
Abstract
Premature chromosome condensation (PCC) is a consequence of early mitotic entry, where mitosis begins before completion of DNA replication. Previously we have identified mutations in MCPH1, a DNA damage response and potential tumor suppressor gene, as a cause of primary microcephaly and PCC. Here we describe a high-throughput assay to identify modifiers of PCC. Reverse transfection of control siRNA followed by a forward transfection of MCPH1 small interfering RNA (siRNA) was performed to induce PCC. Condensin II subunits CAPG2 and CAPH2 were validated as PCC modifiers and therefore positive controls. Cell nuclei were detected by DAPI staining using an Operetta imaging system. PCC and nuclei number were determined using Columbus analysis software. Two batches of nine plates were used to determine assay efficacy. Each plate contained four negative (nontargeting) and eight positive control siRNAs. Mean % PCC was 12.35% (n = 72) for negative controls and 4.25% (n = 144) for positive controls. Overall false-positive and false-negative rates were 0% (n = 72) and 2.1% (n = 144), respectively. This assay is currently being used to screen a human druggable genome siRNA library to identify novel therapeutic targets for cancer treatment. The assay can also be used to identify novel compounds and genes that induce PCC.
Introduction
Premature chromosome condensation (PCC) occurs when cells begin mitosis before completing DNA replication. Previously, we identified mutations in the MCPH1 gene as a cause of PCC.1,2 MCPH1 (Microcephalin) is a multifunctional protein involved in cell cycle checkpoint control, the DNA damage response pathway, and chromatin remodeling.3,4 This identifies the gene as a candidate tumor suppressor that may also play a role in resistance to chemotherapy drugs.
In support of this, previous work in our and other laboratories has demonstrated loss or reduction of MCPH1 expression in breast and ovarian tumors, correlating with increases in tumor stage and aggression.5–7 Furthermore, MCPH1 has been identified as a transcriptional regulator of the BRCA1 and hTERT genes, further implicating it in the control of cell proliferation and tumorigenesis.8,9
The PCC phenotype, seen previously in ovarian, breast, and colon cancers, is a reliable readout of a loss of MCPH1 function.2,10 Here we exploit this phenotype to present the first report of a high-throughput reverse genetics study aimed at identifying both modifiers of PCC and inducers of synthetic lethality in MCPH1-deficient cells. The assay could also be used for small-molecule screening to identify potential lead compounds that either rescue the normal functions of MCPH1 or selectively kill cancer cells deficient in MCPH1. The identification of these targets and compounds, alongside studies of the cellular pathways they are involved in, could enhance drug discovery for breast and ovarian cancers resistant to current chemotherapies. Currently, versions of the PCC assay described here are also being applied to small-molecule and whole-genome small interfering RNA (siRNA) screens aimed at identifying compounds and genes that induce PCC in the presence of functional MCPH1, with the aim of increasing our understanding of the global regulation of DNA condensation.
Materials and Methods
Cell culture
Human osteosarcoma (U2OS) cells from the American Type Culture Collection (ATCC, Manassas, VA) were maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal calf serum (FCS) under standard culture conditions (37 °C, 5% CO2). Cells were passaged at a split ratio of 1:8 twice a week. Cells were obtained from ATCC at passage 8 and were used for screening purposes until passage 25.
siRNA
Dharmacon siGENOME SMARTpool siRNAs (Thermo Fisher Scientific, Pittsburgh, PA) were supplied as lyophilized pellets containing four duplexes. Pellets were reconstituted with 1× RNA buffer (Thermo Fisher Scientific) to a concentration of 2 µM using a Fluid-X XRD-384 dispenser, followed by 90 min of agitation on a rotary shaker. Reconstituted siRNA were stored in polypropylene plates (Matrix, Thermo Fisher Scientific, Pittsburgh, PA) at −80 °C. Working plates (View Plates; PerkinElmer, Waltham, MA) were prepared and stored at −20 °C.
siRNA Transfections
Control siRNAs (2 µM) were directly aliquoted into 96-well working plates (View Plates; PerkinElmer) to columns 1 and 12. Human PLK1 siRNA (M-003290) was used as a positive transfection control with reduced cell number as a functional readout. Human INCENP (M-006823) was used as a positive control for effects on cell number. Human CAPG2 (M-018283) and CAPH2 (M-016186) were used as positive controls for modification of MCPH1-induced PCC. Four wells containing nontargeting siRNAs (D-001206) per plate were used as negative controls. The final concentration of control and library siRNA was 50 nM.
