclonal antibodies (e.g., bevacizumab), antibody derivatives (e.g., ranibizumab), and small molecules (e.g., sunitinib, pazopanib), have been developed and shown to have clinical benefit in patients, either alone or in combination with cytotoxic or other tumor cell–targeted therapy. 4 The utility of these agents in the clinic has been limited as patients progress through acquired resistance or are nonresponsive to such VEGF-based therapies. 5 Therapeutics to other angiogenic pathways are also being developed, but for optimal clinical use, it is important to develop greater insight into the complexity of signaling involved in the angiogenesis. This insight may help prioritize specific agents, identify potential combination partners for optimal efficacy, or identify novel targets or compounds with therapeutic potential.
A number of phenotypic in vitro and in vivo models of angiogenesis have been applied to select compounds to progress through preclinical development. 6 Some of these assays used endothelial cell lines to measure compound effects on specific end points, including proliferation, migration, or endothelial cord formation, and relied on non–high-content approaches to deliver data. Others used more complex organotypic models of angiogenesis that more adequately modeled multiple steps within the angiogenic process but were less amenable to automated HTS. Among the most physiologically relevant 2D models are co-culture models of endothelial cells with fibroblasts, 7 vascular smooth muscle cells, 8 or stromal precursor cells.9,10 In this model, autocrine and paracrine signaling between endothelial cells, stromal cells, and physical intercellular interactions is involved. Several studies by independent groups demonstrated inhibition of tubule formation following treatment in such co-culture models and demonstrated utility for higher throughput imaging-based screening.8–11 The co-culture assay has been used previously as a standard platform to identify and develop therapeutics targeting different angiogenic drivers.12,13 However, information-rich high-content screening analysis across multiple parameters enabling broader mechanism-of-action profiling in a more unbiased and systematic way, to our knowledge, has not been undertaken in a complex co-culture angiogenesis model. Such screens would enable rapid and cost-effective exploitation of multiparametric image analysis readouts and multivariate statistical tools to profile phenotypic response following chemical perturbation. The lack of such studies is likely due at least in part to the bottlenecks and challenges associated with higher throughput high-content imaging in complex primary cell assays, including automated dosing of long-term culture models, high-throughput multiparametric image analysis, and large-scale screening in primary cells where donor origin and passage of cells can contribute significant variability to assay results. Downstream data processing and interpretation of multiparametric phenotypic data are not routine and present unique informatics and statistical challenges. Most published examples of multiparametric high-content phenotypic profiling efforts have used standard 2D culture systems with readouts readily amenable to quantification but nonetheless have demonstrated their value to drug discovery in confirming cellular activity and defining mechanism. For example, high-content analysis (HCA) has been used to elucidate drug mode of action by profiling phenotypic signatures against a library of known phenotypes.14–16 The integration of advanced physiologically relevant culture models with HCA and multivariate analytics offers a robust approach for phenotypic screening and methodology to compare or differentiate different therapeutics, prioritize agents for in vivo testing, or reduce the need for in vivo screening systems.
Here, we combine an organotypic model system with a suite of dedicated tools and novel HCA workflows to map the landscape of phenotypic responses to cellular perturbations for the angiogenic process. A screen using a human primary endothelial cell/stromal fibroblast co-culture model 7 was developed to model multiple aspects of angiogenic signaling, including stromal-endothelial cell interactions, endothelial cell proliferation, migration, and differentiation into tubule structures. Quantitative descriptors of multiple image-based phenotypic parameters, including area of tubules, length, thickness, and branch points, were derived from automated microscopy using custom-designed algorithms created using Metamorph (Molecular Devices, Sunnyvale, CA) and Definiens Cellenger (Definiens, Munich, Germany) cognition network image analysis to generate multiparametric phenotypic profiles. Multiparametric data extracted from this assay were collated using a bespoke informatics platform that integrates data-processing, statistical, and feature display tools to provide a streamlined and robust workflow for building and interrogating phenotypic fingerprints describing compound mechanism of action. This work demonstrates application of a multiparametric phenotypic profiling approach to profile a set of 90 well-characterized compounds and relate distinct clusters to morphological features defining modes of action based on their antiangiogenic and angiogenic phenotypes. Several distinct tubule morphologies were observed that mapped to perturbation of functionally linked angiogenic signaling pathways. This chemical genetic screen provided insight into the molecular pathways contributing to different phenotypic responses in a co-culture of angiogenesis and uncovered novel drug-mechanism-phenotypic response relationships—for example, the potentiation of tubule differentiation by both a cyclooxygenase 2 (COX-2) pathway inhibitor (DuP 697) and a Raf-1 inhibitor (GW5074). To our knowledge, this is the first application of a multivariate high-content analysis approach to a human primary co-culture model of angiogenesis. Our integration of multivariate hit stratification and clustering algorithms into the screening method is robust and capable of supporting analyses of a wide range of phenotypic screens at scale.
