
Abstract
Spleen tyrosine kinase (SYK) and Bruton’s tyrosine kinase (BTK) are key mediators in coupling cell surface receptors, such as the B-cell receptor (BCR), to downstream signaling events affecting diverse biological functions. There is therefore tremendous interest in the development of pharmacological inhibitors targeting the SYK-BTK axis for the treatment of inflammatory disorders and hematological malignancies. A good pharmacodynamic (PD) assay, ideally a blood-based assay that measures proximal events, is warranted for evaluation of such inhibitors. In platelets, collagen-induced activation of membrane glycoprotein GPVI is dependent on the SYK-BTK axis. Here, we report the development of a novel immunoassay that uses the dissociation-enhanced lanthanide fluorescent immunoassay (DELFIA) to measure GPVI-mediated phosphorylation of phospholipase C γ2 (PLCγ2), a direct substrate of SYK and BTK, in platelets. The assay was validated using SYK or BTK inhibitors and generated IC50 correlated with those from the BCR-induced B-cell activation assay. Furthermore, this assay showed good stability and uniformity over a period of 24 h in different donors. Interestingly, compound IC50 values using blood from patients with rheumatoid arthritis were slightly higher compared with those produced using samples from healthy donors. This novel platelet PLCγ2 phosphorylation-based immunoassay should serve as a promising PD assay for preclinical and clinical development of inhibitors targeting the SYK-BTK axis.
Introduction
Spleen tyrosine kinase (SYK) and its downstream effector, Bruton’s tyrosine kinase (BTK), are both nonreceptor cytoplasmic tyrosine kinases that mediate cell surface receptor-induced signal transduction in various immune and pseudo-immune cells types. Predominantly expressed in, but not limited to B cells, monocytes/macrophages, mast cells, osteoclasts, and platelets, the two enzymes are critical components of a signaling axis that mediates autoantibody production, innate immune responses, osteoclast differentiation, and platelet cell functions upon activation of the respective cell surface receptor. 1 As such, pharmacological inhibition of SYK or BTK is expected to affect multiple pathophysiological pathways that contribute to various immunological and hematological diseases. Not surprisingly, the SYK-BTK axis has emerged as a particularly attractive drug target.1–5
The SYK/BTK signaling pathway has been best elucidated in the context of B-cell receptor (BCR) signaling. Briefly, BCR engagement leads to the enzymatic activation of receptor-bound Src family nonreceptor tyrosine kinases—namely, LYN and FYN. These kinases phosphorylate tyrosine residues that are present in the cytoplasmic immunoreceptor tyrosine-based activation motifs (ITAMs) found in the signaling subunits of the receptor. Tyrosine-phosphorylated ITAMs, in turn, recruit SYK through Src homology 2 (SH2) domain-phosphotyrosine interactions, whereby SYK undergoes protein conformational change-dependent activation and phosphorylates the adaptor protein, B-cell linker (BLNK). Tyrosine-phosphorylated BLNK binds to the SH2 domains of both phospholipase C γ2 (PLCγ2) and BTK. PLCγ2–dependent hydrolysis of phosphatidylinositol 4,5-bisphosphate yields diacylglycerol (DAG) and inositol 1,4,5-trisphosphate (IP3). BTK also contains a Pleckstrin homology domain (PH domain) that binds IP3; IP3 binding also induces BTK to phosphorylate PLCγ2. Thus, SYK cooperates with BTK to phosphorylate and thus activate PLCγ2. The generation of DAG and IP3 promotes activation of protein kinase C (PKC) and the release of Ca2+ from intracellular storage sites, respectively. Calcium release upregulates mitogen-activated protein kinase and phosphoinositide 3-kinase (PI3K)–dependent downstream signaling cascades that ultimately regulate B-cell activation, differentiation, and maturation.1,3,6
Recently, the important role of the SYK-BTK signaling axis in platelet activation and the formation of platelet microparticles (small membrane-coated vesicles that are released from the plasma membrane upon platelet activation) and its physiological relevance in promoting inflammatory responses have been appreciated.1,7 Platelets are small, irregular anuclear cell fragments that circulate in the blood. Although they are involved in hemostasis, leading to the formation of blood clots, platelets are also potent mediators of a range of effector responses in both innate and adaptive immunity. 8 For instance, in patients with rheumatoid arthritis (RA), interleukin 1 (IL-1)–containing platelet microparticles are abundant in arthritic joint fluid and can elicit the production of proinflammatory chemokines and cytokines.9,10 Furthermore, platelet microparticles are elevated in inflammatory diseases and correlated with disease severity.11,12 Platelet activation via the platelet-specific collagen receptor glycoprotein VI (GPVI) signals through the recruitment of SYK, activation of BTK, and phosphorylation of PLCγ2. 13 Indeed, we recently showed that selective inhibition of BTK kinase function abrogated agonist-induced GPVI-mediated PLCγ2 phosphorylation and generation of platelet microparticles. 7 With a specific BTK inhibitor, RN486, 14 we have demonstrated that GPVI receptor-mediated phosphorylation of PLCγ2 in platelets is SYK and BTK dependent, 7 which could be used to develop pharmacodynamic (PD) assays. Furthermore, platelets are abundant in the blood and easy to enrich by a simple centrifugation, providing convenience in the preclinical and clinical development.
