
Abstract
The present study describes a novel methodology for the detection of reactive compounds using in vitro peptide-trapping and liquid chromatography–high-resolution accurate mass spectrometry (LC-HRMS). Compounds that contain electrophilic groups can covalently bind to nucleophilic moieties in proteins and form adducts. Such adducts are thought to be associated with drug-mediated toxicity and therefore represent potential liabilities in drug discovery programs. In addition, reactive compounds identified in biological screening can be associated with data that can be misinterpreted if the reactive nature of the compound is not appreciated. In this work, to facilitate the triage of hits from high-throughput screening (HTS), a novel assay was developed to monitor the formation of covalent peptide adducts by compounds suspected to be chemically reactive. The assay consists of in vitro incubations of test compounds (under conditions of physiological pH) with synthetically prepared peptides presenting a variety of nucleophilic moieties such as cysteine, lysine, histidine, arginine, serine, and tyrosine. Reaction mixtures were analyzed using full-scan LC-HRMS, the data were interrogated using postacquisition data mining, and modified amino acids were identified by subsequent LC-HRMS/mass spectrometry. The study demonstrated that in vitro nucleophilic peptide trapping followed by LC-HRMS analysis is a useful approach for screening of intrinsically reactive compounds identified from HTS exercises, which are then removed from follow-up processes, thus obviating the generation of data from biochemical activity assays.
Keywords
Introduction
The high rate of attrition in drug development is largely due to drug-mediated toxicity. It has been recognized that covalent modification of nucleic acid (DNA) and cellular proteins resulting from bioactivation of drugs plays an important role in drug-induced toxicity such as genotoxicity and idiosyncratic toxicity. 1 Of particular concern are low-incidence, idiosyncratic events that are not detected in preclinical animal testing or in clinical trials with inadequate numbers of test subjects. 2 Although the mechanisms are not fully understood, idiosyncratic toxicity is thought to involve the initial formation of reactive metabolites from the parent drug followed by covalent binding to macromolecules, leading to the formation of peptide/protein-compound conjugates.3–5 Subsequent proteolytic processing of drug-modified proteins may lead to the generation of peptides that are recognized as foreign and consequently produce either tolerance or initiation of an active immune response in vivo. 2 Thus, strategies are needed to reduce such attrition in drug discovery.
Most of the current in vitro assays for detecting reactive metabolites, such as glutathione (GSH) trapping assays,6–8 are conducted in the context of mature medicinal chemistry programs and focus on the reactivity of metabolites instead of the drug candidates themselves, although the use of similar assays conducted in the absence of reduced β-nicotinamide adenine dinucleotide 2′-phosphate (NADPH) can often determine if the compound itself is reactive. However, of more interest to the current work is the issue of identifying intrinsically reactive compounds that can be identified in high-throughput screening (HTS) campaigns, as typically employed in early drug discovery programs. The reactivities of such compounds are often recognized by medicinal chemists conducting hit triage exercises and can also be identified using a number of in silico methods. 9 However, compounds are often identified in screens with unknown or suspect chemical reactivity. Experimental protocols are therefore needed to profile the intrinsic reactivity of such hits and provide data to support or reject their further progression through the hit triage process. Not only does this obviate the progression of undesirable reactive series, but it also prevents medicinal chemists following potential artifactual biological activities. In addition, the data generated from these reactivity studies can be used to annotate structurally related compounds and thereby flag these for additional scrutiny in whichever assay they appear as hits.
Correspondingly, to facilitate the triage of hits from HTS, a novel peptide-based in vitro assay was developed and is described here for the detection of reactive compounds using liquid chromatography–high-resolution accurate mass spectrometry (LC-HRMS). Very limited mass spectrometric methods have been reported for this application. An LC–multiple-reaction monitoring (MRM)/mass spectrometry (MS) method was reported for the detection of reactive chemicals with known skin-sensitizing activity using a single nucleophile-containing peptide. 10 Moreover, there are also few reports describing the methodology for screening drug-peptide adducts.2,11 For example, a surface-enhanced laser desorption ionization–time-of-flight (TOF) mass spectrometry system was described to detect covalently bound adducts from microsomal incubations with peptide-trapping agents of custom-synthesized peptides consisting of cysteine or other nucleophilic amino acid residues. 2
The assay reported in the current work consists of in vitro incubations of test compounds (under conditions of physiological pH) with synthetically prepared peptides presenting a variety of nucleophilic moieties. The custom peptides are classified into four types, containing a thiol moiety (Cys), amine moieties (Lys, His, Arg), hydroxyl moieties (Ser, Tyr), or a mixture of these nucleophilic moieties (underlined in
Table 1
and
Summary of Reactions Observed between Compounds 1 to 4 and the Novel Synthetic Peptides Used as Trapping Agents to Assess Reactivity.
