
Abstract
Genetics is one of the various approaches adopted to understand and control mammalian sleep. Reverse genetics, which is usually applied to analyze sleep in gene-deficient mice, has been the mainstream field of genetic studies on sleep for the past three decades and has revealed that various molecules, including orexin, are involved in sleep regulation. Recently, forward genetic studies in humans and mice have identified gene mutations responsible for heritable sleep abnormalities, such as SIK3, NALCN, DEC2, the neuropeptide S receptor, and β1 adrenergic receptor. Furthermore, the protein kinase A-SIK3 pathway was shown to represent the intracellular neural signaling for sleep need. Large-scale genome-wide analyses of human sleep have been conducted, and many gene loci associated with individual differences in sleep have been found. The development of genome-editing technology and gene transfer by an adeno-associated virus has updated and expanded the genetic studies on mammals. These efforts are expected to elucidate the mechanisms of sleep–wake regulation and develop new therapeutic interventions for sleep disorders.
1 Introduction
A good night’s sleep is essential for an active and healthy life. After learning, exercising, or when experiencing a cold, sleep helps to form memories, refreshes the body, and supports the immune response, respectively. A lack of sleep can lead to a variety of mental, physical, and social problems [1 –4]. Insomnia occurs in 10%–20% of the general population and is often comorbid with other physical and psychiatric disorders [5].
Why is sleep important for our health, and how is it regulated? Although we cannot fully answer these simple questions at this time, the discovery of orexin and the development of optogenetics and pharmacogenetics have advanced our understanding of the neural circuits that control sleep and wakefulness [6 –8]. Previous studies have identified multiple neuronal networks that can shift vigilance stages between wakefulness, rapid eye movement (REM) sleep, and non-REM (NREM) sleep and that exhibit activity changes related to sleep/wakefulness. However, how these neuronal circuitries for sleep/wake switching are regulated according to the homeostatic sleep need remains unclear.
To fundamentally understand sleep, genetic studies have been conducted on mammals and key molecules have been identified. Genetic studies of sleep using animal models can be broadly divided into forward and reverse genetics. In the former, two screening methods are adopted, namely, the dominant and recessive screenings. Genome-wide association studies of large numbers of the general population and next generation sequencing analysis of familial sleep deviations have been conducted to understand the genetic factors underlying individual differences in sleep. Reverse genetic approach usually examines gene-deficient animals. This review summarizes the results of forward and reverses genetic studies on mammalian sleep and sleep-regulating molecules such as salt-inducible kinase (SIK3) discovered using genetic approaches.
2 Reverse genetic studies in gene-deficient mice
The majority of genetic studies on sleep conducted in mice are reverse genetics studies. This research field enables the investigation of whether target genes are involved in sleep regulation through the observation of the sleep/wakefulness of genetically modified mice. Most reverse genetics studies use candidate gene approaches, in which target genes are selected based on the known function of gene products, their expression patterns, and hypotheses linking genes to sleep. The earliest experiments on sleep using genetics were performed on genetically modified mice lacking the prion protein [9] and transgenic mice having a rat tyrosine hydroxylase promoter linked to a human growth hormone gene [10]. Since then, sleep has been studied in mice with altered genes related to circadian behavior, inflammation, and neurotransmission [11]. However, careful observation of orexin-deficient mice, which were expected to change their feeding behavior, serendipitously led to one of the most important discoveries about sleep [12]. In particular, it was shown that the enigmatic sleep disorder known as narcolepsy was caused by an orexin deficiency [13], and the orexin system was found to constitute the core of the wake-promoting neural network [6, 8].
The International Mouse Phenotyping Consortium, an international project to which multiple institutions collaborate to identify the function of every protein-coding gene, investigates sleep using a piezoelectric floor in some facilities [14, 15] (https://www.mousephenotype.org). As of March 2022, 553 genes have been tested, and 63 of them were associated with abnormal sleep behavior.
