
Abstract
It has now been almost 20 years since the discovery of p38 MAP kinase and its role in inflammatory cytokine synthesis through reverse pharmacology and its subsequent exploration as a potential target for autoimmune and other diseases. At the time of its discovery, the use of cell-based phenotypic screens to identify new molecular targets was at its infancy, and while p38 MAP kinase was not the first target to be identified this way, it provides a useful model for reviewing the pros and cons of this approach and the subsequent impact it can have on discovering new medicines.
Background
In the late 1980s, SmithKline researchers had been interested in dual inhibitors of the synthesis of the proinflammatory leukotrienes and prostaglandins from arachidonic acid.1–3 While these pathways were not yet fully characterized molecularly, they were known to be mediated by 5-lipoxygenase (5-LO) and cyclooxygenase (COX) enzymes. It was during a study of some of these compounds that it was noted that they inhibited the production of the inflammatory cytokines, interleukin (IL)–1β and tumor necrosis factor (TNF)–α, from lipopolysaccharide (LPS)–activated human monocytes. At the time, considerable evidence had accumulated that these cytokines were stimulated in a number of inflammatory conditions, including some autoimmune diseases, and protein therapies that blocked their activity were being evaluated clinically. When a number of compounds with cytokine-suppressive activity were assessed for their ability to inhibit COX or 5-LO, there was no correlation with their ability to inhibit cytokine production. This was the first indication that the cytokine-suppressive activity of these compounds was mediated through an independent molecular target. Furthermore, the fact that some of these compounds were known to be active in a rat adjuvant arthritis model added additional impetus to an effort to isolate and identify this target to further study its biology and develop additional assays to support drug discovery.
It was at this point that a group of scientists, spearheaded by John Lee, Jerry Adams, and myself, came together to find a way to isolate and identify the target. Our options for identifying the target in the pre-proteomics and genomics era were limited. We very much influenced by the experience of Merck in identifying FLAP, the 5-LO activating protein, through the use of reversible and irreversible radioactively labeled compounds. 4 There were three critical steps to applying these methods. First, we needed to identify a suitable reversible inhibitor that retained its function in cell-based assays; second, we needed to identify a cell line that replicated the cytokine suppression that we saw in primary human monocytes isolated from whole blood to be able to scale up the levels of protein extract needed to isolate the target; and third, we needed to demonstrate that the molecular target identified by binding of the radioactively labeled compound mediated the cytokine-suppressive effects of the compound. We settled on THP.1 cells, which closely replicated our findings in primary human monocytes. After a false start with one compound, we were able to identify 3H SB202190 as a suitable compound that demonstrated binding to a protein in THP.1 extracts using a gel filtration technique. Binding could be competed with nonradioactive SB202190 and many additional compounds that suppressed cytokine production over a wide range of potencies. Critically, binding affinity demonstrated a pharmacological relationship with functional activity, indicating that we had identified a relevant molecular target. 5 This was an important discovery because it enabled us to screen for potent compounds by binding, and this allowed us to continue to move the drug discovery program forward.