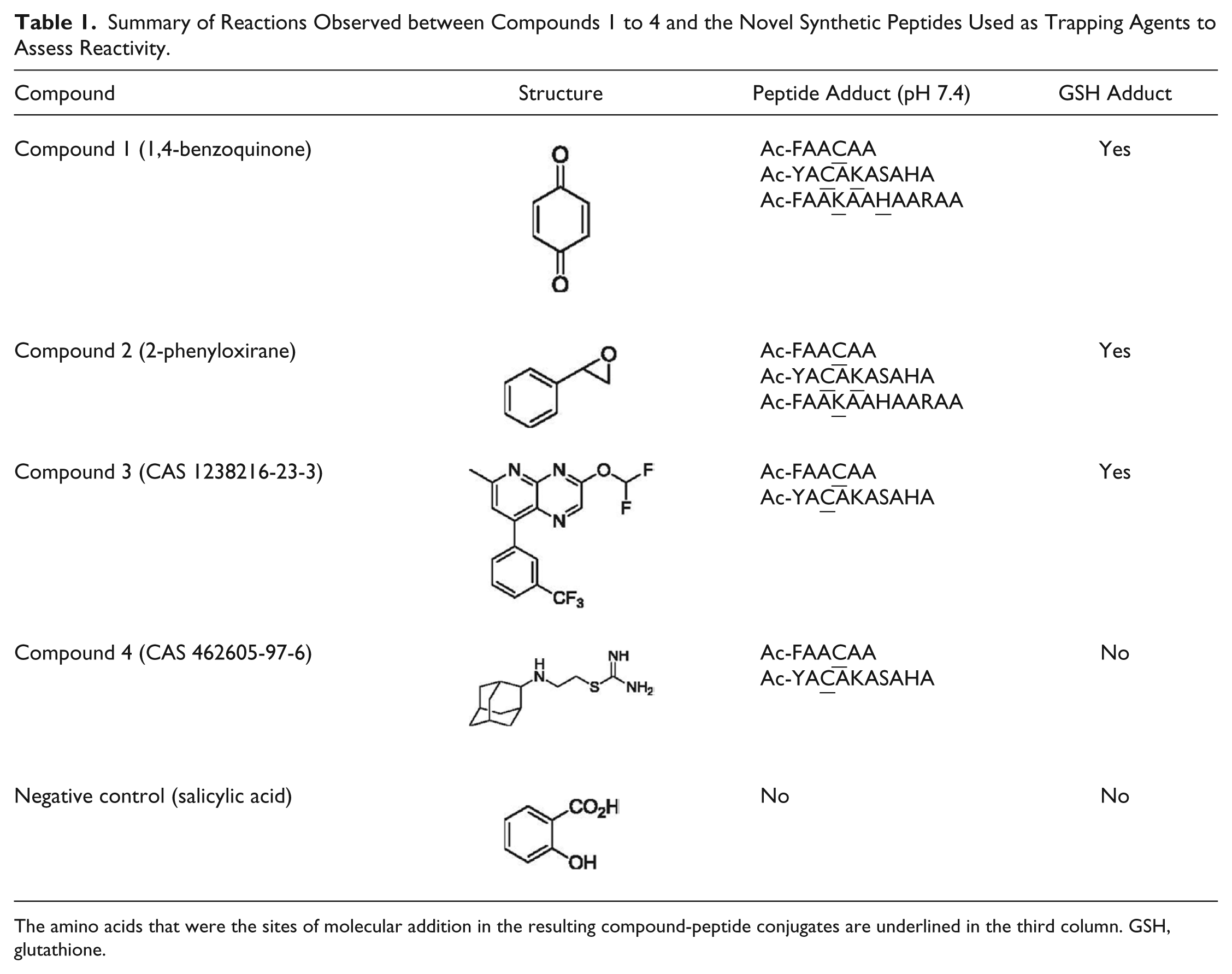
The amino acids that were the sites of molecular addition in the resulting compound-peptide conjugates are underlined in the third column. GSH, glutathione.
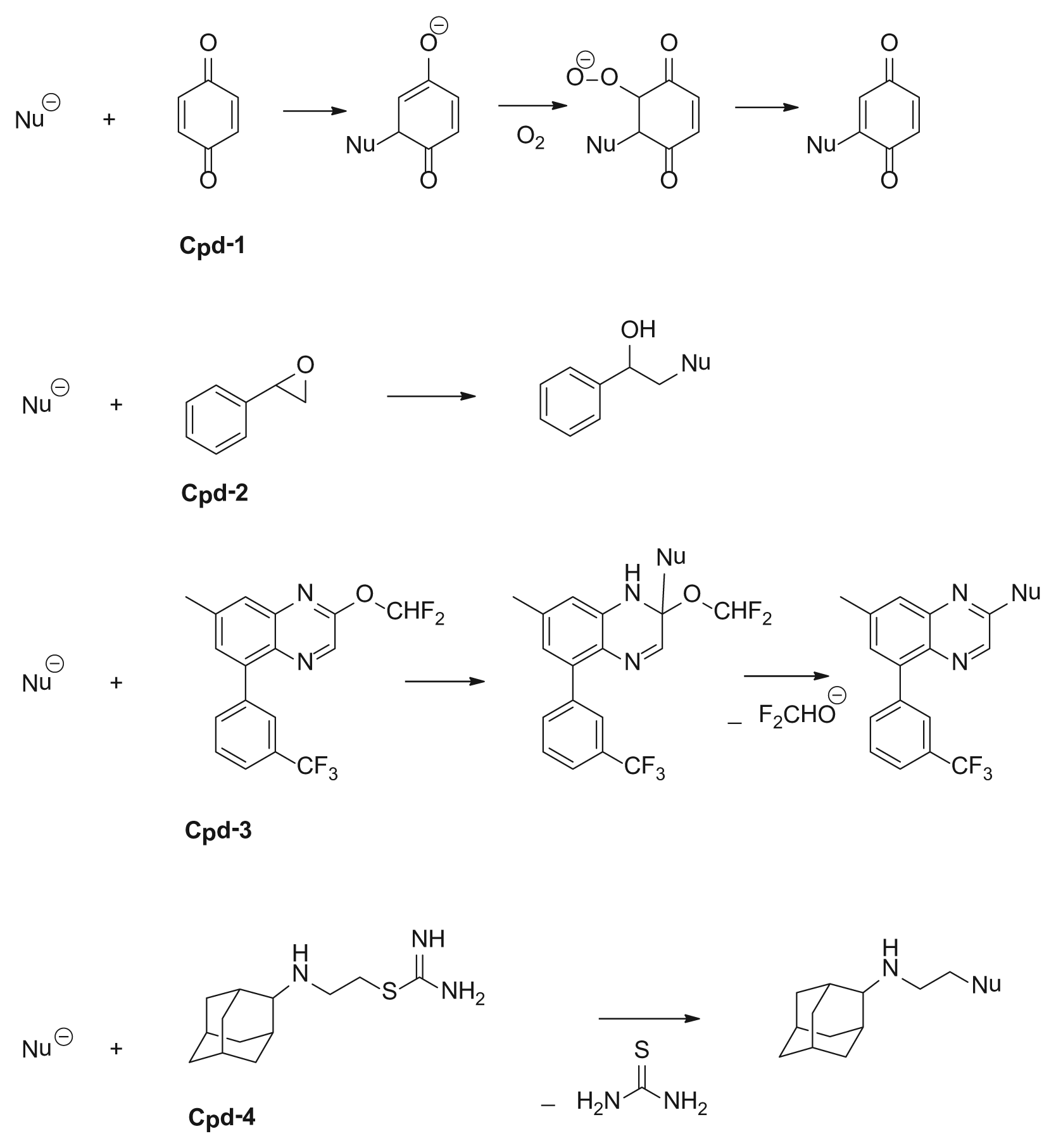
Reaction mechanisms for nucleophiles (peptides) with the electrophilic compounds (compounds 1–4) evaluated.