Clustered regularly interspaced short palindromic repeats (CRISPR) technology can produce gene knockout mice more easily and quickly than the conventional gene targeting using homologous recombination in embryonic stem cells. CRISPR applying three guide RNAs to three different regions of a single gene can produce nearly complete knockout mice from the first generation. Combined with respiration-based sleep/wake staging, Ueda and his colleagues advanced the reverse genetic strategy in mice and identified the glutamate ionotropic receptor NMDA type subunit 3A (Grin3a) gene as a short sleeper gene [16], CamKIIα/β genes for the regulation of sleep duration [17], and cholinergic receptor muscarinic (Chrm) 1 and 3 as regulators of REM sleep [18] (Fig. 1).
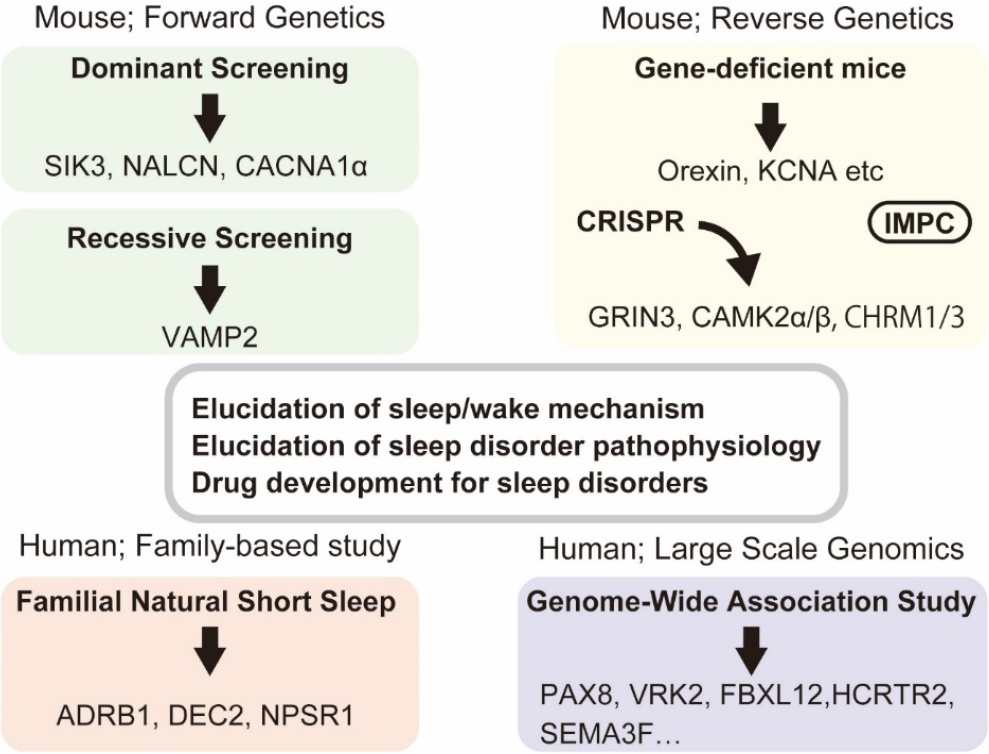
Genetic approaches to mammalian sleep and detection of sleep-associated genes.
However, the reverse genetics approach based on germline lineage has the following several limitations: 1) detailed testing of sleep and wakefulness by EEG and EMG cannot be applied to a comprehensive project, 2) lethal genes cannot be examined, 3) the loss-of-function may be compensated by other genes, and 4) sleep changes not physiologically but as a secondary effect of other organ malfunctions.
3 Forward genetic studies in randomly mutagenized animals
Contrary to reverse genetics, forward genetics allows researchers to identify mutated genes that are responsible for, or associated with, heritable phenotypes. In early 1900, Thomas Hunt Morgan and his colleagues began forward genetic studies in laboratories using the fruit fly Drosophila melanogaster. Ron Konopka and Seymour Benzer established mutant fly lines with abnormal circadian behavior [19], which led to the identification of the transcription–translation feedback loop of clock genes as the molecular mechanism of circadian rhythm, which is also conserved in mammals [20, 21].