Materials and Methods
Cell Culture and Reagents
Human umbilical vein endothelial cells (HUVECs) (pooled), normal human dermal fibroblasts (HDFs), and EGM-2 and FGM-2 culture medium were purchased from Promocell (Heidelberg, Germany). HUVECs and HDFs were maintained in EGM-2 and FGM-2, respectively, and used at passage 3. Compounds were purchased from Tocris Bioscience (Bristol, UK), with the exception of: anti-hVEGF neutralizing antibody (AF293-NA), normal goat immunoglobulin G (IgG) (AB-108C), IFNα2a (R&D Systems, Minneapolis, MN), anti-α2β1 (HA2.1) and anti-αvβ3 (LM609) (Merck Millipore, Billerica, MA), levamisol, perhexilene, bFGF, VEGF, and PDGF (Sigma-Aldrich, Gillingham, UK); cyclic RGD and negative control, ALLN (Peptides International, Louisville, KY), angiostatin, caveolin 1 scaffold domain peptide positive and negative (Calbiochem, Billerica, MA, USA); and the compound used as a positive control, AZ514 (AstraZeneca, UK), a small-molecule inhibitor of vascular endothelial growth factor receptors (VEGFR) 1, 2, and 3 with a pKDR IC50 less than 5 nM.
In Vitro Angiogenesis Model
The in vitro HUVEC tubule formation assay was performed in house as previously published 7 but adapted for a 96-well microplate format. Briefly, HUVECs were co-cultured onto HDF feeder layers in optimized medium for 10 days at an initial seeding ratio of between 2:1 and 8:1. Compounds were dosed at day 0 of cells in co-culture and replenished with media changes every 72 h. Co-cultures were grown in black-walled, clear-bottom, collagen-coated 96-well microplates (BD Biosciences, Franklin Lakes, NJ) suitable for automated fluorescent imaging.
Compound Treatment
All compounds were prepared and titrated as 8-point half-log concentration response in 96-well plates in 100% DMSO and at 1000× final drug concentration using a Hamilton Starlet (Hamilton Robotics, Reno, NV). An intermediate compound plate was prepared containing cell culture media (250 µL) and drug at a 20× final concentration. Then, 5 µL was transferred from the intermediate compound plate to each cell plate (95 µL media). Cell plates were treated in triplicate. The highest tested concentration for each compound was 10 µM, with the exception of concanamycin (0.1 µM), caveolin (1 µM), angiostatin (0.1 µM), and VEGF, PDGF, αvβ1, and αvβ3 (10 µg/mL). Final DMSO concentration was 0.1%.
Immunocytochemistry
Cells were fixed with 70% cold ethanol and labeled with a nucleic acid binding dye, Hoechst, and an antibody directed toward the endothelial cell–specific marker, CD31. Cells were incubated in blocking buffer (phosphate-buffered saline [PBS] containing 0.1% bovine serum albumin) for 30 min. Primary antibody solution diluted in blocking buffer (1:5000) (mouse anti-CD31, ab24590; Abcam, Cambridge, UK) was added at 40 µL per well and incubated for 1 h. Cells were washed three times in blocking buffer for 30 min. Secondary antibody solution diluted in blocking buffer (1:5000) (Alexa Fluor 488 goat anti-mouse; Invitrogen, Glasgow, UK) and Hoechst 33342 (H-1399; Invitrogen, Glasgow, UK) (2 µM) was added at 40 µL per well and incubated for 1 h. Cells were washed three times in blocking buffer for 30 min and PBS (100 µL) added to each well before plates were sealed with black plate sealers prior to imaging.
Fluorescent Image Acquisition and Image Analysis
Cell images were captured using the ImageXpress (IX5000) (Molecular Devices) using a 10xS Fluor objective reading in two channels (Hoechst, CD31). Sixteen fields of view per well were acquired. Metamorph angiogenesis tube formation application (Molecular Devices) was used to quantify tubule morphology (14 parameters) using thresholds for minimum and maximum tubule width (µm) and tubule intensity above local background (gray level) (
Data Processing
Data from Definiens and Metamorph tubule formation algorithms were output as csv files and collated using a customized automated data-processing workflow created using Pipeline Pilot (Accelrys, San Diego, CA) (
Statistical Analysis
Before the high-content screen was analyzed, multisite data were aggregated to a single value per well by either mean or sum of a calculated parameter. Subsequently, well-level data were normalized by subtracting the median of the positive controls from all samples and then dividing treatment data points with the median of DMSO-treated controls per plate. For concentration-response relationships, this was then expressed as % inhibition: [1 − (X − Median of Positive Controls/Median of DMSO Controls − Median of Positive Controls) × 100]. Outliers within DMSO-treated controls were flagged for visual inspection where the value for at least one parameter fell outside the mean by a confidence interval (CI) of 99.9% (26 of 384 controls). Following visual inspection, if the image suggested experimental artifact, the observation was removed from the data set prior to normalization (13 of 384 controls). Data have been analyzed using a principal components analysis run in R programming language and applied to all available data, including all compound- and DMSO-treated samples. This included a method that transforms the data to stabilize the variance found in the results. A Mahalanobis distance metric was applied to calculate the similarity of each drug and dose to controls. Those treatments significantly dissimilar to those in the ellipsoidal DMSO control cloud were identified by calculating an associated p value for each distance (adjusted for false discovery rate) and thresholded at a 0.1% significance level. Only those compounds and doses at which significant difference was observed were selected for hierarchical clustering after exclusion of those doses identified as toxic using the nuclei count. An agglomerative hierarchical clustering approach was then used to identify groups of active compounds by morphology. Dose-response curves were fitted using the four-parameter logistic regression model available in the Spotfire Decision Site (TIBCO, Palo Alto, CA): [y = min + (max – min)/(1 + (X50/x)Hill)].