Materials and Methods
Chemicals and Materials
High-performance liquid chromatography (LC) grade water and acetonitrile were from Thermo Fisher Scientific (Waltham, MA). 1,4-Benzoquinone, salicylic acid, NADPH, GSH, and MOPS (3-[N-morpholino]propanesulfonic acid) were purchased from Sigma-Aldrich (St. Louis, MO). 2-Phenyloxirane was from Alfa Aesar (Ward Hill, MA). 3-(Difluoromethoxy)-6-methyl-8-(3-(trifluoromethyl)phenyl)pyrido[3,2-b]pyrazine (CAS 1238216-23-3) was synthesized according to Selby, 12 and 2-((1r,3r,5r,7r)-adamant-2-ylamino)ethyl carbamimidothioate (CAS 462605-97-6) was purchased from Aurora Fine Chemicals LLC (San Diego, CA). The synthetic peptides were custom synthesized by Cambridge Research Biochemicals (Billingham, Cleveland, UK). Liver S9 fractions (human and rat, 20 mg/mL) were from BD Biosciences (Sparks, MD).
Sample Incubation and Processing
Peptide Incubation
Test compounds, as well as salicylic acid as the negative control (80 µM or 8 mM, final concentration), were incubated with the custom peptides (0.4 mM, final concentration) at 37 °C in MOPS buffer (50 mM, pH 7.4) for 14 h. The MOPS buffer and the incubation mixture were purged with nitrogen before the incubation. The incubation was carried out in an incubator with constant shaking. After the incubation, the samples were centrifuged at 5000 g for 10 min at 4 °C. The supernatant was removed and then diluted 10 times with water/acetonitrile (95:5, v/v). The diluted supernatant was analyzed using LC-HRMS without further sample processing. All samples were prepared in duplicate.
Liver S9 Fraction Incubation
The S9 fraction is most frequently used in assays that measure the metabolism of drugs and other xenobiotics. It is defined by the U.S. National Library of Medicine’s “IUPAC Glossary of Terms Used in Toxicology” as the “supernatant fraction obtained from an organ (usually liver) homogenate by centrifuging at 9000 g for 20 minutes in a suitable medium; this fraction contains cytosol and microsomes.” 13 The microsomes component of the S9 fraction contains cytochrome P450 isoforms (phase I metabolism) and other enzyme activities. The cytosolic portion contains the major part of the activities of transferases (phase II metabolism). The S9 fraction is easier to prepare than purified microsomes. In the current study, test compounds (50 µM) except compound 3 (10 µM) were incubated with human or rat liver S9 fractions (1.0 mg/mL), NADPH (1.0 mM), and GSH (5.0 mM) in potassium phosphate buffer (100 mM, pH 7.4) for 1 h. The total incubation volume was 0.5 mL. After 3-min preincubations at 37 °C, reactions were initiated by addition of a solution containing NADPH or NADPH + GSH. Samples were incubated in a Tecan system (Freedom EVO model; Tecan, Durham, NC) at 37 °C with continuous shaking. Aliquots were removed at 0, 15, 30, and 60 min; quenched with equal volumes of acetonitrile; and centrifuged at 2000 g for 2 min at 4 °C. The supernatants were analyzed using LC-HRMS.
Analytical Details
The LC-MS system 1 for identifying the compound-peptide conjugates consisted of an Eksigent UHPLC system (AB Sciex, Foster City, CA) and an Exactive mass spectrometer (Thermo Scientific, San Jose, CA) affording HRMS data. The mass spectrometer was operated in positive electrospray ionization mode in 25,000 full-width at half-height maximum resolution with mass range m/z 100 to 1500. The operation conditions were as follows: spray voltage at 4.5 kV, capillary voltage at 42.5 V, capillary temperature at 300 °C, tube lens voltage at 105 V, skimmer voltage at 20 V, automatic gain control (AGC) at 1×106, and maximum injection time at 50 ms. Nitrogen was used as the sheath gas, auxiliary gas, and sweep gas, set at 50, 10, and 2, respectively (arbitrary units). The external calibration for mass accuracy was performed daily in positive ion mode according to the manufacturer’s guidelines. The chromatographic separation was performed using an Acquity UPLC BEH300 C18 column (2.1 × 50 mm, 1.7 µm; Waters, Milford, MA) with the mobile phase delivered at a flow rate of 0.2 mL/min. The mobile phase consisted of water (0.1% formic acid, v/v, MP-A) and acetonitrile (0.1% formic acid, v/v, MP-B). The linear gradient conditions for MP-B were as follows: 5% isocratic for 0.3 min, 5% to 75% over 2 min, 75% isocratic for 0.2 min, 75% to 5% over 0.1 min, and 5% isocratic for 0.9 min to reequilibrate, for a total runtime of 3.5 min. A volume of 5 µL of sample was injected, and full-scan HRMS data were acquired in duplicate per sample. The exact mass-to-charge ratios of molecular ions of the predicted compound-peptide conjugates were calculated and used to extract the ions from the full-scan total ion current (TIC). Data within a window of 20 ± 10 mDa of the accurate mass-to-charge ratios of each analyte were extracted and processed. Data were analyzed with Thermo Scientific Xcalibur software.