Soon after the behavioral definition of sleep in fruit flies was established [22, 23], Cirelli et al. first identified Shaker mutants through behavioral screening of randomly mutagenized flies [24]. Subsequently, many of the fruit fly sleep mutants were found to have mutations in the Shaker potassium channel or related molecules. Therefore, it was concluded that potassium channels may play an important role in the regulation of sleep in mammals. There are approximately 40 voltage-gated potassium channels in mice, which are classified into 12 subfamilies; Kv1.1 (KCNA1) to Kv1.8 (KCNA10) is mammalian Shaker-related channels. Among these channels, mice lacking the voltage-gated potassium channel subunit Kv1.2 (KCNA2) showed an increased waking time and decreased NREM sleep.
Hence, in both fruit flies and mammals, the deletion of Shaker-related channels results in prolonged wakefulness. Unfortunately, these deletion in mice causes embryonic lethality and developmental abnormalities, making these mutants unsuitable for studying sleep/wakefulness. Please consult our previous review on this topic for more details [21].
While the research results on circadian behavior in fruit flies have been successfully applied to mammals, the same could not easily be done for the results of sleep research. This is probably because the brain structure and genetic makeup of the two types of organisms are very different. Therefore, forward genetics in mice was chosen as a potential application to discover novel sleep-regulating genes. In early 2010, we began screening randomly mutagenized mice for sleep abnormalities using EEG/EMG which allows unequivocal staging of wakefulness, NREM sleep, and REM sleep and examined not only quantitative but also qualitative data obtained from EEG spectrum analysis.
4 Dominant screening of sleep-regulating genes
The dominant screening method examines the dominant effect of gene mutations on sleep in heterozygous mutant mice. As loss-of-function or null alleles are usually recessive to wild-type alleles, loss-of-function variants are generally identified by recessive screening. A strong heritable phenotype is required to identify gene mutations. However, in general, a heterozygous loss-of-function mutation results in mild changes in sleep/wakefulness and, thus, dominant screening usually leads to heterozygous gain-of-function gene mutations. In fact, this method also detected a loss-of-function mutation of the calcium voltage-gated channel subunit α1A (Cacna 1a) gene in the long sleeper pedigree Drowsy [25]. As the number of genes critically involved in the target phenotype is not expected to be very large, the genes found by genetic screening may be actually those that are already known to regulate the target phenotype. For example, dominant screening for obesity led to the detection of the melanocortin 4 receptor (MC4R) gene and Sim1 gene, which are well known to regulate energy metabolism [26]. The reason why dominant screening failed to discover novel genes regulating energy metabolism is probably because there are few genes left to be discovered.
In forward genetics studies, linkage analysis is generally used to narrow down the chromosomal regions associated with sleep abnormalities. For linkage analysis, the mutated inbred lines need to be backcrossed with close, but different, lines. We used C57BL/6J (B6J) and C57BL/6N (B6N) as a mutagenized strain and counter strain, respectively. Originally, C.C. Little inbred the C57BL/6 strain in 1921 and maintained it in the Jackson laboratory (Maine, USA). In 1951, the C57BL/6 strain was introduced in the National Institute of Health and was renamed C57BL/6N, whereas the C57BL/6 strain was renamed C57BL/6J.
Previous studies showed that Intraperitoneal administration of a chemical mutagen, ethyl nitrosourea (ENU), to male B6J mice causes numerous point mutations in spermatogonia, and subsequently in the sperm, at a rate of approximately one mutation per million bases [27, 28] To obtain the next generation, the ENU-treated B6J mice or their sperm were subjected to natural mating or in vitro fertilization with wild-type B6N females. The offspring (F1 generation) were evaluated for sleep abnormalities using EEG/EMG-based sleep staging [25, 29].
Based on our examination of the reproducibility of sleep parameters, we used simple parameters, such as total wake time, as an indicator to detect mice with sleep abnormalities [25]. When the abnormalities were shown in F1 mice, these were crossed with wild-type female B6N mice to obtain the next generation, namely, N2 mice. By examining N2 mice, we determined whether the abnormal phenotype is heritable. If they showed the same sleep abnormalities as their father F1 mouse, these mice were further used for quantitative trait locus analysis and whole-exome sequencing. Because of the large difference in sleep between males and females, only male mice were used for sleep analysis [30]. This approach identified the genetic mutations responsible for heritable sleep abnormalities, such as the Sik3 gene mutation in the Sleepy mutant pedigree and the Nalcn gene mutation in the Dreamless mutant pedigree [31] (Fig. 1).