Pharmacodynamic biomarkers specifically refer to time-associated measurements to establish the relationship between drug concentration and pharmacologic responses. 15 The availability of a robust PD assay, which can be reliably used to demonstrate target engagement, confirm mechanism of action, and assess tolerability and potential toxicity of compounds, will greatly facilitate both preclinical and clinical drug development of small-molecule inhibitors targeting the SYK-BTK axis. Ideally, such a PD assay should be based on measurement of events proximal to SYK and BTK activation, be relatively high throughput, be technically easy to implement, and use blood samples. Currently, a couple of potential PD assays could be developed for assessing SYK/BTK activity in the whole-blood setting, including measurement of BCR-mediated B-cell activation, either by flow cytometric analysis of cell surface CD69 expression or by Western blot analysis of phosphorylation of BTK or ERK.16,17 Alternatively, Fcϵ receptor-mediated basophil activation, as measured by flow cytometric analysis of cell surface CD63 expression, has been demonstrated. 18 However, CD69 and CD63 expression are relatively distal downstream events following SYK-BTK activation, which occur hours after stimulation of B cells and basophils, respectively. Furthermore, both assays require flow cytometric analysis, which is time-consuming and requires specific expertise to run and manage in clinical settings. Measurement of BTK or ERK phosphorylation by immunoblot analysis is labor intensive and low throughput. Given the essential role of the SYK-BTK axis in platelet activation by GPVI, the abundance of platelets in the blood, and the ease of platelet enrichment by simple centrifugation, we developed a PD assay that measures GPVI-mediated phosphorylation of PLCγ2 in human platelets. The assay was optimized for a 96-well format, sandwich-based immunodetection system and validated using SYK and BTK inhibitors.
Materials and Methods
Antibodies
Rabbit antibodies against the phosphorylated form of PLCγ2 (p-PLCγ2) and β-tubulin were obtained from Cell Signaling Technology (Danvers, MA). Mouse and rabbit antibodies against total PLCγ2 (t-PLCγ2) were obtained from Santa Cruz Biotechnology (Santa Cruz, CA) and Novus Biologicals (Littleton, CO), respectively. Biotin-conjugated anti–rabbit antibody was obtained from Jackson ImmunoResearch (West Grove, PA). The dissociation-enhanced lanthanide fluorescent assay (DELFIA) Eu-labeled streptavidin tracer solution and DELFIA enhancement solution were obtained from PerkinElmer (Waltham, MA).
Compounds and Human Whole-Blood CD69 Assay
BTK-selective inhibitor RN486 14 and other proprietary BTK 19 and SYK 20 inhibitors were developed and synthesized by Hoffmann-La Roche (Nutley, NJ). PI3K family inhibitors21,22 were synthesized by Hoffmann-La Roche. The IC50 potency of each compound was determined in a human whole-blood assay measuring anti-IgM antibody-induced B-cell activation by flow cytometric analysis of surface CD69 expression in CD20+ B cells, as previously described. 14
Human Platelet Enrichment, Activation, and Preparation of Protein Lysates
Plasma-rich platelets (PRPs) were prepared by a 15-min centrifugation (140 g) of human whole blood from healthy volunteers or RA patients collected in Vacutainer tubes with acid citrate dextrose (ACD) additives (BD, Franklin Lakes, NJ). PRPs were incubated with GDGRS peptide (Calbiochem, Billerica, MA; 1 mM) at room temperature for 15 min. In a 96-well plate (BD Bioscience, San Jose, CA), each well containing 90 µL PRP was pretreated with test compound at a desired concentration or DMSO (vehicle control) for 15 min followed by stimulation with convulxin (Enzo Life Sciences, Farmingdale, NY) at a final concentration of 0.125 µg/mL for 15 min at 37 °C. The platelets were pelleted by centrifugation at 2400 g for 10 min at 4 °C and resuspended with 80 µL of lysis buffer composed of M-PER protein extraction reagent (Thermo, Rockford, IL) supplemented with 1 mM sodium orthovanadate (NEB, Ipswich, MA) and protease inhibitor cocktail (Roche, Indianapolis, IN). Platelet lysates were stored at −80 °C or analyzed by using Western blot or the sandwich-based DELFIA.