However, the binding assay itself proved inadequate for purifying the protein sufficiently to get a sequence, and so we designed a related, iodinated compound, SB206718, which could be cross-linked to the target protein upon UV irradiation. Using this technique, we were able to identify a single band in a gel and hence could follow this labeled protein through subsequent 2D electrophoresis. However, we were initially unlucky with this approach, since the isoelectric point and molecular weight coincided with actin and precluded separation. Further separation of the target from actin on gels required proteolytic or cyanogen bromide cleavage of the target protein. From this procedure, were we eventually able to get the peptide sequence and use this to design degenerate oligonucleotides to screen a complementary DNA (cDNA) library and eventually to isolate a cDNA that encoded the target. 5
The sequence obtained demonstrated that the molecular target was a new member of the MAP kinase family. We designated it CSBP, which stands for cytokine suppressive anti-inflammatory drug binding protein by analogy to FKBP, but it was separately discovered as RK 6 and p38, 7 and it is the latter terminology that has remained the preferred nomenclature along with MAPK14, despite the fact that the protein actually has a molecular weight of 41 to 42 kD! The MAP kinases are proline-directed serine threonine kinases that are activated through the phosphorylation of both a threonine and tyrosine in an activation loop proximal to the adenosine triphosphate (ATP) binding site. Based on its homology with a yeast stress-activated protein kinase Hog1, p38 MAP kinase was originally often thought of as a stress-activated protein kinase, but subsequent studies indicated that it could be activated by many nonstress stimuli ( Fig. 1 ). We were able to demonstrate that the SB compounds inhibited the kinase activity of p38 MAP kinase and hence demonstrated a linkage between the suppression of inflammatory cytokine production and the inhibition of a single protein kinase. 5
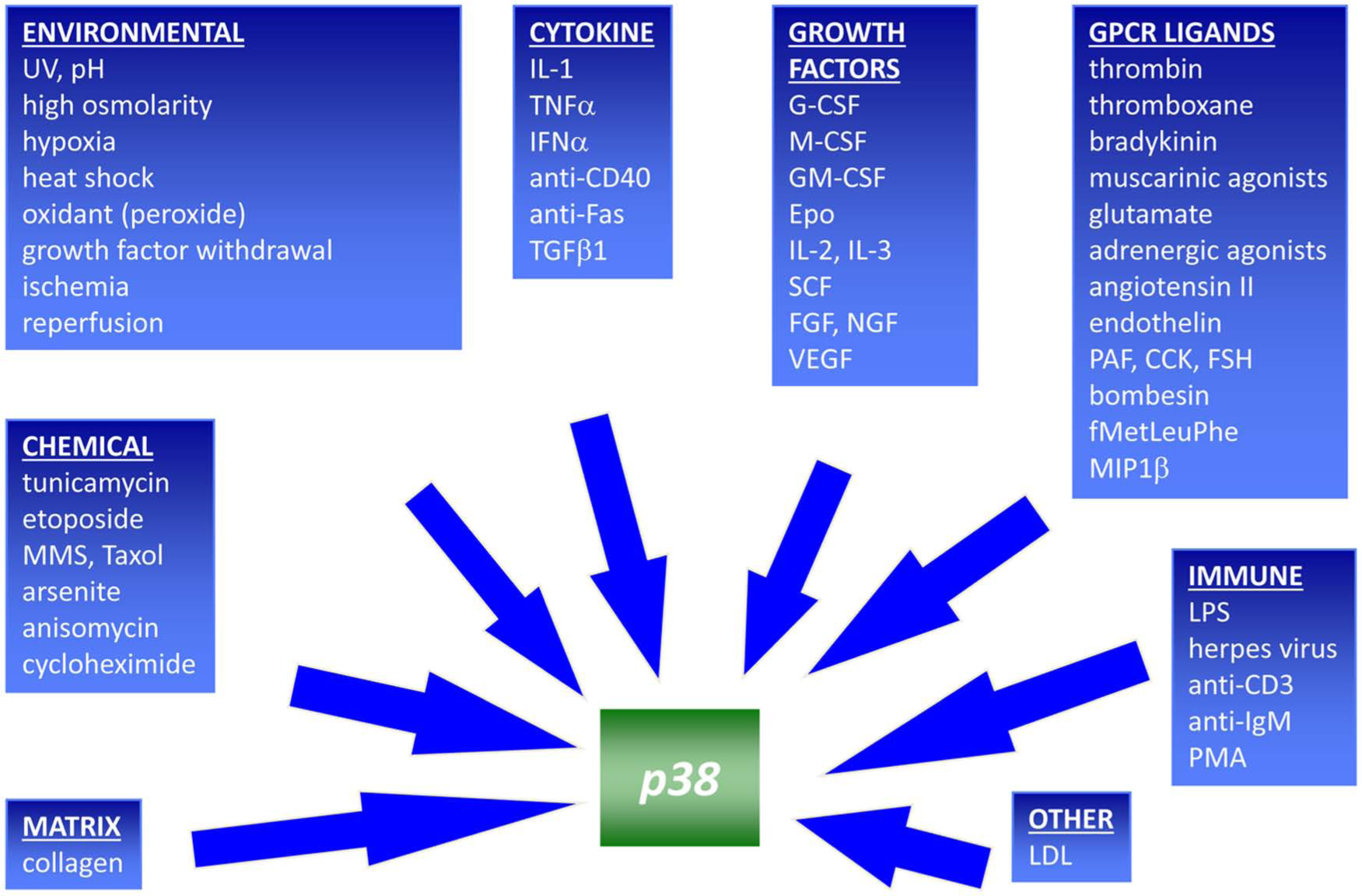
Stimuli known to activate p38 MAP kinase.