For reverse transfection, a Fluid-X XRD-384 dispenser (at 300 rpm) was used to add the transfection mixture: 0.1 µL Lipofectamine RNAiMAX transfection reagent (Invitrogen, Carlsbad, CA) suspended in 17.5 µL Opti-MEM-1 (1×) low-serum media (Invitrogen) per well. Plates were gently mixed on a rotary shaker at room temperature for a minimum of 20 min. Then, 80 µL U2OS cells per well, at a density of 7.5 × 104 cells/mL in DMEM/10% FCS, in a uniform suspension maintained by constant magnetic stirring, was added to plates using a Fluid-X XRD-384 dispenser at 300 rpm.
To minimize edge effects, cells and transfection complexes were left to rest in a laminar flow hood for 1 h at room temperature before returning to a 37 °C 5% CO2 incubator. 11
After a 24-h incubation, a forward transfection of MCPH1 siRNA with the sense strand sequence 5′-CUCU–CUGUGUGAAGCACCAdTdT-3′ was performed to induce PCC. 2 For each well, 0.25 µL MCPH1 siRNA (40 µM) was mixed with 16.25 µL Opti-MEM-1 (1×) low-serum media (Invitrogen). Alongside this, 0.2 µL Lipofectamine RNAiMAX transfection reagent (Invitrogen) was mixed with 3.3 µL Opti-MEM-1 (1×) low-serum media and incubated at room temperature for 5 min. The siRNA solution was then mixed with the transfection reagent solution and incubated at room temperature for a minimum of 20 min. The siRNA/transfection reagent mix was added to the well using a Fluid-X XRD-384 dispenser at 300 rpm to a final concentration of 100 nM.
For large-scale transfections of MCPH1, CAPG2, and CAPH2, forward siRNA transfections were performed in 25-cm2 flasks. Cells were seeded at 7.5 × 104 cells per milliliter in 5-mL culture media and allowed to adhere overnight. For each siRNA, a solution of 240 µL Opti-MEM-1 was prepared with 14.6 µL RNAiMAX. In three separate tubes, 1170 µL Opti-MEM-1 was mixed with 17.5 µL of CAPG2, CAPH2 (20 µM stock), or MCPH1 (40 µM stock) siRNA and incubated for 5 min at room temperature. The siRNA and RNAiMAX preparations were combined and incubated at room temperature for 20 min. Cells were rinsed with phosphate-buffered saline (PBS) and 5.8 mL Opti-MEM-1 added to each flask. The siRNA suspension was added to the cells at a final concentration of 100 nM for MCPH1 and 50 nM for CAPH2 and CAPG2 and incubated for 4 to 6 h before DMEM supplemented with 30% FCS was added (10% FCS final concentration).
Quantitative Real-Time PCR
RNA was extracted using the RNeasy Plus Mini Kit (QIAGEN, Valencia, CA) according to the manufacturer’s protocol 48 h following the large-scale forward transfection. Complementary DNA (cDNA) synthesis was performed using Superscript III (Invitrogen). Briefly, RNA, random hexamers, and dNTPs were heated to 65 °C for 5 min and cooled on ice for 2 min. A master mix of first-strand buffer, dithiothreitol (DTT), and Superscript III was added and incubated at 25 °C for 5 min before heating at 50 °C for 50 min and finally 70 °C for 15 min. Duplicate real-time PCR analysis was performed using the SYBR Green PCR Master Mix and 7500HT machine (Applied Biosystems, Foster City, CA). Details of primer sequences for quantitative real-time PCR (qRT-PCR) are listed in
Western Blotting
After 72 h, transfected cells were lysed in RIPA buffer (1% NP-40, 0.1% sodium dodecyl sulfate [SDS], 50 mM Tris-HCl [pH 7.4], 150 mM NaCl, 0.5% sodium deoxycholate, 1 mM EDTA) and quantified by the Bradford assay. Protein samples (40 µg) were prepared with 4× LDS sample buffer (Invitrogen) and 50 mM DTT and electrophorosed on a NuPAGE Novex 4-12% Bis-Tris gel (Invitrogen) in MES buffer (Invitrogen). Protein was transferred onto a PVDF membrane, blocked for 30 min in 10% nonfat milk in TBST (Tris-buffered saline [TBS]/0.1% Tween-20), and incubated at 4 °C overnight with a rabbit anti-MCPH1 (Abcam, Cambridge, UK; 1:1000 ab2612) or mouse anti–β-actin (Sigma, St. Louis, MO; 1:5000 100M4789) antibody. Horseradish peroxidase (HRP)–conjugated secondary antibodies (DAKO, Glostrup, Denmark; 1:500) were added for an hour incubation at room temperature, followed by washes and visualization with Supersignal Pico/Femto (Thermo Fisher Scientific).