Western Blot Analysis
Proteins from total platelet lysates were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) using NuPAGE Bis-Tris precast gels (Invitrogen, Carlsbad, CA) and then transferred onto PVDF membranes. The membranes were incubated with primary antibodies (p-PLCγ2, t-PLCγ2, or β-tubulin, 1:1000 dilution) overnight at 4 °C followed by a 1-h incubation at room temperature with 1:4000 diluted horseradish peroxidase (HRP)–conjugated secondary antibodies. Membranes were washed before protein visualization and quantification using ECL-Plus (GE Healthcare Life Science, Pittsburgh, PA). The images were acquired by a Typhoon Imager (GE Healthcare Life Science), and band intensity was quantitated by ImageQuant (GE Healthcare Life Science).
DELFIA Assay
DELFIA yellow 96-well plates (PerkinElmer), which have a high protein-binding capacity and very low fluorescence background, were used for assay development to increase assay sensitivity. The plates were coated overnight at 4 °C with 4 µg/mL mouse anti–t-PLCγ2 antibody (Santa Cruz Biotechnology, sc-5283) in Tris-buffered saline (TBS) buffer (25 mM Tris, 137 mM NaCl [pH 7.6]), then washed with TBS-T buffer (TBS buffer plus 0.1% Tween-20) and blocked with 3% bovine serum albumin (BSA) in TBS-T for 1 h. The plates were incubated with 50 µL/well of samples for 1 h, followed by incubation with 50 µL of detection antibody cocktail consisting of 0.5 µg/mL rabbit anti–p-PLCγ2 antibody (Cell Signaling Technology, 3871) for phospho-PLCγ2 detection or anti–t-PLCγ2 antibody (Novus Biologic, H00005336-D01P) for total PLCγ2 detection, respectively, and 0.5 µg/mL biotin-conjugate anti–rabbit antibody in TBS-T with 1% BSA for 1 h. The plates were washed with TBS-T and incubated with 50 µL tracer solution consisting of 1:1000 DELFIA Eu-labeled streptavidin (PerkinElmer) diluted in TBS-T with 1% BSA for 20 min. The plates were then subjected to a final wash with TBS-T followed by the addition of 100 µL DELFIA enhancement solution (PerkinElmer) for 15 min and detection using an EnVision reader (PerkinElmer), measuring the emission wavelength at 615 nm. All incubation steps were done at room temperature unless stated otherwise.
IC50, IC75, and IC90 Determination and Statistical Analysis
IC50, IC75, and IC90 values were determined based on sigmoidal concentration-response curve fitting by using Prism (GraphPad Software, La Jolla, CA). In most studies, the values reported were the averages from at least two studies conducted with samples in replicates. The statistical analysis was performed using the Student t test (*p < .05; **p < .01; ns, not statistically significant).
Results and Discussion
Selection of Sandwich Antibody Pair
Commercially available antibodies against human PLCγ2 and its phosphorylated form were screened for positive hybridization by Western blot analysis. Human platelets were pretreated with a selective BTK inhibitor, RN486, or vehicle control and subsequently stimulated with convulxin, a specific and strong ligand for the GPVI receptor on platelets, to trigger the phosphorylation of PLCγ2. Stimulated and unstimulated cell lysates were subjected to Western blotting with antibodies against total PLCγ2 (t-PLCγ2) or phospho-PLCγ2 (p-PLCγ2).