Significance
There are many reasons why this discovery has had significance in not only our understanding of fundamental signaling pathways within cells but also in the drug discovery industry. Briefly, these include (1) the use of phenotypic screens for identifying new drug targets, (2) the viability of protein kinases as drug targets, and (3) the application of chemical biology for biological characterization and target validation. Analysis of each of these areas will constitute the rest of this article.
Use of Phenotypic Assays
The value of using a phenotypic screen for identifying a druggable pathway for regulating a particular gene or combination of genes was exemplified by the discovery of p38 MAP kinase. While there are potentially multiple targets that regulate inflammatory cytokine production in monocytes, it is possible to argue that a target discovered using a phenotypic approach achieves two goals: it identifies a node in the signaling pathway that is directly tied to the altered function, and it demonstrates that this may be targeted by a small molecule. Hence, it bypasses some of the criticisms of the molecular target approach, which often lead to the discovery of potential drugs in search of a disease due to inadequate validation of the molecular target. This was especially true in the years following the broad sequencing of the human genome and expressed messenger RNAs (mRNAs) (expressed sequence tags [ESTs]),8–10 where it was common to pursue homologues of known drug targets with the belief that they would yield useful biological insights and novel therapeutic options. In contrast, there are numerous examples of successful drugs whose targets were discovered subsequent to their introduction into the clinic and/or the characterization of their pharmacological activity in vitro and in animal models. Examples include cyclosporine,11,12 FK506, 13 rapamycin,14–16 metformin, 17 and the PPAR ligands such as the fibrates and thiazolidinediones.18,19 In the case of the p38 discovery, there was evidence that compounds that bound to the target, but which were devoid of COX or 5-LO activity, nevertheless retained activity in animal models of rheumatoid arthritis, 20 so in a sense there already was some validation for the targeted mechanism.
Another feature of targets discovered in phenotypic screens is that they can identify a class of targets that were previously thought to be intractable. While the lack of a molecular target has not precluded the development of some highly successful or promising medicines (e.g., niacin, 21 dimethyl fumarate,22,23 IMIDs [thalidomide, lenalidomide]), 24 there is clearly value to identifying the target to spur the generation of improved analogues. Also, many protein targets or targeted motifs belong to families, and so this becomes important in defining the target selectivity of compounds and relating this to efficacy and safety.
At the time of the p38 MAP kinase discovery, there was considerable skepticism that the reverse pharmacology approach could be easily applied to multiple additional projects due to technological, resource, and time constraints. However, the methods available for isolating the molecular target from phenotypic screens have clearly evolved since p38 was discovered such that these concerns have been considerably, but not completely, reduced. There has been significant advancement in the fields of proteomics and genomics that allow other approaches. For example, it is now possible to develop affinity reagents that can be used to isolate the target through affinity chromatography, provided that a suitable site on the ligand can be found to add a biotin or other group that can be used to conjugate the molecule to a column.25,26 Furthermore, the availability of mass spectrometric sequencing methods combined with a database of all the potential human protein sequences derived from translation of the human genome has made identification of potential candidate target proteins considerably easier.9,10,27,28 The list of potential candidates may be further trimmed through the use of small interfering RNAs (siRNAs) directed toward candidate genes, in the case of inhibition, or overexpression in the case of activation. Methodologies for knockdown with siRNA are well established, and many cDNAs and expression vectors can now be purchased, thus obviating the need to clone these from scratch. However, it is still important to ensure that every available tool is used to ensure the specificity of the target selected through appropriate use of related, inactive control compounds and critical that a pharmacological relationship between binding affinity for the target and cell-based activity established. 5 An excellent recent example of this is the discovery that BET proteins are the target of benzodiazepine compounds shown to stimulate ApoA-1 production in a human hepatocyte cell line. 29 Hence, there is now a strong argument that this approach should take its position alongside the more established target-driven approach, especially when it is considered that phenotypic approaches to drug discovery have been more productive in driving drug discovery. 30