Fixation and Staining
Forty-eight hours after transfection with MCPH1 siRNA, media were removed by plate inversion and blotting onto clean paper towels to remove excess liquid. Cells were washed three times with 80 µL PBS, using a Fluid-X XRD-384 dispenser at 150 rpm, dispensing to the left side of the well to minimize disturbance of cells. All reagents subsequently added to cells used these settings.
Cells were fixed for 5 min in ice-cold methanol. Methanol was removed from a −20 °C freezer and kept on ice while it was passed through the dispenser tubing for 1 min to ensure the temperature of methanol being dispensed into plates was maintained. Ice-cold methanol (80 µL) was then dispensed into each well. Plates were placed in a −20 °C freezer for 5 min. After fixation, each plate was inverted and blotted to remove the methanol and rehydrated with 80 µL PBS per well.
Plates were blocked with 100 µL 1% nonfat milk powder/PBS (w/v) previously clarified of particulates by centrifugation at 3000 g for 20 min. DNA/nuclei were stained with DAPI (5 µg/mL) diluted in blocking solution. Plates were incubated for 1 h at room temperature in the dark.
High-Throughput Imaging
Plates were imaged with a PerkinElmer Operetta high-content wide-field fluorescence imaging system, coupled to Harmony software. Plates were automatically loaded onto the Operetta using a PerkinElmer Plate Handler II robotic arm, operated through Plateworks software. Plates were barcoded with specific information and barcodes were read before loading onto the Operetta. Wells were imaged using a 20× objective lens, in a single focal plane across each plate. The bottom of each well was detected automatically by the Operetta focusing laser and the focal plane calculated relative to this value. DAPI emission (455 nm) was imaged for 50 ms. Fifteen fields of view (each 510 × 675 µm) were imaged per well, with an identical pattern of fields used in every well. This pattern was designed to avoid imaging cells in the area targeted by the dispensers.
Image Analysis
Modified Columbus (PerkinElmer) image analysis algorithms were used throughout. Nuclei were detected using a modified “find nuclei” algorithm as blue (DAPI) fluorescent regions >100 µm2, with a split factor of 7.0, an individual threshold of 0.40, and a contrast >0.10 (method B). Border objects were excluded to ensure only whole nuclei were analyzed. The phenotypic nuclear spots characteristic of PCC were detected using a modified “find spots” algorithm, as well as fluorescent spots with a relative spot intensity >0.030 and a splitting coefficient of 1.0 (method A). PCC was defined as a nucleus containing 14 or more spots. Key output parameters were number of whole nuclei and percentage of nuclei with PCC.
Statistical Analysis
Statistical analysis was performed on a plate-by-plate basis using the z score calculation with a k value of 2, using CAPG2 and CAPH2 as the positive reference. 12 The strictly standardized mean difference (SSMD) was chosen as the quality control metric to evaluate the variability of and the difference between the control siRNA populations.
Small-Scale siRNA Screen
To test the assay performance, an initial screen was performed in duplicate on a batch of nine plates comprising the Dharmacon siGENOME SMARTpool Human Protein Kinase siRNA Library (G-003505).
Results and Discussion
Imaging Optimization
To identify modifiers of PCC, the PCC phenotype needed to be reliably and accurately induced and identified in a sufficient number of cells to generate a statistically powerful assay. Initial experiments aimed to determine whether the Operetta high-throughput screening (HTS) microscope and Harmony/Columbus software could achieve this. Previously, nuclei displaying PCC were imaged with a confocal microscope using a 63× objective lens, and the proportion of cells with PCC was manually assessed. Although this gave accurate resolution of chromosome condensation and a reproducible PCC phenotype, the method was low throughput and logistically unsuitable for application to a high-throughput screen. Therefore, our initial aim was to adapt these existing methods to a high-throughput protocol and determine the reliability of siRNA knockdown under these conditions.
We have previously shown that low levels of MCPH1 are linked with PCC in microcephaly patient lymphoblastoid cells and that siRNA targeting the MCPH1 gene significantly increased the proportion of HeLa cells with PCC.