Results
Human Primary Co-culture Model of Angiogenesis as a High-Content 96-Well Screen
It is well described that HUVECs form a network structure when co-cultured with HDFs in vitro.
7
For the study presented here, these methods were applied in a 96-well format amenable to HTS and adapted to a high-content multiparametric image analysis platform (
Fig. 1A
). The endothelial networks formed over a period of 10 days, with co-cultures treated with compound and/or media replenishment at days 0, 4, and 7 after co-culture. After 11 days, co-cultures were fixed, immunolabeled, and imaged using a high-throughput fluorescent imaging microscope. Multisite data were captured for each well to ensure appropriate well area for accurate quantification of relatively large structures (
Fig. 1B
,
C
). Distinct phenotypes were identified using automated image analysis to classify cells based on tubule and nuclei staining. A Metamorph angiogenesis tube formation application was used to obtain 14 readouts of tubule morphology such as total tube area, length, and number of branch points (
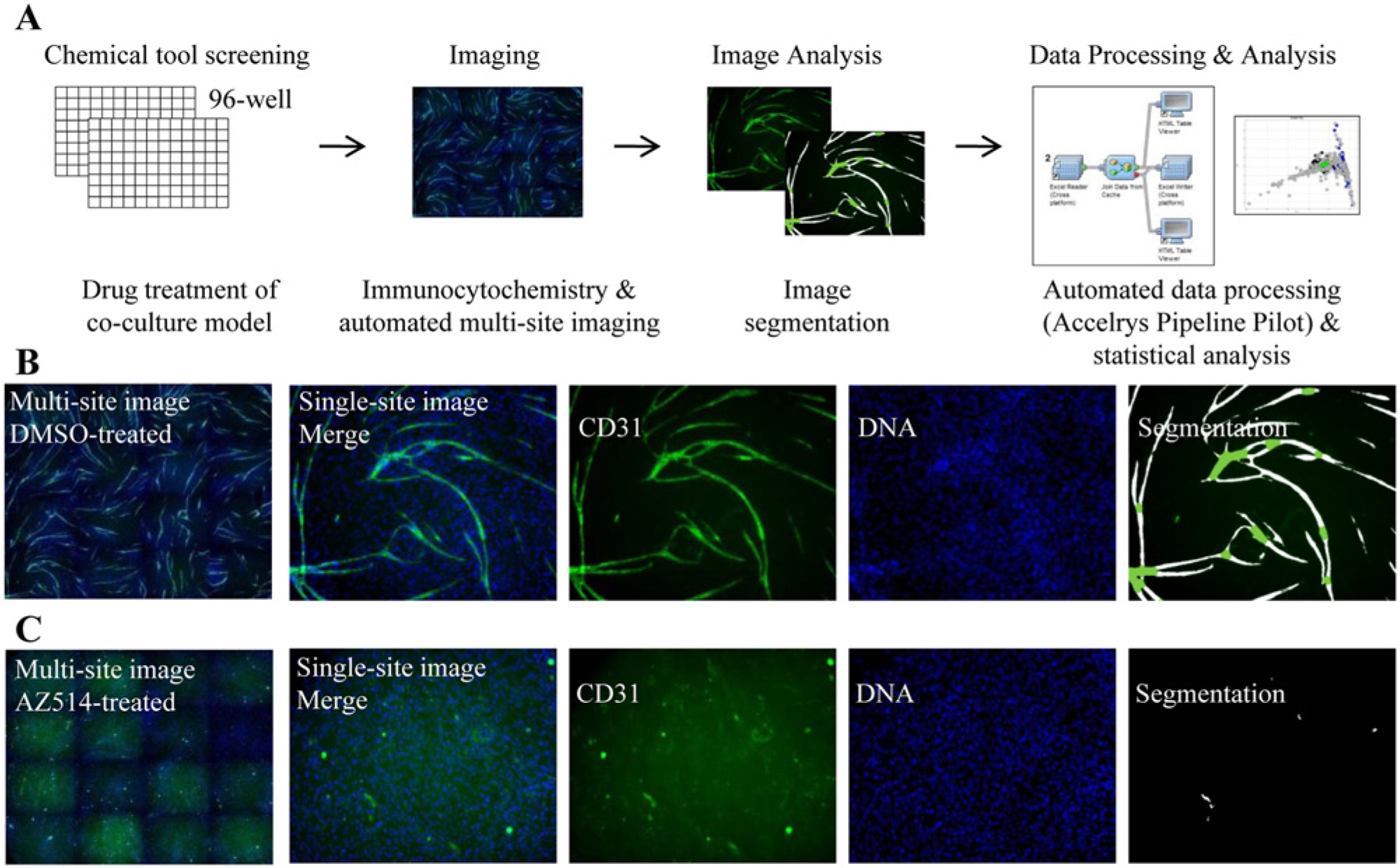
High-content imaging and analysis of tubule formation in co-culture. (
Compound Screening in a High-Content Cell-Based Model of Angiogenesis
The assay was used to test the phenotypic effects of 90 well-characterized agents comprising small molecules, antibodies, and growth factors. Compounds were selected based on either an established effect in angiogenesis-related conditions and/or a known activity against an angiogenic process-related target or pathway. The compounds are listed together with their known principal molecular targets (
Hit Selection by Multivariate Analysis of Phenotypic Outcomes in the Tubule Formation Assay
To better understand the effects of active compounds and related pathways on tubule phenotype, we performed multivariate analysis using all 19 measured features (