The LC-MS system 2 for characterizing the compound-peptide conjugates consisted of a Shimadzu LC-20AD HPLC system (Shimadzu Electronic, Kyoto, Japan) and a Thermo Scientific Q-Exactive mass spectrometer. The Q-Exactive mass spectrometer was equipped with a heated electrospray ionization source and operated in the positive ionization mode. The operation conditions were as follows: spray voltage at 4.5 kV, capillary temperature at 300 °C, vaporizer temperature at 300 °C, AGC at 1 × 106, and maximum injection time at 120 ms. Nitrogen was used as the sheath gas, auxiliary gas, and sweep gas, set at 65, 25, and 2, respectively (arbitrary units). The external calibration for mass accuracy was performed daily in positive mode according to the manufacturer’s guidelines. The mass spectrometer acquired MS full scans at a resolution of 35,000, with a mass range of m/z 133 to 2000, and acquired targeted MS/MS scans at a resolution of 17,500. When using the MS/MS scan mode, the precursor ions were selected in quadrupole using a specified m/z window (m/z 2.5) of the theoretical m/z value and subsequently fragmented with normalized collision energy of 35 eV applied to the collision cell. A full scan of the product ions originating from the precursor ions was performed. The chromatographic separation condition was the same as in the LC-MS system 1. A volume of 5 µL of sample was injected, and the targeted HRMS/MS data were acquired in duplicate per sample. Data were analyzed with Xcalibur software.
The LC-MS system 3 for identifying and characterizing GSH conjugates consisted of a Waters Acquity UPLC system equipped with a photodiode array detector and a Waters Xevo G2 MS quadrupole-TOF mass spectrometer. The chromatographic separation was performed using an Acquity UPLC HSS T3 column (2.1 × 100 mm, 1.8 µm) with the mobile phases delivered at a flow rate of 0.3 mL/min. The mobile phases for compounds 1, 2, and 3 were water (0.1% formic acid, v/v, MP-A) and acetonitrile (0.1% formic acid, v/v, MP-B). The mobile phase for compound 4 was water (5 mM ammonium acetate with 0.1% formic acid, v/v, MP-A) and acetonitrile (MP-B). For compounds 1 and 2, the linear gradient conditions for MP-B were as follows: 5% isocratic for 0.5 min, 5% to 30% over 15 min, 30% to 100% over 1 min, and 100% isocratic for 1.5 min, followed by reequilibration. The total runtime was 20 min. For compound 3, the linear gradient conditions for MP-B were as follows: 5% isocratic for 0.5 min, 5% to 70% over 14 min, 70% to 100% over 2 min, and 100% isocratic for 1.5 min, followed by reequilibration. The total runtime was 20 min. For compound 4, the linear gradient conditions for MP-B were 2% isocratic for 0.5 min, 2% to 15% over 15 min, 15% to 100% over 2 min, and 100% isocratic for 1.5 min, followed by reequilibration. The total runtime was 20 min. A volume of 8 µL of sample was injected. The Xevo G2 MS quadrupole-TOF mass spectrometer was fitted with an electrospray ionization source and operated in the positive ionization mode. The external mass calibration for mass accuracy was performed according to the manufacturer’s guidelines. Leucine enkephalin (2.0 mg/L) in water/acetonitrile (1/1 v/v) with 0.1% formic acid (v/v) was infused (7 µL/min) as the reference solution for mass correction, and the 13C isotope of this compound (m/z 557.2805) was used as the reference signal. A capillary voltage of 2.8 kV was applied to the source, and the nanolockspray capillary