2
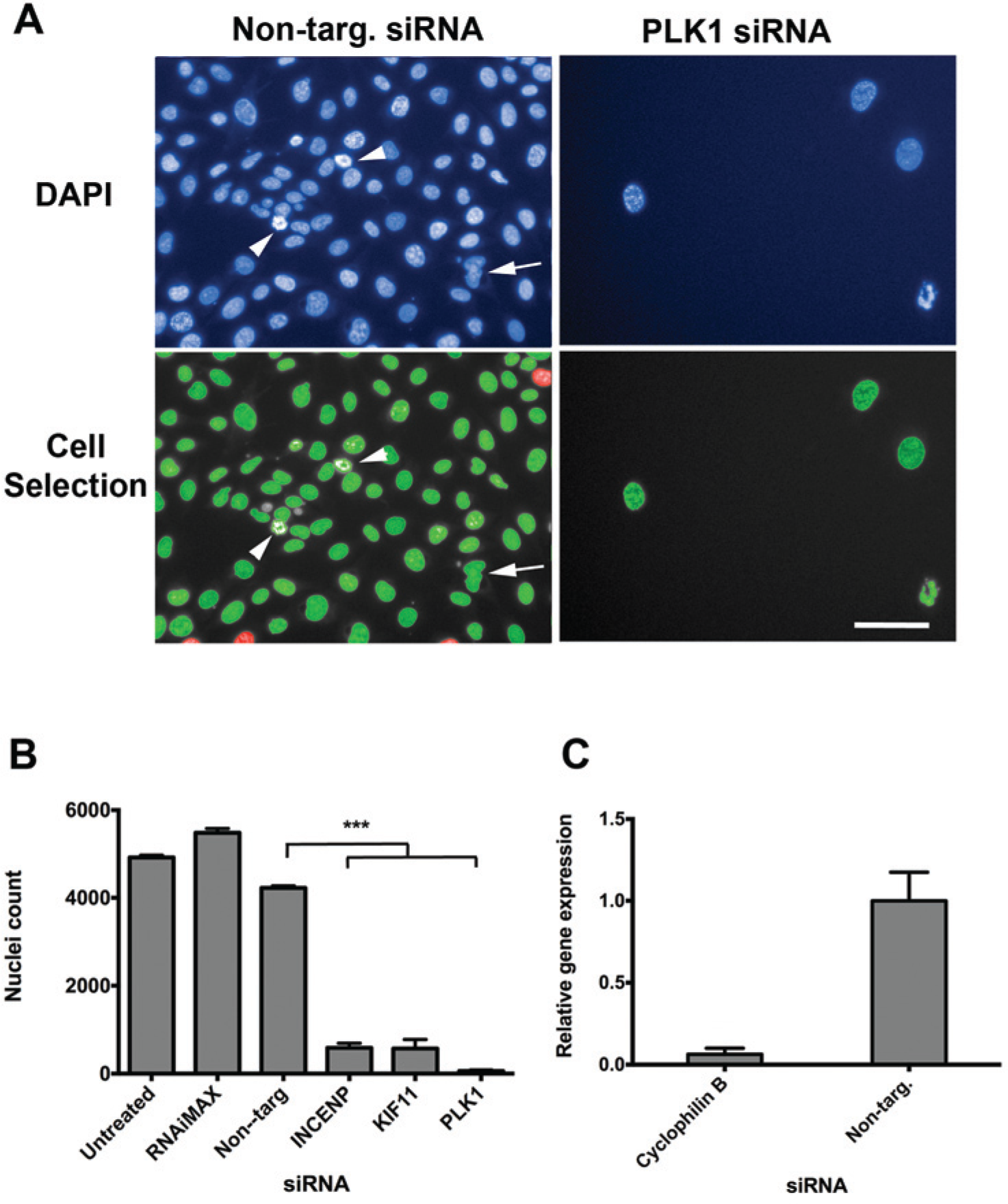
For the current assay, we chose the U2OS osteosarcoma cell line, which has a flat morphology with a large cytoplasmic area and grows as a consistent monolayer allowing discrete resolution of nuclei. Cells were seeded at a range of densities across a 96-well plate and incubated under standard conditions for 72 h before fixation with ice-cold methanol. Nuclei were stained with DAPI and imaged with the PerkinElmer Operetta system (
Fig. 1A
). Cells seeded at 6000 cells/well reached 80% to 90% confluence at 72 h, and nuclei were identified as discrete objects (
Fig. 1A
). Images were taken at a single focal plane across the 96-well plate in the position that encompassed the widest part of the nucleus. Using a 20× objective lens, each field of view contained approximately 500 to 600 whole nuclei. Incomplete nuclei at the edge of the image were excluded from further analysis (highlighted in red;
Fig. 1A
). Nuclei were determined as objects greater than 100 µm2 (highlighted in green;
Fig. 1A
), which excluded cell debris but included irregular bright nuclei likely to be cells undergoing mitosis or apoptosis (arrowhead in
Fig. 1A
; for more detail, see
Nuclei detection using Columbus and assay control validation. (
Determining Transfection Efficiency
We next determined the transfection efficiency of U2OS cells and investigated a number of potential targets for use as “transfection controls.” A transfection control provides a surrogate measure for transfection efficiency that does not rely on the assay end point (in this case % cells with PCC) to provide that information. Initial assessment of transfection efficiency with a fluorescently labeled siRNA (Dharmacon) required a separate assay to determine success and was considered logistically impractical for monitoring transfection efficiency throughout the screening process (not shown). The Harmony/Columbus software that provides automated analysis of Operetta data requires independent recognition of the nucleus ( Fig. 1A ). Therefore, any analysis protocol will automatically evaluate the number of nuclei within an image, providing a surrogate value for cell number. Hence, targeting a gene whose expression is crucial for cell growth and proliferation and using cell number as a marker is an ideal choice for a transfection control.