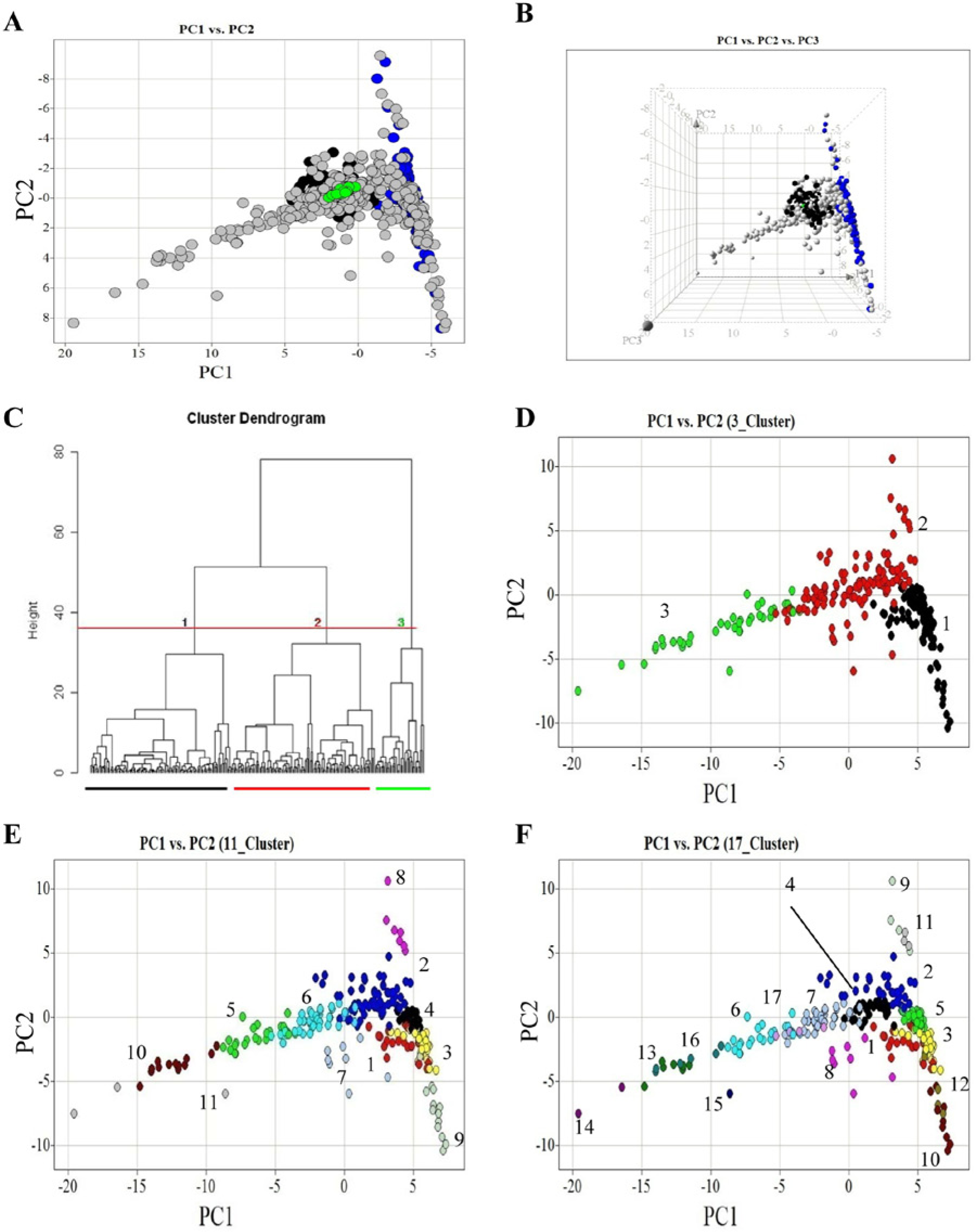
Multiparametric analysis and clustering of a chemical genetic screen in an angiogenesis model. Nineteen quantified measures of co-culture morphology were scaled and analyzed by principal components analysis (PCA). PCA uses a transformation to convert a set of measures of possibly correlated variables into a set of values of uncorrelated variables called principal components. PCA is a method to reduce dimensionality in multivariate data. The first three principal components accounted for ~80% of the variance and were selected for plotting (
Using the Mahalanobis method, 68 compounds were identified as having a significant effect on angiogenesis with at least one dose (
Global Analysis of Represented Signaling Pathways
To interrogate the effects of targeting specific pathways on tubule morphology, we used hierarchical clustering to group compounds by phenotypic profile. Only those compounds and doses defined as active were selected for clustering. Agglomerative clustering of phenotype metrics identified three top-level groups of morphologies within the data set with further division of groups within ( Fig. 2C ). Tubule morphologies in identified group 1 ( Fig. 2C , D ) were broadly characterized by discrete undifferentiated endothelial cells with small total tubule area and length measures ( Fig. 3B – D ). Conversely, phenotypes in group 3 had comparatively high total tubule area, length, and branch point measures. Associated images revealed extensive and altered tubule branching patterns ( Fig. 3I – L ). Group 2 contained the largest number of compounds. In this group, although tubules did form, in general the tubule length and extent of branching were limited relative to negative controls ( Fig. 3E – G ).