Protein Kinases as Drug Targets
Another feature of targets discovered in phenotypic screens is that they can identify a class of targets that were previously thought to be intractable. Such was the case in the early 1990s when p38 MAP kinase was first identified. Accepted wisdom had suggested that protein kinases were likely to be poor targets since they all bind ATP and that any inhibitor that competes with ATP would necessarily have the potential to bind to a lot of targets, leading to a high potential for off-target toxicity. The discovery of potent inhibitors of p38 MAP kinase as well as the contemporaneous identification of potent inhibitors of some tyrosine kinases associated with cancer (e.g., imatinib for inhibition of bcr-abl in chronic myelogenous leukemia) 31 began to question this premise. Early attempts to address the selectivity issue began by isolating and cloning close homologues and evaluating selectivity, but it has become clear that the issue of selectivity is driven not so much by the homology at the protein sequence level but more by the conservation of the binding feature in the binding pocket. It has really taken more than 10 years to fully address the selectivity of protein kinase inhibitors, and there are now multiple kinase panels available through service organizations that address selectivity across the majority of the 518 kinases that constitute the human kinome. 32 With the availability of kinase panels, it is now straightforward for any kinase-directed program to use this information early on to select preferred scaffolds to work on and to identify particular off-targets that may need to be specifically monitored during the lead optimization process.
Based on profiling across hundreds of protein kinases, the two earliest p38 MAP kinase inhibitors, SB202190 and SB203580, have actually proved to be reasonably selective, inhibiting few kinases outside of the MAP kinases. 32 This has meant that they are excellent tools for teasing apart different signaling pathways. However, they did not enter the clinic due to their effects on P450 enzymes and associated liver safety issues. 33 Subsequently, even more selective inhibitors have been discovered and have progressed into clinical trials. 34 Unfortunately, none of these has proved to show efficacy in rheumatoid arthritis for reasons that are not entirely clear, and most have shown some evidence for common adverse events. 35 However, there is still promise for use of p38 inhibitors in the treatment of pain 36 and cancer.37,38
Why is SB203580 as selective as it is? One possibility is that the compound preferentially binds to the inactive form of p38 MAP kinase. This was a very early but perhaps not wholly appreciated aspect of p38 inhibition by this compound. In the original isolation of p38 MAP kinase detailed above, we used nonstimulated THP-1 cellular extracts, suggesting that SB203580 might bind to the inactive form of p38 MAP kinase. 39 This was further demonstrated by the x-ray crystal structure of SB203580 bound to the inactive, nonphosphorylated form of p38 MAP kinase. 40 Furthermore, the binding affinity of SB203580 does not differ between the active and inactive forms, whereas ATP binds much more weakly to the inactive form, giving SB203580 a significant competitive advantage by binding to the inactive form. 39 Hence, a reasonable conclusion from these studies was that SB203580 may lock the kinase in an inactive form, which could contribute to its selectivity. Indeed, a similar observation was made with imatinib in its binding to the bcr-abl kinase 41 and established a general principal by which selective inhibitors might be found. Again, we see how the approach of using phenotypic screening and reverse pharmacology contributed to this serendipitous finding by highlighting a way to inhibit a previously perceived “undruggable” target.
Many other p38 MAP kinase inhibitors have since been subsequently described that inhibit p38 MAP kinase by binding directly at the ATP site, at an adjacent “DFG out” site, or through other allosteric sites, resulting in highly potent and selective inhibitors, with some also illustrating substrate selectivity.42,43
Another feature contributing to the specificity of p38 inhibition of SB203580 was highlighted by the discovery of three closely related homologues, p38β, γ, and δ. SB203580 inhibits the original p38/CSBP, now named p38α, and p38β but not the γ or δ homologues. 44 The γ and δ kinases differ from α and β in the absence of a small amino acid (Thr106) in the loop at the back of the ATP and SB203580 binding pocket, which is replaced by a methionine. When the Thr is changed to Met in p38α, this confers resistance, and vice versa, p38γ and δ become sensitive to SB203580 when Met is changed to Thr.45,46 Indeed, this residue is only found in a few other kinases, including c-Raf, suggesting that it is a “gatekeeper” for inhibitors such as SB203580. 47 The role of a gatekeeper residue in determining the inhibitor selectivity of protein kinases, first characterized by Garske et al., 48 is the basis for new approaches to inhibiting this target class.