5 Recessive screening of sleep-regulating genes
Contrary to dominant screening, recessive screening examines the recessive effect of gene mutations on sleep in homozygous mutant mice. This type of screening usually requires a larger generation before gene mutations are identified. As loss-of-function or null alleles are usually recessive to wild-type alleles, loss-of-function variants are generally identified by recessive screening. Through this method, it is possible to identify genetic variants that have a mild to moderate effect on sleep and cannot be detected by dominant screening.
Through the Harwell Ageing Screen, recessive screening has been conducted for metabolic, sensory, and behavioral abnormalities including sleep, which identified Vamp2 gene mutations in G3 generation mice showing sleep abnormalities [15] (Fig. 1).
6 Genetic studies of human sleep
There have been several large genome-wide association studies for self-reported sleep duration [32 –39]. A large UK biobank study (453,379 participants) identified 57 genomic loci for self-reported insomnia symptoms [37]. PAX8, VRK2, and FBXL12 loci were repeatedly found in two independent studies [32, 38] (Fig. 1). HCRTR2 (orexin receptor type 2) loci were associated with short sleep duration [38] and daytime sleepiness [40], and a missense variant of the HCRTC2 gene was associated with daytime napping, morning preference, and ease of awakening [39], which suggests that individual differences in the orexin system due to genetic variances may be involved in individual differences in sleep habits. Hence, multiple differences in many genes contribute to individual differences in sleep. However, causality is rarely proven, and the interpretation of results depends on the known functions of gene products studied in other animal models or in cultured cells; usually, it does not provide new insights into specific mechanisms.
Dr. Fu and colleagues have found gene mutations in individuals with familial natural short sleep who are healthy short sleepers and sleep 4–6 h a day, which they consider sufficient (Fig. 1). The study also found a DEC2 (or BHLHE41) P384R mutation in human short sleepers and showed that mice carrying the Dec2 P384R transgene exhibited short sleep duration, proving the causality between the human gene mutation and short sleep duration [41]. DEC2 is a basic helix-loop-helix protein, and P384 is a well-conserved amino acid located in a proline-rich domain near the C-terminus. Another research group found the DEC2 Y362H mutation in people with familial natural short sleep [42].
Recently, Dr. Ptáček and Dr. Fu found a neuropeptide S receptor (Npsr1) gene mutation in familial natural short sleep families [41]. The mutation causes the substitution of the 206th tyrosine residue with histidine (Y206H), which is located in the well-conserved extracellular region of the G protein-coupled receptor for neuropeptide S. Then, to prove the causality, Npsr1-Y206H mice were generated, and their sleep/wake behavior was examined. The mice showed shorter total sleep time, consistent with observations in the human short sleeper family with the same mutation [43]. Npsr1-Y206H mice are resistant to the loss of memory performance induced by sleep deprivation, similarly to natural short sleepers in humans [43]. Similarly, a missense mutation was found in the β1 adrenergic receptor (Adrb1) gene in other familial natural short sleep families [44]. Mice with the same mutation, Adrb1-A187V, showed short sleep, consistent with the human family, and exhibited increased activity in the Adrb1-positive neurons of the dorsal pons [44]. These findings indicate that combining human genetic findings with mouse studies increases the possibility of determining causality rather than mere correlation, and it enables the validation of results obtained from human genetics at the level of molecular pathways and neural circuits.