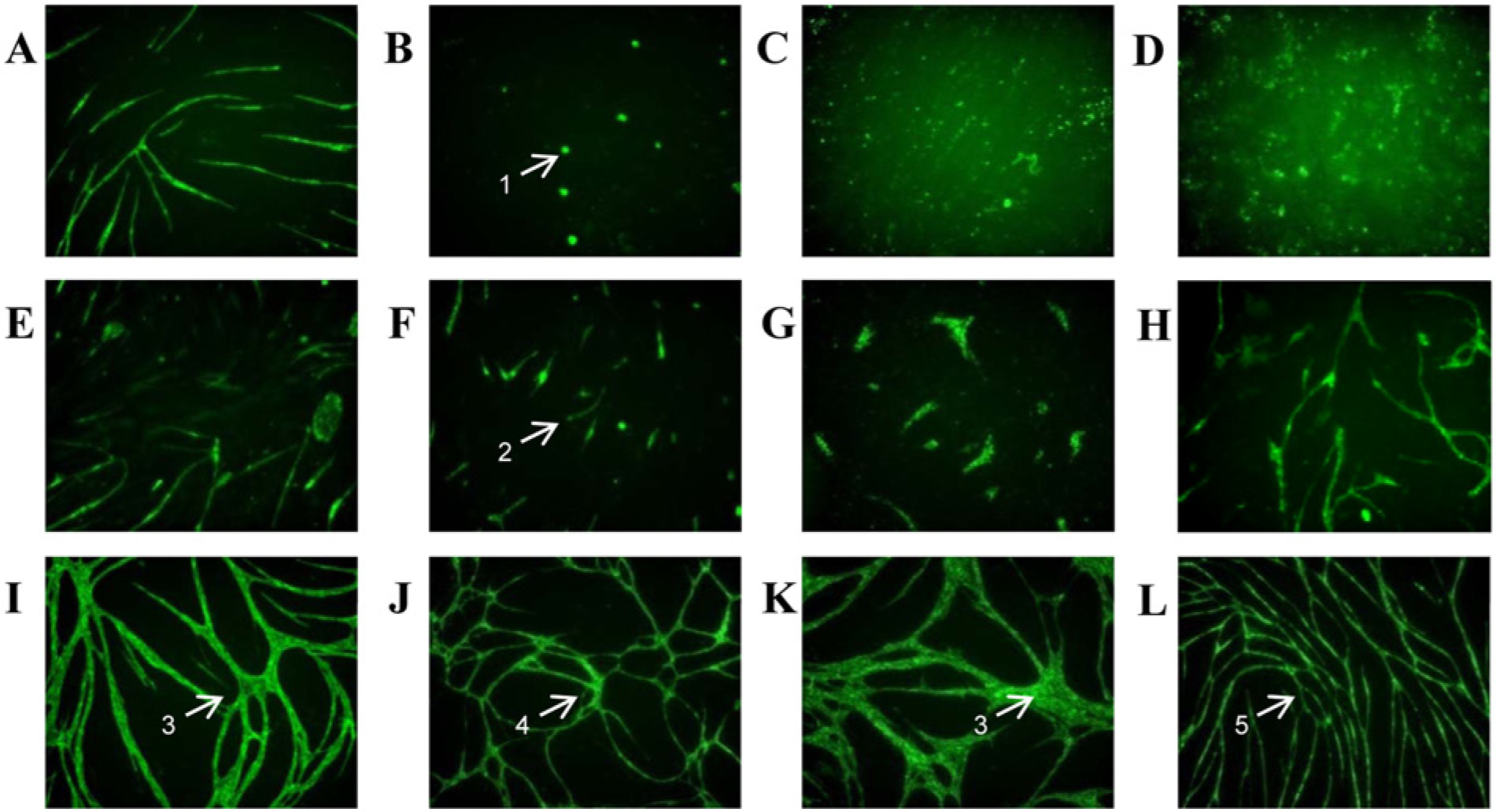
Tubule phenotypes resulting from different chemical treatments. Example images of cells treated with different perturbing agents and labeled with CD31. (
Annotation of chemical probes with their primary gene target and functional association enabled assessment of whether agents with similar biological fingerprints targeted the same or related signaling networks. Clusters of compounds with similar phenotypes that were also similar in function were identified from the data set. For example, cluster 1 ( Fig. 2C , D ) contains several compounds known to target genes involved in cell survival or proliferation ( Table 1 ). All screened VEGF inhibitors grouped in cluster 1, perhaps unsurprising given published data demonstrating a role for VEGF in endothelial proliferation. 2 Other agents overrepresented in cluster 1 include the seven screened cytoskeletal disrupting agents, three compounds targeting PI3K, two targeting Akt/PKB, and those directed toward DNA topoisomerase and mTOR ( Table 1 ). All of these pathways have well-characterized roles in cell proliferation and survival18–20 and include a class of agents distinct from VEGFR inhibitors, the vascular disrupting agents (e.g., combrestatin 4A). VEGFR inhibitors clustered together in a distinct group (cluster 4 within cluster 1; Table 1 ), helping to distinguish them from the microtubule disrupters (clusters 1, 3, and 9 within cluster 1; Table 1 ). A few exceptions to the clustering of mechanisms described were observed, generally at the top dose of compounds tested (e.g., JTE013, A205804, and PP1) and it seems likely that at such doses, nonspecific effects on cell survival may be present.