Use of Chemical Tools for Biological Characterization and Target Validation
When it was clear that SB203580 was not going to be a clinical candidate, we had a large supply of the compound in hand. Given the high level of interest in the target, a decision was made to make SB203580 readily available to academic researchers to stimulate a productive effort to understand the role of this new target in disease. So in many ways, this was an early example of the “open innovation” model that is very much in vogue these days.49,50 In this model, tool compounds and molecular reagents are shared between academia and industry to foster precompetitive research to rapidly understand the role of a potential new target in many different biological systems, something that would be beyond the resources and expertise of any one laboratory.
A survey of PubMed shows that since the publication of SB203580/SB202190 in 1994, these compounds have been used in approximately 400 studies every year since 1997. So what has been the value of these studies? Well, the experience is clearly mixed. Several studies used concentrations that were well above those needed to inhibit p38 MAP kinase in cells,51,52 did not show inhibition of the downstream substrate MAPKAP kinase-2, and did not use a complementary molecular approach such as the use of dominant negative proteins. Of course, we now know much more about the selectivity of this compound, and the likelihood that studies at higher concentrations could inhibit off-target kinases, including the closely related MAP kinases JNK2 and JNK3, 32 but this certainly affected the quality of the early studies. One of the common mistakes in these early studies was to use the inhibitor at concentrations without regard to whether these were relevant to inhibition of the pathway in cells. Because one of the substrates of p38 MAPK, MAPKAP kinase-2, was already known, it was possible to directly measure the inhibition of p38 MAP kinase in cells through inhibition of the phosphorylation of MAPKAP kinase 2. 39 Hence, this could serve as an internal control and also was an early example of a phosphobiomarker for measuring the pharmacological effects of a protein kinase inhibitor, 53 which was subsequently used in clinical studies. 54
While most of the studies supported the role of p38 MAP kinase in inhibiting various inflammatory-driven processes, one could nevertheless walk away from the early in vitro cell line studies with a view that inhibiting p38 MAP kinase could influence multiple differentiation steps55–58 and key signaling pathways, 59 leading to pleiotropic effects in multiple organ systems and a high potential for toxicity. However, the advancement of multiple candidate inhibitors into the clinic without consistent evidence for some of these potential mechanism-based toxicities suggested that there was significantly more complexity in vivo, likely due to considerable redundancy. Hence, a certain caution and emphasis on in vivo studies is important in sifting through these data. The increased focus on characterizing the known off-target activities and ADME properties of tool compounds in the current open innovation efforts is to be applauded as a way to add more rigor to these studies. Also, the ready availability of molecular tools such as siRNA and lentiviral technologies will continue to expand the opportunities for complementing compound-driven inhibition studies.
On a more positive note, the p38 inhibitors, along with other molecular techniques, enabled the delineation of various downstream signaling pathways and molecular events that are briefly summarized in Figures 2 and 3 but detailed in other reviews. 60

Effects of p38 MAP kinase inhibition. Left panel: Molecular mechanisms regulated by p38 MAP kinase inhibition. Right panel: Direct or indirect substrates of p38 MAP kinase, whose phosphorylation is altered by p38 MAP kinase inhibition.