voltage on the reference sprayer was 1.5 kV. The sampling cone voltage was 24 eV, the source temperature was 100 °C, and the desolvation temperature was 350 °C. Scans were acquired over m/z 100 to 1200 using a scan time of 1.0 s. Data-independent MSe spectra were acquired using a collision voltage of 40 V and argon as the collision gas. Waters MetaboLynx software was used for identification of GSH conjugates based on expected or otherwise common shifts in the mass-to-charge ratios of precursor ions with respect to that of the parent compounds.
Results
The formation of compound-peptide conjugates through the reactions of the four electrophilic test compounds and the custom nucleophilic peptides is depicted in Figure 2 . The exact masses of the resulting mono-, di-, and tri-protonated compound-peptide conjugates assessed in this study are also shown. Full-scan LC-HRMS analysis, followed by postacquisition ion extraction, was used to identify peptide adducts formed by compounds 1 to 4. All four compounds were observed to react with at least one peptide in a 1:1 ratio (one peptide conjugated with one compound). In addition, compounds 1 and 2 formed adducts in a 1:2 ratio (one peptide conjugated with two compounds) with Cys- and Lys-containing or Lys- and His-containing peptides ( Fig. 2A-2, A-3, B-2 ). Compounds 3 and 4 only reacted with Cys-containing peptides ( Fig. 2C-1, C-2, D-1, D-2 ).
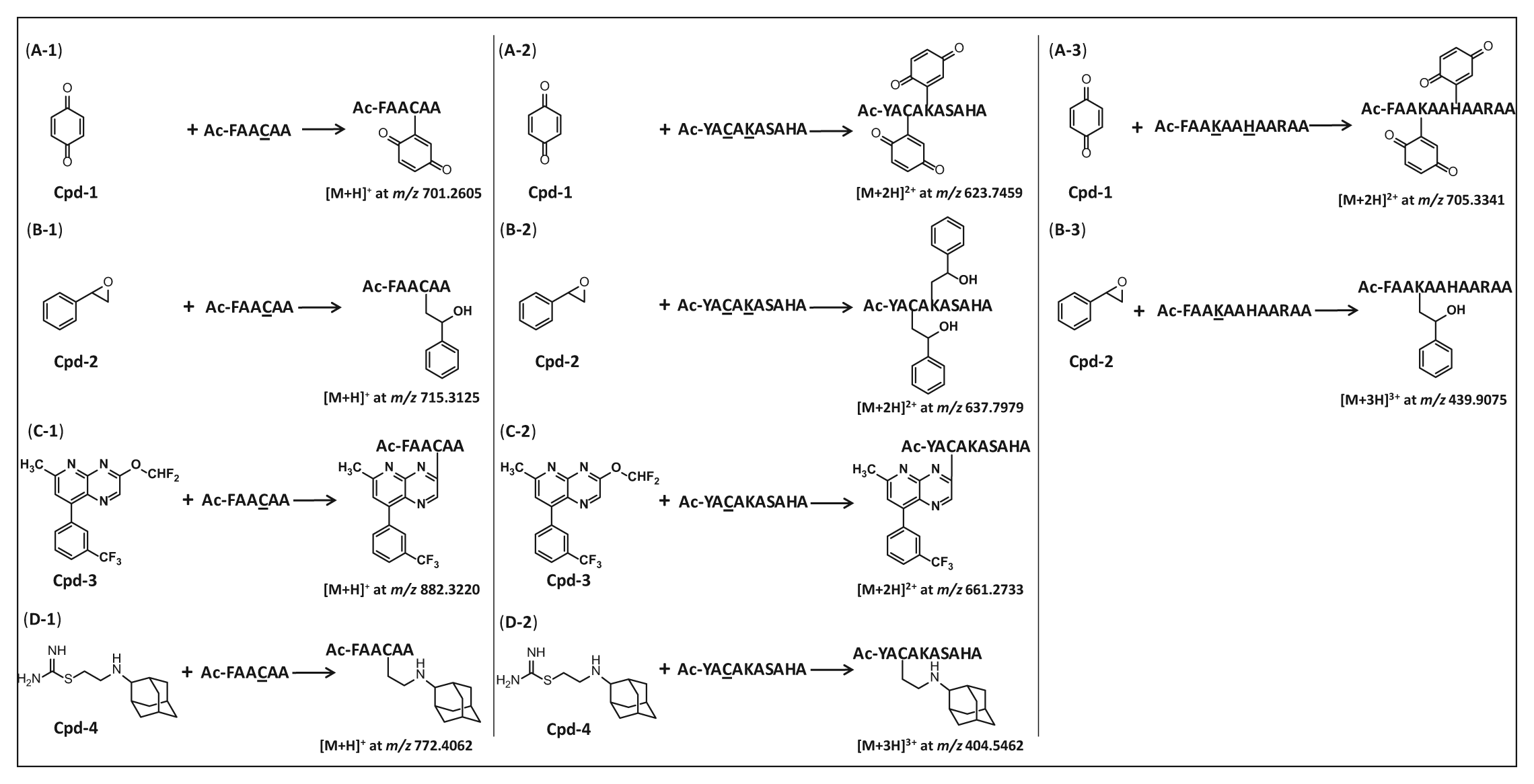
Schemes illustrating reactions between the example compounds and test nucleophilic peptides. The amino acids in the peptides that reacted with the compounds are underlined. (
As an example, the Cys-containing peptide (Ac-FAACAA) was incubated with compound 4, as well as with salicylic acid as a negative control, at pH 7.4 for 14 h. The extracted ion chromatogram (XIC) of the unreacted peptide is illustrated in Figure 3A . Figure 3C shows the XIC of both the unreacted peptide and the peptide–compound 4 adduct. Compared with the unreacted peptide (retention time [RT] 1.51 min), the appearance of a new chromatographic peak at 1.72 min demonstrated the formation of the compound 4–peptide conjugate ( Fig. 3C ). The measured accurate mass (m/z 772.4040) of the peak at RT 1.72 min matched the calculated exact mass of a conjugate of compound 4 and a single peptide (m/z 772.4062), suggesting the compound and the peptide formed a compound-peptide conjugate in a 1:1 ratio ( Fig. 3C , D ). About one-third of the peptide remained unconjugated in the incubation with compound 4, by comparing the signal intensities of the unreacted peptide between the test and the negative control ( Fig. 3A , C ). The corresponding ion spectra of the mono-protonated unreacted peptide and compound 4–peptide conjugate are shown in Figure 3B , D . The mass accuracy for each peptide or compound-peptide conjugate was within 5 ppm of the corresponding theoretical m/z value.
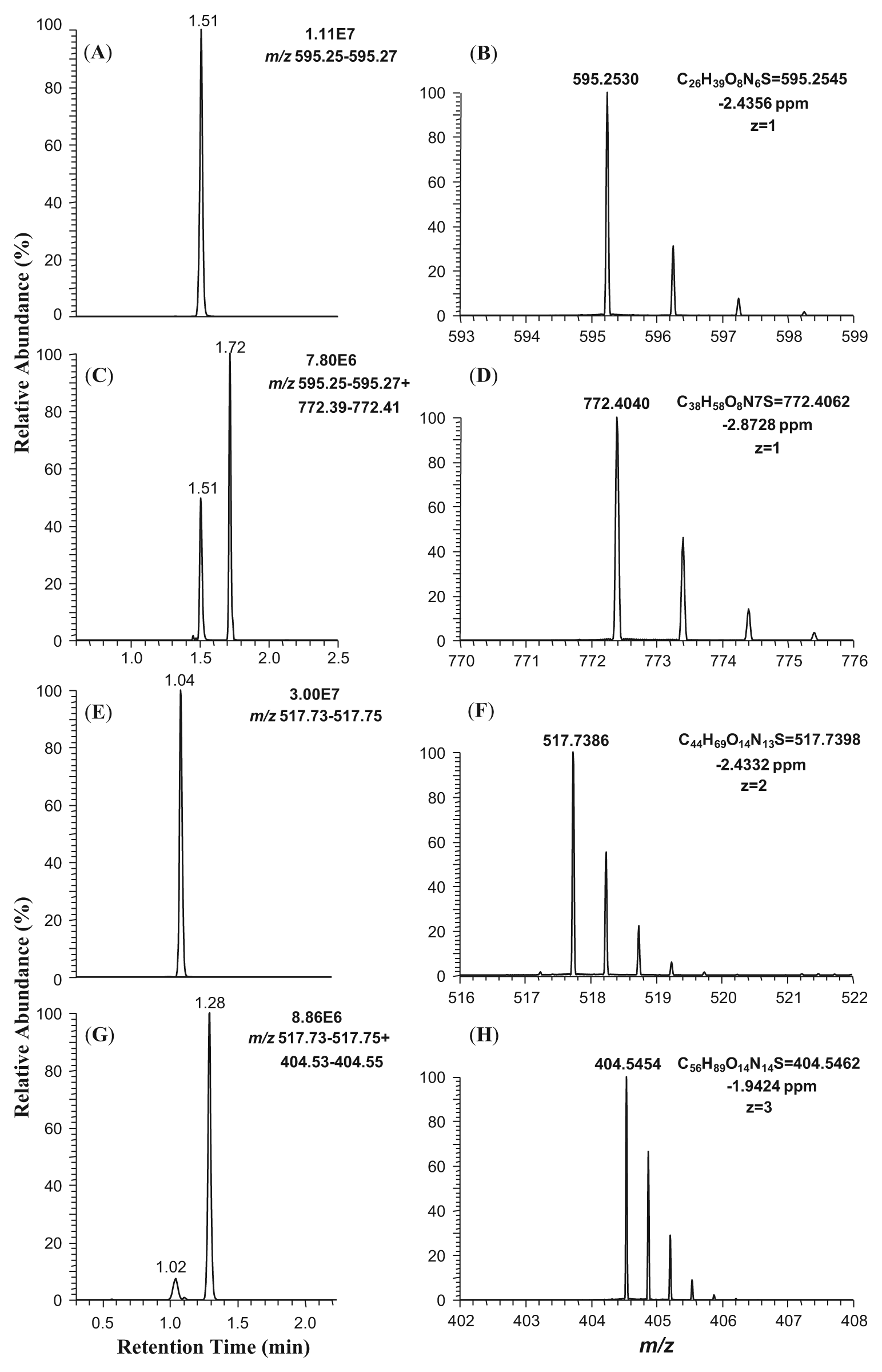
Extracted ion chromatograms and ion spectra of Ac-FAACAA and its conjugate with compound 4 acquired using full-scan liquid chromatography–high-resolution accurate mass spectrometry. (