Active Compounds Arranged by Phenotypic Cluster and Annotated with Known Function and Targeted Pathway.

Functional annotations assigned based on a priori knowledge from literature: P, proliferation/survival; Ac, activation; M, migration; A, adhesion; D, differentiation. Those with uncharacterized function are left unlabeled. COX-2, cyclooxygenase 2; EGFR, endothelial growth factor receptor; FGF, fibroblast growth factor; IFN, interferon; MMP, metalloproteinase; NO, nitric oxide; PDGF, platelet-derived growth factor; PDGFR, platelet-derived growth factor receptor; TGFBR, transforming growth factor β receptor; TNFα, tumor necrosis factor α; VEGFR, vascular endothelial growth factor receptor
Where multiple active doses exist.
In the largest cluster of phenotypes (cluster 2; Fig. 2C , D ), clustering by function is less clear, but it is apparent that most compounds tested with known effects on cellular migration and/or adhesion exist within this phenotypic grouping ( Table 1 ). There are some compounds with known effects on migration that cluster in group 1 or 3 (i.e., Src, integrin, and calpain). Such classified phenotypes, although present in the large cluster 3, if observed at the level of 17 clusters also exist in close proximity to cluster 2. These observations serve as a reminder that clustered groups are not discrete but give an indication of similarity across a spectrum of phenotypic responses.
Cluster 3 phenotypes exhibited increased and/or altered branching (cluster 3; Fig. 2C , D ) and were enriched for agents targeting activation of angiogenesis (e.g., VEGF) and cellular differentiation (e.g., inhibitors of TGFBR signaling) (cluster 3; Table 1 ). The latter group may be indicative of compounds that, if used therapeutically, could lead to nonfunctional vessels. All three TGFBR signaling inhibitors—A83-01, LY364947, and SB431542—exhibited bell-shaped dose relationships with total tubule area. Taking A83-01 as an example, from the multiparametric analysis, it is clear that at higher doses, the altered branching structure is not observed, and morphologies are more similar to those in cluster 2 ( Fig. 4A – D ). A less pronounced but similar effect was observed with LY364947. This could be indicative of a loss of specificity at higher doses or could be a feature of TGFB signaling. Either way, this assay demonstrates the ability to detect early in drug discovery polypharmacological modes of action of compounds. Other compounds in this group that resulted in altered branching structure are those targeting p38, COX-2, and Raf-mediated signaling. P38 perturbation has been shown previously 21 to enhance tubule formation in an FGF-driven tubule formation model of bovine endothelial cells in collagen, and here too we observe similar effects with SB203580 and, to a lesser extent, with SB202190. Matsumoto et al. 21 correlated the increased tubule complexity observed in the presence of p38 inhibition with an increase in expression of Jagged-1, an intermediate in Notch signaling. The results presented here confirm a clustering of p38 perturbed phenotypes with other pathways known to be involved in differentiation. Also identified by the assay is a novel proangiogenic effect elicited by COX-2 inhibition with DuP 697. This is in contrast with previous results proposing a role for COX-2–mediated release of prostaglandin (PGE2) in stimulating proangiogenic factors such as VEGF 22 and experimental data demonstrating that DuP 697 induced HUVEC apoptosis and inhibition of HUVEC tube formation in a single-cell tube formation Matrigel assay. 23 Furthermore, perturbation of Raf-mediated signaling with GW5074 demonstrated proangiogenic properties in our co-culture model that contrast with previous reports demonstrating that GW5074 represses lumen formation by HUVECs in a 3D collagen assay. 24 As considered further in the Discussion, such examples of conflicting efficacy in the co-culture model used in this study with previous in vitro studies performed on single-cell HUVEC assays may highlight the impact that stromal cell types have on the drug-induced phenotypic response of target cell populations and, as such, may better model in vivo drug responses.