7 The SIK3 pathway regulates sleep need
Lastly, we introduce the role of SIK3 in the regulation of sleep need, which was originally identified by dominant screening [31]. The Sleepy (Slp) mutant pedigree with longer NREM sleep duration and greater EEG delta density during NREM sleep was established using an EEG/EMG-based screen for sleep abnormalities in randomly mutated mice. Linkage analysis showed that there was a single peak of the logarithm of the odds score on chromosome 9. Whole-genome sequencing revealed a genetic mutation in the salt-inducible kinase (Sik) 3 gene on this chromosome, which correlated perfectly with the long NREM sleep phenotype. Given a large number of mutations in the offspring of ENU-treated mice, we cannot rule out the possibility that the long NREM sleep of Sleepy mutant mice is due to a genuine mutation close to the SIK3 gene, as most genomic regions have not been examined by whole-genome sequencing. When Sik3 gene-modified mice with the same mutation were generated using the CRISPR/ Cas9 system, the Sik3 (Slp) mice showed long NREM sleep as the original Sleepy mutant pedigree did [31], proving the causality between the Sik3 (Slp) allele and increased sleep need. The Slp mutation is an in-flame splice mutation that causes the skipping of exon 13 [Fig. 2(A)]. The region encoded by exon 13 contains serine 551 (S551), a well-conserved protein kinase A (PKA)-phosphorylated serine residue. The substitution of S551 with alanine (S551A) or aspartic acid (S551D) resulted in a decrease in NREM sleep time comparable with that observed in Sleepy mutant mice [45] [Fig. 2(B) and (C)]. Although the cell type and organ involved in the increased sleep need in Sik3 (Slp) mice were not determined, the induction of Sik3 (Slp) in neurons increased sleep, indicating that neurons, and not glia or other cells, are the primary site where SIK3 regulates sleep need [46]. Sik3 orthologues are also involved in sleep in fruit flies and roundworms [31, 47], suggesting that the role of this gene in sleep is conserved in diverse animals, including invertebrates.
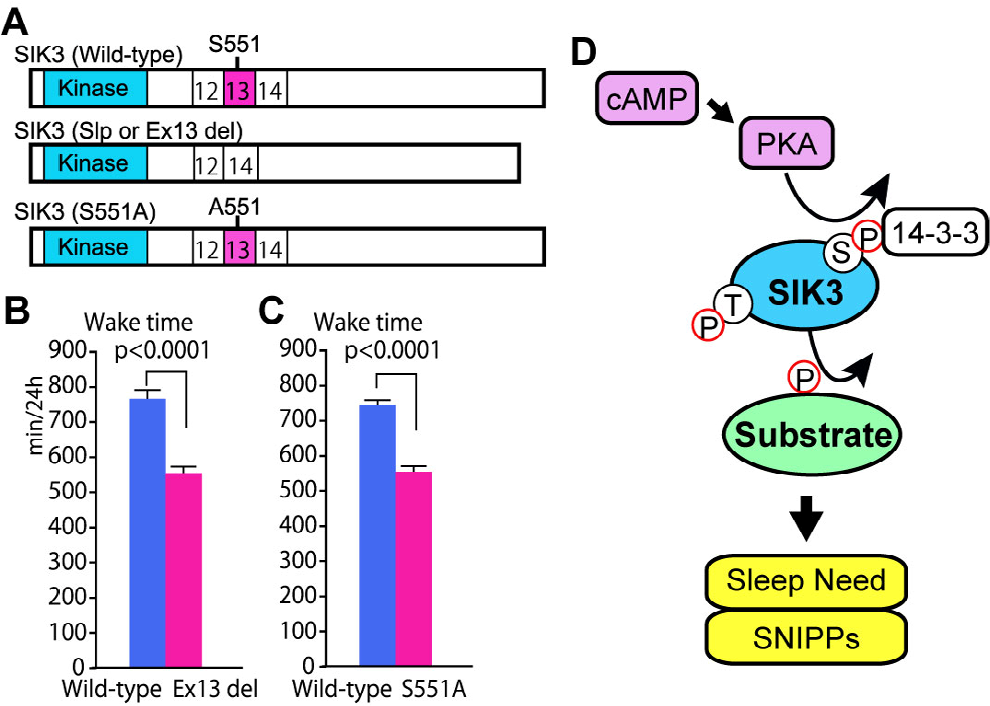
Regulation of sleep need via PKA-SIK3 signaling. (A) Scheme of wild-type, exon 13-encoded region-deleted (Ex13 del), and S551A-type SIK3 proteins. (B) and (C) Heterozygous Sik3 ex13 del mice (B) and Sik3 S551A mice (C) showed a shorter total wake time compared to wild-type mice. (D) PKA-SIK3 signaling enhances sleep need and the phosphorylation of sleep-need-index phosphoproteins (SNIPPs).