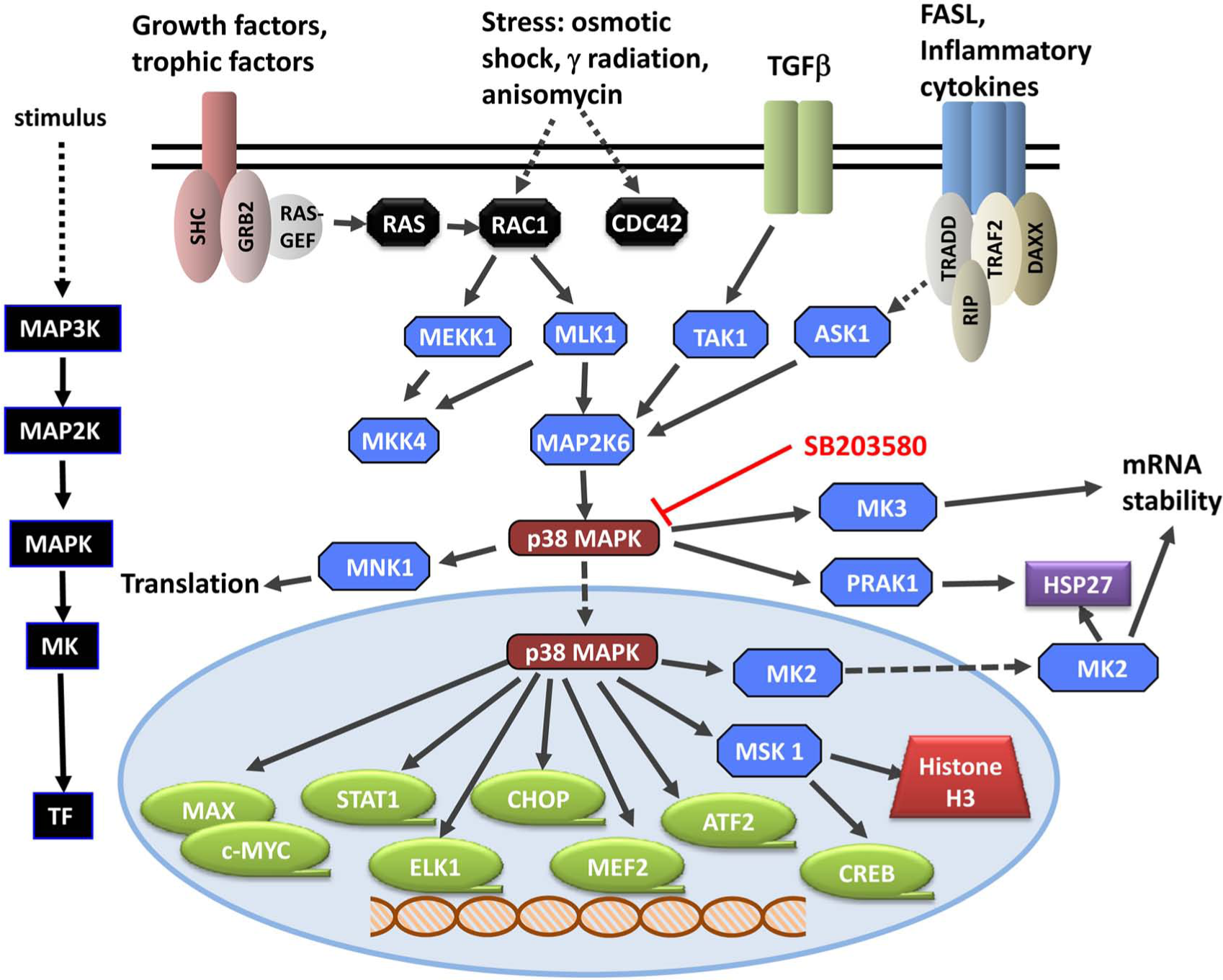
p38 MAP kinase signaling pathways in response to several stimuli. Inhibition with SB203580 complemented with other molecular techniques has enabled the delineation of several substrates and signaling pathways, resulting in altered transcription, translation, and cell physiology. On the left is highlighted the generic MAP kinase signaling cascade, which is illustrated in detail on the right. Dotted lines refer to one or more yet to be defined signaling steps. The dashed lines refer to translocation of the kinase in response to activation by upstream kinases.
In conclusion, this brief perspective has demonstrated how the discovery of p38 MAP kinase helped set off a revolution in approaches to drug discovery, embracing three different avenues of drug discovery: phenotypic screening followed by reverse pharmacology, protein kinases as targets, and open innovation. Without the subsequent -omics revolution and the development of associated technologies, this would not have happened. However, it is reasonable to conclude that these areas are now well-established drug discovery paradigms. p38 MAP kinase inhibition has yet to prove itself as a viable clinical approach to disease, and serine-threonine kinases have trailed their tyrosine kinase brethren as a platform for the identification of new medicines. Nevertheless, there are now marketed examples of inhibitors that target this family, 61 and this will likely continue well into the future. It is also likely that we will see the druggable target space continue to expand and provide unexpected novel approaches to drug discovery.
Footnotes
Declaration of Conflicting Interests
The author declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author received no financial support for the research, authorship, and/or publication of this article.