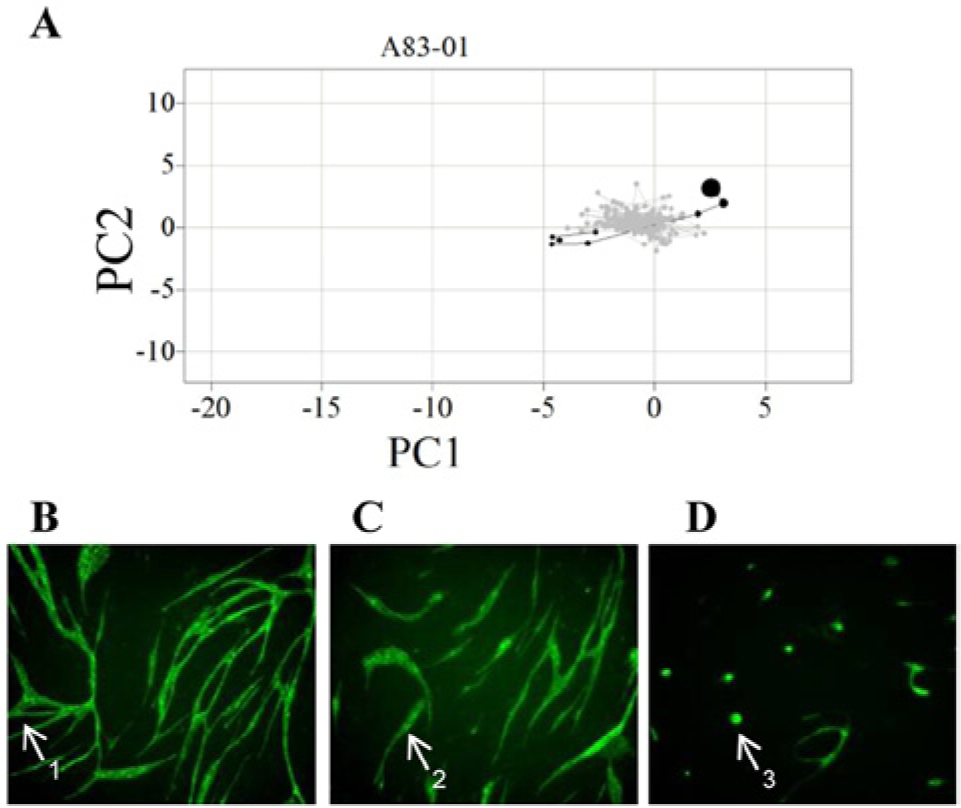
Phenotypic analysis of TGFβR signaling inhibition with A83-01. (
Discussion
Many diseases that require new therapeutics today are multigenic and complex. In such cases, the link between therapeutic targets and clinical outcomes is difficult to predict, partly because of a lack of understanding of target biology within the context of in vivo pathophysiology. For complex disease traits where disease progression is driven by multiple mechanisms and pathway networks, it is challenging to identify optimal therapeutic targets. In such cases, there is likely to be a need for therapeutics with selective polypharmacology or rational drug combination strategies to attain significant efficacy in the clinical setting. Information-rich analysis of physiologically relevant assays offers a potential solution to implementation of phenotypic profiling in early discovery. Such approaches could complement target-directed screening by addressing the challenges of identifying novel targets and the most optimal target selectivity profiles for complex disorders. We describe a high-content multicellular phenotypic approach to rapidly and robustly measure phenotypic effects of compounds on angiogenesis. In this chemical genetic screen, multiple features were used to generate multiparametric phenotypic profiles informing mechanism of action. The workflow developed for the study incorporated a bespoke informatics platform that integrated processing, quality control, statistics, and feature display (
Angiogenesis is a multistage process that is regulated by autocrine and paracrine interactions involving multiple cell types. Such complexity in the underlying signaling mechanisms that govern angiogenesis presents challenges to the discovery of effective and sustained treatment of angiogenic disorders. Although the role of anti-VEGF therapy in the treatment of angiogenesis-related conditions such as tumor angiogenesis and macular degeneration of the eye is proven, it is also apparent that angiogenesis signaling is multimodal. A significant number of patients do not respond to anti-VEGF therapy. 5 This warrants further investigation of novel pathways that could be targeted for therapeutic intervention, and imperative to this goal is the use of model systems for phenotypic screening that recapitulate in vivo processes as closely as possible. Several reported studies used single cell-type assays to measure phenotypes such as endothelial proliferation, migration, apoptosis, and tube formation in 2D and 3D to evaluate putative antiangiogenic therapies. 6 While such assays provide valuable information on mechanisms of endothelial function, they do not account for the influence of the angiogenic microenvironment on pharmacological response. We and others8–11 have used a human primary co-culture model of angiogenesis to profile the effects of compounds on autocrine and paracrine signaling across multiple cell types. Earlier studies8–11 demonstrate that both anti- and proangiogenic compound effects could be quantified via traditional single end-point screening methods and dose-response relationships obtained. Parallel assays of endothelial cell proliferation, migration, and tubule formation were used to identify compound mode of action from single end-point screening. In this study, it is shown that by multiparametric analysis of tubule morphology from a single robust 96-well co-culture assay, it is possible to delineate compound mode of action from the acquired image-based phenotypic fingerprint.
The approach in this study resulted in novel and previously unreported observations of angiogenic signaling alongside validation of compounds with known effects. Of those compounds active in this assay, at least some have been identified as active and clinically efficacious. Data from this study confirm reported 8 antiangiogenic effects of vinblastine and Taxol and further associate all such microtubule modulating agents with a characteristic phenotype. This includes the vascular disrupting agent, combrestatin 4A, one member of an important class of compounds with efficacy on established vessels. This result demonstrates the utility of the model to identify not just those compounds that act on the angiogenic process itself but also on established vessels. This group of compounds clusters in close proximity to the established VEGFR inhibitor molecules, the mainstay of antiangiogenic therapy for both tumors and macular degeneration. Perturbation of a number of other signaling pathways was also shown to lead to distinct tubule morphologies. For example, inhibition of p38 MAPK, previously reported 21 to increase tubule morphogenesis, led to compound and dose-dependent phenotypes distinct from either vascular disrupting agents (VDAs) or VEGFR inhibitors. Such mechanistic characterization can be used to identify modulators of angiogenesis that act independently of currently targeted signaling pathways as a strategy to develop complementary therapies efficacious in VEGF-independent and/or VEGF-resistant patient populations. A range of other perturbations, including inhibition of COX-2, Raf, TGFBR, and γ-secretase signaling, was also identified as leading to increased and altered tubule morphology. The effects observed with inhibitors of TGFRB and γ-secretase are in line with results reported previously.12,25 To our knowledge, this is the first in vitro demonstration that inhibition of Raf and COX-2 induces an increase in tubule density. Conversely, previous studies employing single endothelial in vitro assays indicate that GW5074 and DuP 697 confer an antiangiogenic effect through demonstrative endothelial dysfunction and perturbation of 3D tube formation.23,24 A distinction between assays in these previous studies and the assay used here is the presence of a stromal fibroblast population supporting tube formation. In the co-culture assay used for this phenotypic characterization, the physical interaction with stromal fibroblast or derived soluble factors or matrix appears to protect endothelial cells from dysfunction and induced tubule density following inhibition of COX-2 or Raf-1–mediated signaling. Indeed, two separate in vivo studies using zebrafish and Xenopus embryo models indicate no antiangiogenic properties of GW5074. In fact, GW5074 moderately increased lymphatic endothelial growth in Xenopus embryos.26,27 Such discrepancy in the translation between in vitro endothelial cultures and in vivo model systems highlights the impact that the in vivo angiogenic niche microenvironment has on pharmacological response, suggesting that the co-culture model may more accurately recapitulate how the microenvironment influences endothelial function and pharmacological responses. The roles of COX-2 and Raf1 targets are worthy of further investigation as antiangiogenic therapy. Although an increase in tubule density was observed with these agents in this in vitro co-culture assay, similar morphologies observed with DLL4 perturbation in vitro translated in vivo to nonfunctional vessels. 28 It was also possible, through use of the co-culture and presence of fibroblasts in the assay, to rapidly identify compounds with efficacy but low toxicity profiles in a single screen, thus circumventing the requirement for parallel toxicity counterscreens.