As SIK3 is a member of the SIK family along with SIK1 and SIK2, and SIK1 S577 and SIK2 S587 are equivalent to SIK3 S551, we examined the sleep behavior of mice in which SIK1 S577 and SIK2 S587 were replaced with alanine. Both SIK1 and SIK2 PKA site-deficient mice showed increased sleep need but this was considerably weaker compared to that observed in SIK3 S551A mice. This finding can be explained by the fact that the expression of SIK1 and SIK2 in the brain is considerably weaker than that of SIK3 [48]. SIK was originally identified and named as a gene expressed in the rat adrenal cortex in response to a high-salt diet, but this type of diet did not increase SIK1 in the mouse adrenal cortex [48].
The SIK3 protein is a kinase with a kinase domain at the amino terminus [Fig. 2(A)] and belongs to the AMP kinase family [49]. There are several isoforms depending on the presence or absence of a C-terminus [48]. A common feature of SIK3 protein mutants that cause long NREM sleep such as Slp, S551A, and S551D is reduced binding to the 14-3-3 protein compared with wild-type SIK3 [45]. Although it is unclear whether binding to the 14-3-3 protein itself is important for sleep regulation, changes in the biochemical properties of the SIK3 protein may lead to shorter waking times. These results indicate that PKA-SIK3 signaling plays a crucial and conserved role in sleep regulation [Fig. 2(D)]. Reverse genetics and pharmacological methods have shown that the kinases CaMKIIα/β and ERK1/2 also act as intracellular signals that regulate sleep [17, 50].
As Sik3 (Slp) mice have an inherently high sleep need, they can be used to reveal the molecular entities of sleep need and sleepiness. Phosphoproteomic analysis using Sik3 (Slp) mice and sleep-deprived wild-type mice revealed increased phosphorylation in 80 phosphoproteins, which may be a molecular signature of sleep need [51]. Sixty-nine out of 80 phosphoproteins were associated with the function or structure of synapses, indicating that these may be the site where sleep need is regulated [52].
8 Future directions
As discussed in this study, several strategies are used in the genetic research of mammalian sleep, and each is characterized by both strengths and limitations. The randomness of mutations and the lack of a need for hypotheses represent the strengths of forward genetics. In contrast, the strength of reverse genetics is that it allows researchers to select candidate genes based on their own ideas and hypotheses about the mechanisms that control sleep. The limitation of ENU-based forward and reverse genetics is that it is often not possible to assess the sleep phenotype due to the lethal effects of mutations or to secondary effects on nontarget organs, such as the brain, because they are germline mutations rather than target organ-specific. However, the combination of tissue-specific Cas9-expressing mice and adeno-associated virus carrying the PHP.eB capsid enables genome-editing in a more spatially- and cell type-specific manner, which can remove the limitations of germline-based strategies and expand reverse genetics.
The use of homology-directed repair allows the application of the CRISPR/Cas9 system to the creation of gain-of-function mutations. The development of CRISPR-mediated base editing, in which cytidine deaminases are fused to Cas9 nickase to substitute a single base without generating indels, has expanded the applicability of the CRISPR-related toolbox to forward and reverse genetics studies [53]. For plant genes, Cas9 nickase fused with cytosine and adenine base editors has been engineered to achieve random mutagenesis and has been used in combination with guide RNA for targeted random mutagenesis [54].
With the development of genome-editing technology, it has become easier to validate findings obtained in humans by conducting tests in animals to prove causality and gain insights into the mechanisms of sleep regulation. Ultimately, these efforts to elucidate the molecular basis of animal sleep will lead to a better understanding of sleep disorders and related psychiatric and neurological disorders.
Footnotes
Conflict of interests
The authors declare that there is no conflict of interests.
Funding
This work was supported by the Ministry of Education, Culture, Sports, Science and Technology World Premier International Research Center Initiative to M.Y., Japan Society for the Promotion of Science KAKENHI (Grant Nos. 17H06095 and 22H04918 to M.Y.and H.F.; Grant Nos. 17H04023, 17H05583, and 20H00567 to H.F.), and by AMED (Grant No. JP21zf0127005 to M.Y.).
Acknowledgments
We thank all Yanagisawa/Funato laboratory members and IIIS members for their kind support, technical assistance, and discussion.
Authors’ contribution
H.F. and M.Y. conceived and wrote the manuscript.