We had previously shown that silencing PLK1 by reverse transfection had a significant effect on cell number when used as a control in a screen to identify genes involved with cell proliferation. 13 In the mouse IMCD3 (inner medullary collecting duct) cell line, a Dharmacon siGenome siRNA duplex pool targeting Plk1 significantly inhibited cell proliferation compared with a nontargeting control (p < 0.0001, Student t test; z score <–6). 13 When PLK1 was silenced in U2OS cells by reverse transfection, a similar effect was observed that functionally confirmed a successful knockdown ( Fig. 1A , B ). A nontargeting siRNA control had no significant effect on cell number ( Fig. 1B ). We also investigated knockdown of INCENP and KIF11 and demonstrated that targeting these genes and PLK1 inhibited cell proliferation such that there were >90% fewer cells compared with nontargeting siRNA ( Fig. 1B ). This indicated a high transfection efficiency with these conditions. To confirm knockdown and high transfection efficiency, we transfected U2OS cells with an siRNA targeting cyclophilin B, the positive control recommended by Dharmacon for more than 20 different cell lines (www.thermoscientificbio.com/resource-library). Following transfection alongside nontargeting siRNA, we extracted RNA and performed qRT-PCR, demonstrating 94% knockdown of cyclophilin B ( Fig. 1C ). This provided quantitative evidence that U2OS cells can be transfected at high levels of efficiency and supported the functional observations from the silencing of PLK1, INCENP, and KIF11 ( Fig. 1B ).
Identification of PCC
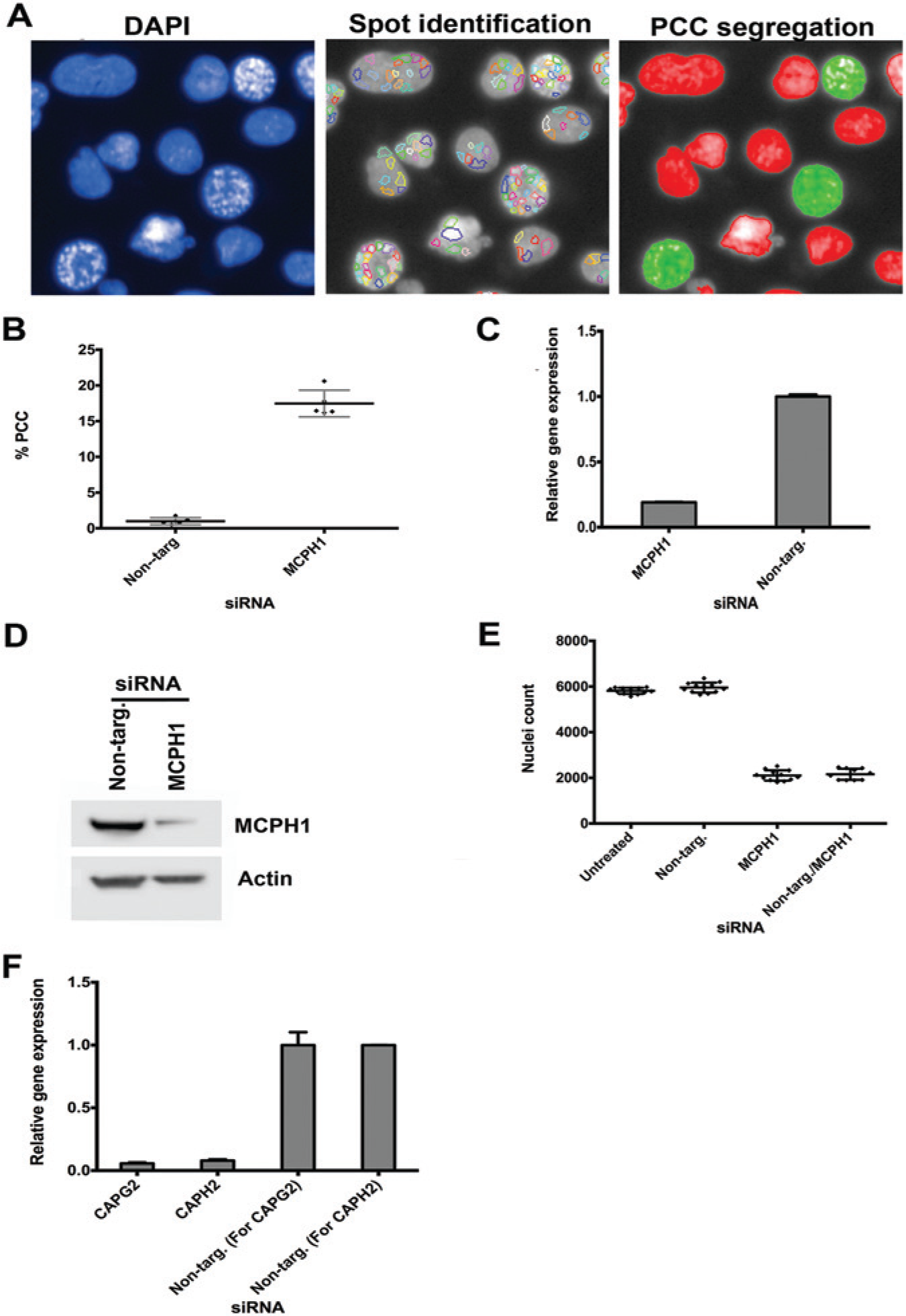