The inactive agents included all negative controls tested and 16 others with diverse targets and functional associations. It is difficult to draw conclusions from the inactives without further screening as any lack of activity could be due to the physiochemical properties of the compound, the dose range evaluated, or a lack of the relevant intact signaling network in the model used. Several inactive agents shared a common primary target with other compounds that tested as active. This possibly indicates that at least in these cases, the signaling pathway in the model was intact. An exception was observed with the three compounds targeting endothelial or inducible nitric oxide synthase (NOS) signaling, all of which were inactive in the assay despite well-characterized involvement of NOS in the angiogenic process. 29 Further screening will help understand the limits of the assay and so drive alternative model development that encompasses additional pathways and target space—for example, mechanical forces imposed by flow, 3D environment, interplay of other cell types such as vascular smooth muscle cells and pericytes, and/or a level of hypoxia, none of which are modeled in the described assay. As imaging technology and associated image analysis and informatics tools reach higher levels of sophistication and capability, it is likely that researchers will be able to model more aspects of biology in high throughput.
This study demonstrates the feasibility of applying co-culture assays in a 96-well format to an automated chemical genetic screen for angiogenesis. Differentiating mode of actions of compounds is difficult to establish from a single parameter in a single assay without manual inspection of several images. Quantitative multiparametric image analysis can help identify the contribution of various signaling pathways to different angiogenic processes, obviating the need for manual image inspection, which soon becomes infeasible for large screens and is largely subjective. A number of high-content multiparametric approaches have already been used in other areas of biology,14–16,30 and all have used multivariate analysis to interpret their data. The high-content workflow developed here integrated multivariate analysis into a standardized process from image acquisition to data visualization to rapidly and robustly monitor phenotypic responses in advanced cellular models. Following acquisition of images using automated microscopy, custom-designed analysis algorithms yielded measurement of features across multiple cell types. The implementation of automated routines using Pipeline Pilot (Accelrys) enabled images, metadata, image analysis output, and compound information to be merged without manual intervention. The use of a custom script within the Spotfire Decision Site (TIBCO) enabled rapid quality control of data by linking data points to raw images. The tools developed further integrated multivariate statistical techniques into Pipeline Pilot, specifically including the use of a Mahalanobis distance metric and associated p value corrected for false discovery rate to classify active compounds from multivariate data. This distance metric, when applied appropriately, enables robust identification of actives for any compound or dose, taking into account multiple features, not just primary end points. Standardized workflows such as that described open the possibility for future screens and extension of capability—for example, incorporation of morphological, functional, and gene expression responses from diverse assay platforms (e.g., cytokine release alongside tubule parameters and potentially next-generation sequencing data). The developed workflow is both flexible and extendible, meaning approaches to use well-characterized training sets of compounds to aid in defining and prioritizing hits from future diversity screens for further downstream de-convolution are possible.
In conclusion, we have been successful in employing automated phenotypic profiling to a complex biological assay to accelerate discovery of novel drugs targeting angiogenesis. Multiple angiogenic modulators were classified by their phenotypic fingerprint, including characterization of VDAs, VEGFR inhibitors, the effect of standard cancer chemotherapeutics on vasculature, and the novel effects resulting from inhibition of COX-2 and Raf. In the future, there may be a shift to using more complex cell-based assays in early drug discovery using a network-based rather than single target-based approach. Exploitation of such approaches in high throughput, the feasibility of which is supported by these data, will enable more predictive screens to be used earlier in drug discovery, leading to reduced use of animal models and improved translation to the clinic.
Footnotes
Acknowledgements
The authors are grateful to TCSCellWorks for access to the co-culture model under a nonexclusive license agreement.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
B. J. Isherwood and S. T. Barry are current employees of AstraZeneca. R. E. Walls, T. M. Houslay, S. R. Brave, and N. O. Carragher are former employees of AstraZeneca.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.