In contrast to the Cys-containing peptide (Ac-FAACAA) that possesses one nucleophilic side chain, the peptide Ac-YACAKASAHA consists of a mixture of nucleophilic amino acids (i.e., Cys, Lys, Ser, and His), and this was used in the next study. In two separate experiments, compound 4 and salicylic acid (negative control) were incubated with the peptide Ac-YACAKASAHA at pH 7.4 overnight (14 h). Subsequent LC-HRMS analysis demonstrated the production of a compound 4–peptide conjugate (RT 1.28 min), as evidenced by the data shown in Figure 3G . This is in contrast to the incubation with salicylic acid that did not result in modification of the peptide ( Fig. 3E ). By comparing the signal intensities of the unreacted peptide between the test compound and the negative control, only 10% of the peptide remained unconjugated in the incubation with compound 4, whereas 90% of the peptide was trapped by compound 4 ( Fig. 3E , G ). The corresponding ion spectra of the bi-protonated unreacted peptide and the tri-protonated compound 4–peptide conjugate are illustrated in Figure 3F , H . Mass accuracies of the unmodified and modified peptides were within 5 ppm of the corresponding theoretical m/z values.
LC-MS/MS can be used to differentiate among potential binding sites within a peptide. As noted above, Cys is the only nucleophilic constituent in the trapping peptide Ac-FAACAA. To confirm that Cys was the amino acid that reacted, LC-HRMS/MS analysis was applied to the compound-peptide conjugate as well as the unreacted peptide from the supernatant of the incubation mixture. The positive ion electrospray product ion MS/MS spectra of both the peptide conjugate of compound 4 and the unmodified peptide (Ac-FAACAA), generated using collision-induced dissociation (CID), are illustrated in
The peptide Ac-YACAKASAHA contains multiple nucleophilic amino acids, any of which may react with compound 4. To identify the amino acid modified in the compound-peptide conjugate, the supernatant of the reaction mixture of compound 4 and Ac-YACAKASAHA was analyzed using LC-MS/MS. Figure 4 illustrates the positive ion electrospray MS/MS product ion scans, acquired using CID, of the compound 4–conjugated peptide ( Fig. 4A ) and the unmodified peptide (Ac-YACAKASAHA) ( Fig. 4B ). The MS/MS fragmentation of the compound 4–conjugated peptide showed an additional mass of m/z 177 in connection with the b3 and y8 ions ( Fig. 4A ), corresponding to the mass of compound 4 less the thiourea moiety, whereas the b3 and y8 ions remained the same as those in the unreacted peptide ( Fig. 4B ). In both the modified and unmodified peptides, the b2 and y7 ions remained the same. Cumulatively, the mass shifts observed in the b3, y8, b4, b6, and y9 ions, as illustrated in Figure 4A , suggest that Cys was the site of conjugation by compound 4, indicating that Cys was the most reactive amino acid toward compound 4 among the nucleophilic residues (Cys, Lys, Ser, and His) in the peptide.
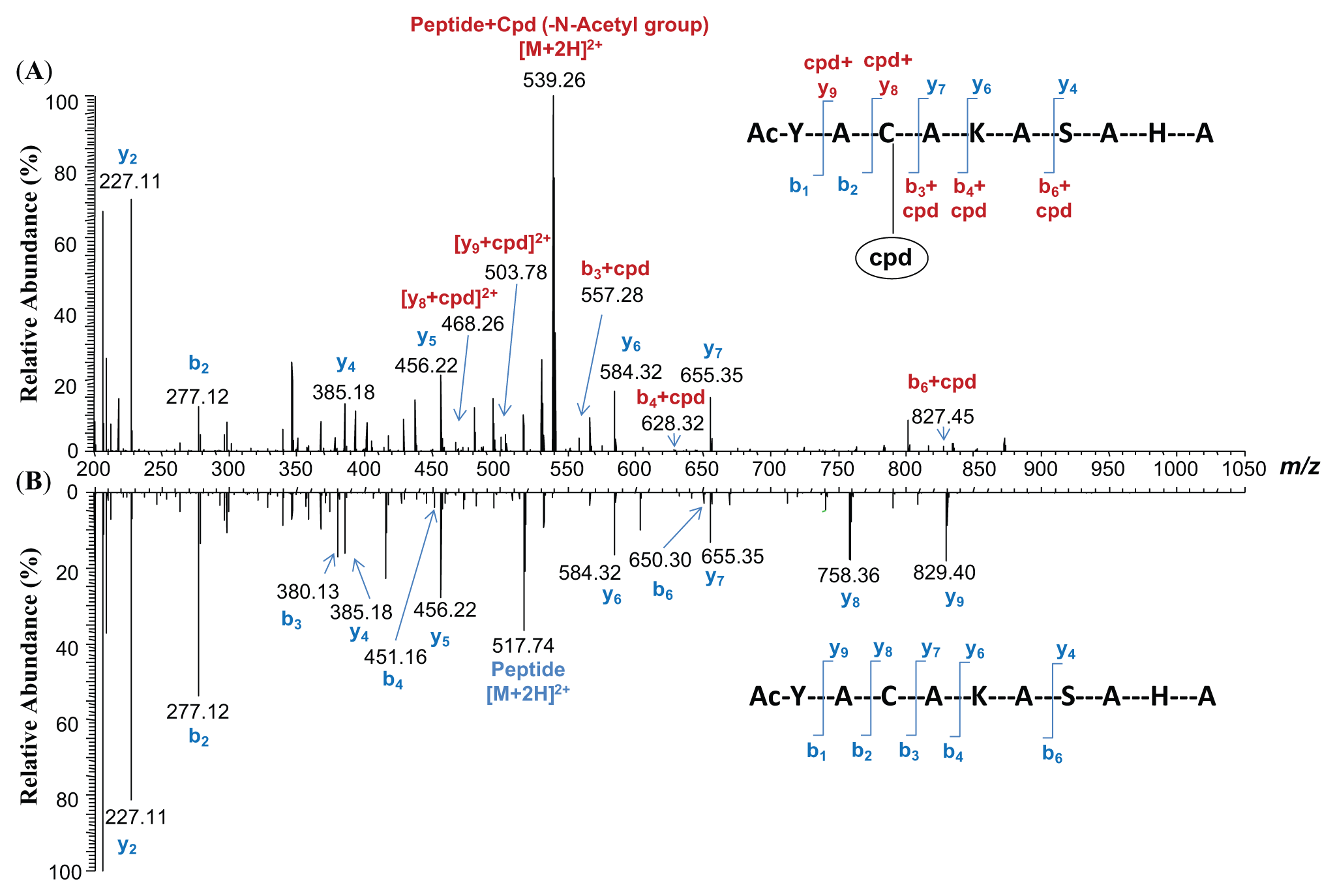
Positive ion electrospray tandem mass spectrometry (MS/MS) product ion spectra of (
The positive ion electropray LC-MS analysis of incubations containing GSH-supplemented human and rat liver S9 fractions revealed that compounds 1, 2, and 3 formed GSH conjugates in these incubations ( Fig. 5 ). Specifically, compound 1 formed a GSH conjugate of m/z 416.1129, which deviated by 0.2 ppm from the theoretical mass-to-charge ratio of the molecular formula C16H22N3O8S ( Fig. 5A , B ). Similarly, compound 2 formed a GSH conjugate of m/z 428.1494 ( Fig. 5C ). This mass differed from that of the expected molecular formula C18H26N3O7S by 0.7 ppm ( Fig. 5D ). A GSH conjugate of compound 3 was also detected ( Fig. 5E ). The measured mass-to-charge ratio of this adduct was m/z 595.1580, suggesting removal of the difluoromethoxyl group and displacement by GSH, yielding a product with the molecular formula C25H26F3N6O6S ( Fig. 5F ). The measured and theoretical masses of this metabolite differed by 1.2 ppm. The positive ion electrospray product ion MS/MS spectra of each GSH conjugate described above exhibited product ions that corresponded to neutral losses of the γ-glutamyl group (129 Da), further confirming the assignments of these metabolites. No GSH conjugates of compound 4 were detected.