Crucial to the development of a PCC modifier assay is the consistent and reproducible generation of PCC in the U2OS cell line. We have previously shown that knockdown of MCPH1 induces PCC in HeLa cells. 2 We repeated the same experiment for U2OS cells, using a forward transfection. PCC was identified using the Columbus image analysis software to detect the number of spots present in the DAPI-stained nuclei ( Fig. 2A ; PCC cells highlighted in green). This assay was repeated on five further passages over a number of weeks investigating different batches of each reagent. PCC was consistently <1% when cells were transfected with nontargeting siRNA, whereas MCPH1 siRNA induced PCC in 15% to 20% of the cell population. Mean values at each passage are shown in Figure 2B (p < 0.001, Student t test; z <–3; n = 5). There was no correlation of passage number with baseline or induced levels of PCC, and using different batches of reagent had no significant effect. Next we confirmed the efficiency of the MCPH1 siRNA knockdown at both the RNA level by qRT-PCR ( Fig. 2C ) and the protein level by Western blotting ( Fig. 2D ).
Premature chromosome condensation (PCC) detection and positive control validation. (
MCPH1 also has a role in cell proliferation and apoptosis. Consequently, alongside assessment of PCC, we determined the effect of MCPH1 silencing on cell number. As previously described, nuclei number was used as a surrogate for cell number and assessed using an automated analysis protocol. MCPH1 siRNA reduced cell numbers by 59% ( Fig. 2E ; p < 0.001, Student t test; z <−3; n = 12), whereby the number of cells within 10 fields of view fell from approximately 6000 to 2000. To ensure that the end cell number was large enough to achieve statistical significance for the PCC modifier assay, we increased the number of fields of view to 15 to improve the signal-to-noise ratio.
Assessment of Screening Controls
To identify modifiers of PCC, our approach was to perform a unique double knockdown. Initially, a reverse transfection of positive and negative controls was performed followed by a forward transfection with MCPH1 siRNA to induce PCC. CAPG2 and CAPH2 regulatory subunits of the condensin II complex are involved in early prophase chromosome condensation. Previously, the PCC phenotype has been reduced by siRNA knockdown of both subunits in microcephaly patient lymphoblastoid cells and MCPH1-depleted HeLa cells, indicating that CAPG2 and CAPH2 are modifiers of the PCC phenotype. 14 Therefore, we validated these genes as positive controls, inhibiting the induction of PCC caused by MCPH1 silencing. In our study, CAPG2 and CAPH2 siRNA knockdown reduced PCC to 3.5% and 5%, respectively, compared with MCPH1 siRNA transfection alone at 12.4%. The efficiency of CAPG2 and CAPH2 siRNA knockdown at the RNA level was confirmed by qRT-PCR ( Fig. 2F ). Dharmacon-scrambled nontargeting siRNAs were included as negative controls. The negative controls had no significant effect on levels of PCC ( Fig. 2B ).
Next we investigated the efficacy of the double-knockdown procedure. We anticipated that the double transfection would have a significant effect on proliferation given our findings of the significant loss in cell numbers caused by MCPH1 siRNA. Thus, we performed a reverse transfection with nontargeting siRNA (negative control) followed 24 h later by MCPH1 knockdown. The reverse transfection had no significant additional effect on cell number compared with MCPH1 transfection alone ( Fig. 2E ).
Validation of Screen
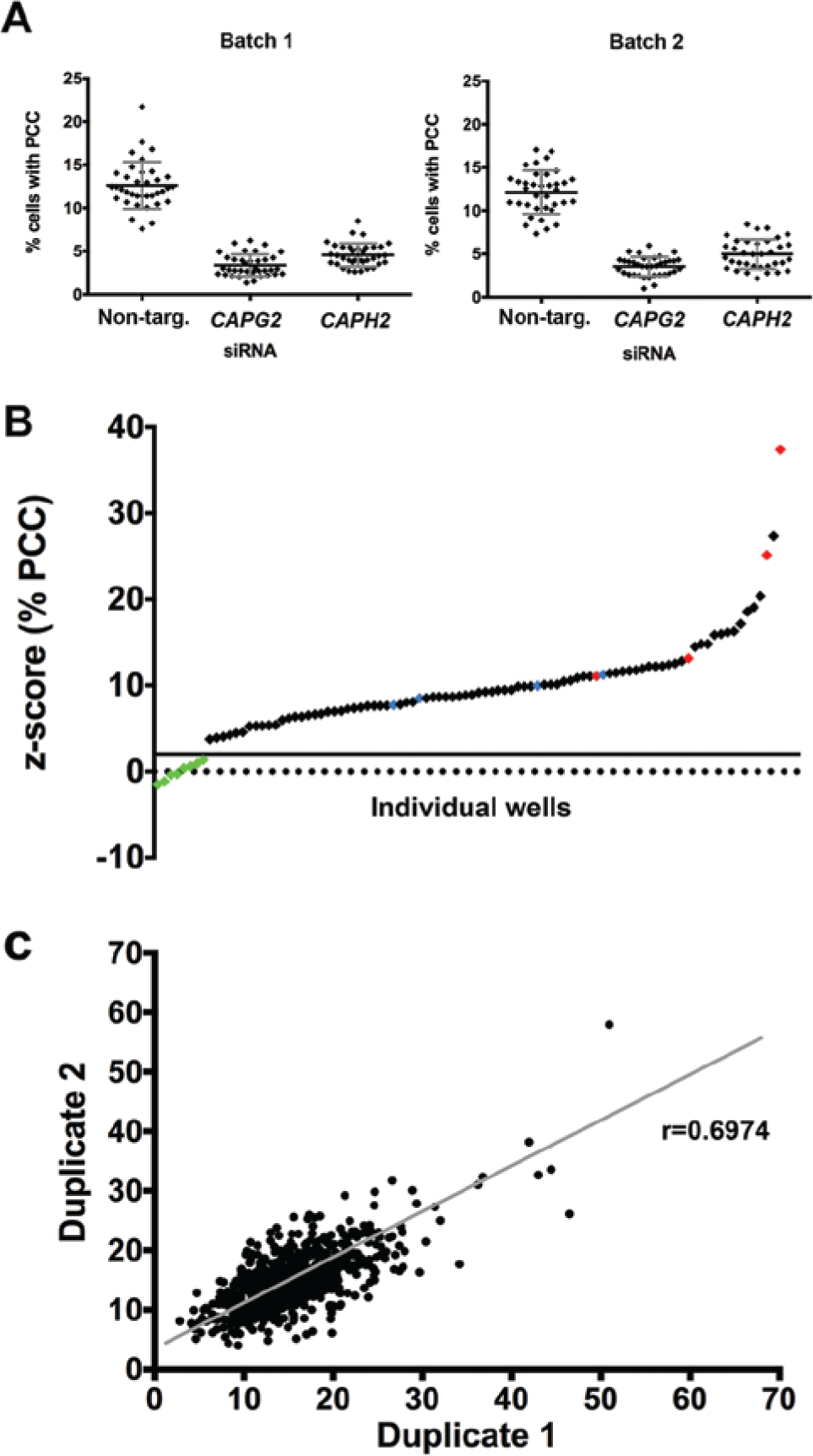
An initial screen of nine plates was performed in duplicate, comprising the Dharmacon siGenome protein kinase sublibrary to determine assay efficacy. Each plate contained eight positive control siRNA (four CAPG2 and four CAPH2) and four negative (nontargeting) controls. The mean (SD) % PCC for positive controls was 4.0% (1.3%) for the first batch and 4.3% (1.4%) for the second batch (total n = 72 wells) ( Fig. 3 ). The mean (SD) % PCC for negative controls was 12.6% (2.7%) for the first batch and 12.1% (2.5%) for the second batch (total n = 36) ( Fig. 3A ). Nuclei counts for the positive controls (mean cell number 3133.7) did not differ significantly from those of the nontargeting negative controls (mean cell number 3173) (p > 0.5, Student t test), demonstrating that CAPG2/CAPH2 transfections did not increase cell death. The reduction in PCC seen with the positive controls was therefore indicative of effective CAPG2/H2 knockdown rather than as a secondary effect due to loss of cells.
Comparison of assay reproducibility. (
Statistical analysis was performed on a platewise basis to exclude plate-specific effects. Due to the robustness of the CAPG2 and CAPH2 data, a z score calculation that substituted positive control values for negative control values was used. Therefore, a z score of <2 for both replicates identified potential hits. The % PCC for the positive controls fell consistently below the +2 z score threshold, equating to a % PCC of around 5% ( Fig. 3B ). The overall false-positive rate was 0% (n = 72), and the overall false-negative rate was 2.1% (n = 144).