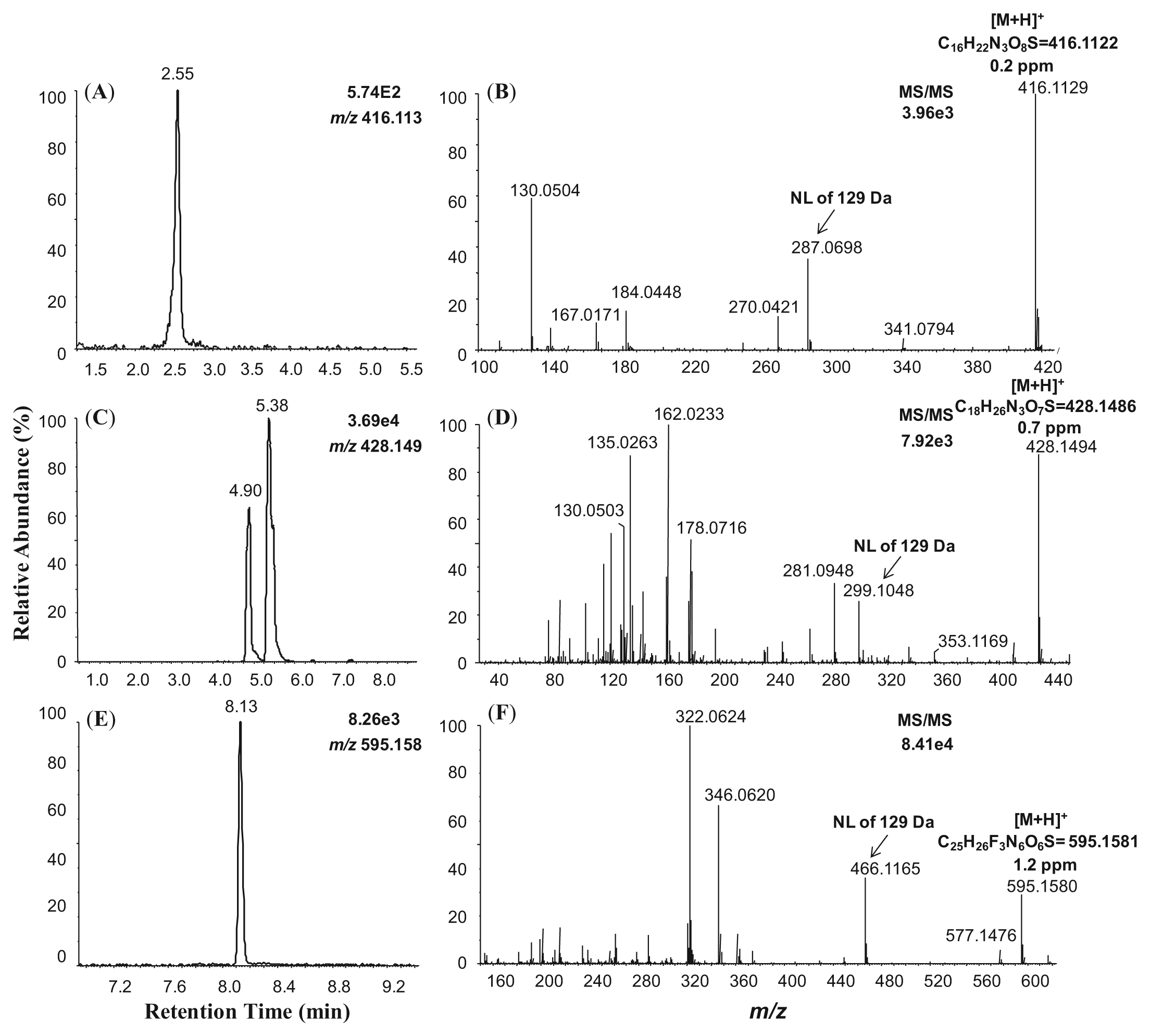
Positive ion electrospray liquid chromatography–mass spectrometry (LC-MS) chromatograms and MS/MS product ion spectra of the glutathione (GSH) conjugates of compound 1 (
Discussion
Described here is a simple and rapid assay for the identification of reactive electrophilic compounds (that might be identified from a biological screening process, as typically conducted in early medicinal chemistry programs). Such compounds can be responsible for artifactual activities in primary assays and have a potential to elicit a toxic response if retained through the lead optimization process. Compounds selected as model electrophiles were shown to react with different custom nucleophilic peptides (
Fig. 2
). The custom peptides were classified into four groups according to their nucleophilic moieties: thiol group (Cys), nucleophilic nitrogen-functionality (Lys, His, Arg), hydroxyl group (Ser, Tyr), and a fourth class that contained all of these nucleophilic entities (
Methods using LC-HRMS and LC-HRMS/MS found great utility in studies of biomarkers,14,15 biotransformation,16,17 and bioanalysis to minimize or eliminate interferences18,19 in drug discovery and development. High-resolution mass analyzers, such as TOF and Orbitrap-based mass spectrometers, have made data acquisition by LC-HRMS analysis routine. HRMS has unique advantages over low-resolution approaches, offering the ability to distinguish among multiply-charged peptides and small-molecule interferences that are often singly charged. This is because multiply charged ions are observed as a cluster of MS peaks with a level of complexity that depends on the charge state of the ion and the number of isotopes that are detectable ( Fig. 3F , H ). Such spectra typically cannot be resolved using unit-resolution mass spectrometry. Accordingly, HRMS offers certain advantages in this assay, since both the peptides and any associated conjugates become multiply protonated following electrospray ionization ( Fig. 3F , H ). Thus, this HRMS assay is able to differentiate among compounds of interest and those with the same nominal mass-to-charge values that might be present as matrix interferences. Furthermore, HRMS analysis used in combination with postacquisition data mining allows narrow mass-to-charge windows (e.g., 20 mDa) of selected ion chromatograms to be visualized and inspected with high mass accuracy (<5 ppm) ( Fig. 3 ). All of these features are helpful in reducing to a minimum the number of clean-up steps that are required during sample preparation. Only incubation and centrifugation are needed prior to analysis, and chromatographic methods employing runtimes of less than 5 min can be used ( Figures 3 and 4 ).
The MS/MS fragmentation by CID is a conventional method for peptide sequencing and/or for determining protein posttranslational modifications, even though it is inefficient in dissociating S-S bonds.
20