The % PCC was compared between duplicate 1 of the screen and duplicate 2. A statistical analysis was performed, and the Pearson’s correlation identified a strong positive correlation: r = 0.6974; p < 0.0001; n = 720 ( Fig. 3C ). This demonstrated the robustness of the duplicate strategy in identifying modifiers of PCC.
The SSMD was calculated for each batch to exclude any batch-specific effects. The SSMD for the first batch of positive controls (CAPG2/CAPH2) for % PCC was 2.89 and for the second batch was 2.68. These values demonstrate that the reduction in % PCC seen with these siRNAs was “strong” compared with that of the nontargeting negative controls, reflecting the suitability of CAPG2/H2 as positive controls for reduced PCC. 15 The SSMDs for transfection efficiency based on a reduction in cell number using PLK1 and INCENP siRNA were 9.27 and 6.38 for the first batch and 12.12 and 7.41 for the second batch, respectively. PLK1 and INCENP are therefore appropriate positive controls for effects on cell number.
To ensure a reduction in % PCC was due to the modification of MCPH1 function rather than a secondary effect due to cell death as a result of siRNA toxicity, the cell number output from a complementary siRNA screen in U2OS cells without MCPH1 siRNA knockdown was used for comparison. Any hits that caused cell death in the complementary screen producing a z score of <–2 were eliminated from further analysis. This method allows the exclusion of false-positive hits and will be applied during the discovery phase of all subsequent screens. Synthetic lethality was also identified applying this approach, whereby single knockdown had no effect on cell number but, in combination with MCPH1, caused significant cell death.
We have identified eight potential modifiers of MCPH1 function in the human protein kinase sublibrary, with one inducing synthetic lethality. Four individual Dharmacon ON-TARGETplus siRNAs that boast a superior silencing mechanism compared with the SMARTpools used in the siRNA screen will be transfected into U2OS cells and three ovarian cancer cell lines to minimize any off-target effects. Modification of MCPH1 function by the specific siRNA under investigation will be confirmed by the PCC readout.
In conclusion, we have developed a novel high-throughput protocol to detect modifiers of PCC. This assay enables the identification of genes whose knockdown affects PCC rate. Similarly, this protocol could also easily be adapted for large-scale small-molecule screens to identify compounds that reduce PCC in MCPH1-deficient cells or cause PCC in normal cells. This assay will facilitate the future elucidation of the function of MCPH1 and chromosome condensation.
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by Yorkshire Cancer Research (grant number L363).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.