In our studies, the incubation buffer was nitrogen purged to prevent the oxidation of the peptides and the formation of disulfide bonds between peptides to ensure thiol exposure of the peptides to the test compounds, thus improving the yield of the compound-peptide conjugates in the samples. The observed MS/MS fragmentation could unambiguously determine the identity of the peptide and reveal the modification sites of the peptide with the mass shift of certain b and y ions (
Fig. 4
and
In parallel studies, three of the four test compounds formed GSH conjugates in a bioactivation assay using GSH-supplemented liver S9 fractions ( Table 1 ), suggesting the peptide-trapping assay could be used orthogonally to a GSH-trapping assay for the identification of reactive compounds and electrophilic metabolites. A potential rationale for the observation that compound 4 reacted with cysteine-containing peptides but not with GSH could be that the first step in the reaction sequence might involve a transient binding to H-bonding elements in the peptide. Such interactions could be expected to both facilitate the cleavage of the thiourea moiety and effectively position this group for attack by the thiol nucleophile. Alternatively, the different incubation conditions employed in the two assays may also account for the observed differences. GSH-trapping assays that use neutral-loss scanning (e.g., 129 Da) for the selective detection of GSH conjugates are amenable to HTS approaches, whereas the current peptide-trapping strategy, which is relatively low throughput and requires compound-dependent data analysis, was developed to facilitate the characterization of structure-liability relationships in early discovery and is applicable in circumstances where direct chemical reactivity is a matter of concern. The current assay is employed in early drug discovery to provide feedback to chemists to eliminate problematic compounds, and in future publications, we plan to describe the expansion of these techniques to significantly increase the throughput of this assay format.
In summary, the study presented here is a novel approach to identifying chemically reactive compounds. The assay was demonstrated by in vitro peptide trapping of four model compounds at physiological pH followed by LC-HRMS analysis. The reactive compounds became conjugated to different nucleophilic peptides, and the amino acids modified in the compound-peptide conjugates were identified by LC-HRMS/MS analysis. By taking advantage of the high-resolution accurate mass feature of MS analyzers, the methodology enabled accurate identification and characterization of reactive compounds to help inform structure-liability relationships. Furthermore, the assay displayed broader reactivity against the selected electrophilic compounds than a standard GSH-trapping assay and was used orthogonally alongside a GSH-trapping assay for the rapid detection of reactive compounds and electrophilic metabolites. The developed assay has influenced several HTS triage exercises on lead compounds with unknown chemical reactivity.
Footnotes
Acknowledgements
We greatly appreciate the generosity of Anthony Paiva and Dr. Wilson Z. Shou for access to the Thermo Q-Exactive instrument in their laboratory and the helpful discussions during the course of the study. We also thank Dr. Nicholas Meanwell for his critical review of the manuscript and valuable scientific discussions.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.