We found many of the commercially available antibodies against human PLCγ2 and its phosphorylated form had poor quality and showed nonspecific cross-reactivity (data not shown). The exception was a rabbit antibody raised against phosphorylated tyrosine at 1217 of PLCγ2 from Cell Signaling Technology (cst-3871), which recognized a convulxin-induced phosphorylated PLCγ2 band (
Fig. 1A
). Importantly, this signal was attenuated with pretreatment of 1 µM RN486 (top panel). The reduced PLCγ2 phosphorylation by the BTK compound was not due to a decrease in total PLCγ2 protein as detected by a mouse antibody recognizing amino acids 826 to 925 (Santa Cruz Biotechnology, sc-5283) and a rabbit antibody generated by immunization with full-length PLCγ2 protein (Novus Biologics, H00005336-D01P) (
Fig. 1A
, middle and bottom panels). Convulxin-induced phosphorylation of PLCγ2 was inhibited by RN486 in a concentration-dependent manner (
Fig. 1B
). The specificity of phospho-PLCγ2 (Y1217) antibody was further demonstrated by using small-molecule inhibitors of the PI3K family members with the DELFIA assay described below, which did not affect convulxin-induced phosphorylation of PLCγ2 (
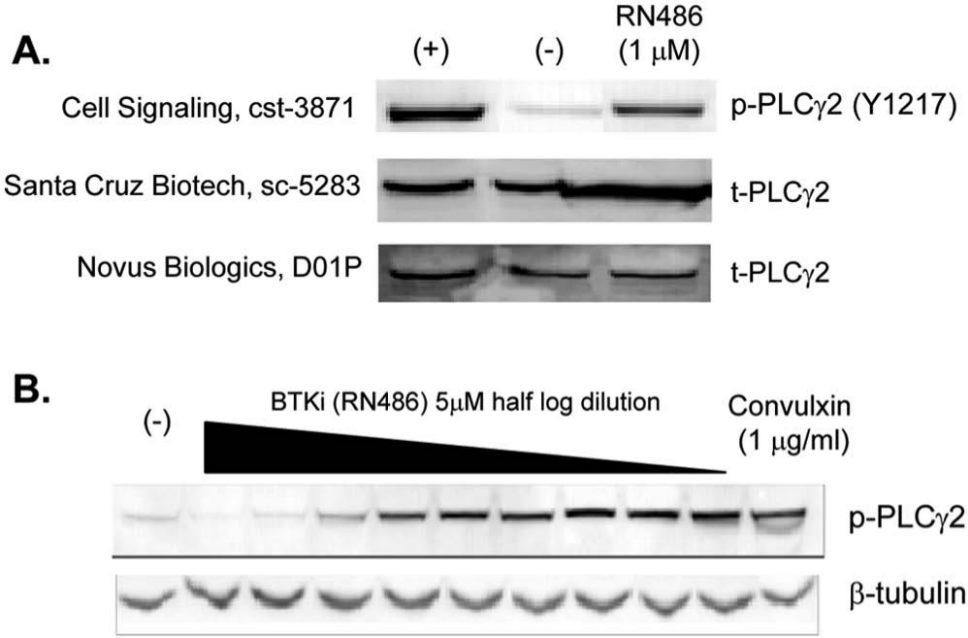
Western blot analysis of phospholipase C γ2 (PLCγ2) and phospho-PLCγ2 (p-PLCγ2) antibodies in human platelets. (
Development of Phospho-PLCγ2 DELFIA Assay
To facilitate clinical PD assay development, we selected the DELFIA technology because it offers a higher sensitivity requiring less reagents and samples, wide dynamic range, superior stability that allows for delayed signal detection and batch processing, and excellent flexibility of converting to a 384-well format 23 compared with the conventional enzyme-linked immunosorbent assay.
To develop the assay in the DELFIA format, we selected the appropriate sandwich antibody pair choices based on the antibodies from different species and different epitope specificity: mouse antibodies sc-5283 from Santa Cruz Biotechnology against PLCγ2 as the capture antibody and the rabbit antibody cst-3871 from Cell Signaling Technology as the detection antibody for quantifying phosphorylated PLCγ2. The pair for quantification of total PLCγ2 was composed of mouse antibody sc-5283 as the capture antibody and a rabbit antibody D01P from Novus Biologics as the detection antibody.
To initially optimize the assay conditions for the concentration of capture and detection antibodies, we used batch-prepared cell lysates from the human B-cell line, Ramos, to help minimize donor-to-donor variations. Ramos cells were stimulated by cross-linking B-cell receptor with anti–human IgM antibodies for 5 min to trigger phosphorylation of PLCγ2, and cell lysates were prepared, serially diluted, and used in subsequent experiments.
The 96-well DELFIA yellow plates were coated with total PLCγ2 capture antibody (Santa Cruz Biotechnology, sc-5283) at different concentrations from 1 to 32 µg/mL. Then, 15 µg of Ramos cell lysate was allowed to bind the capture plates followed by incubation with 1 µg/mL of detection antibody and 0.5 µg/mL of biotin-labeled secondary antibody. The induction fold in PLCγ2 phosphorylation of each antibody combination was calculated by dividing the measured relative fluorescence units (RFU) from stimulated samples by that of unstimulated samples. Significant induction (about 8-fold) of PLCγ2 phosphorylation was observed with 4 µg/mL of capture antibody ( Fig. 2A ). Maximal fold in induction was reached with 16 µg/mL of capture antibody. For economic purposes as a screening assay and to avoid potential nonspecific cross-reactivity, we selected the 4-µg/mL concentration for capture antibody for further assay optimization.
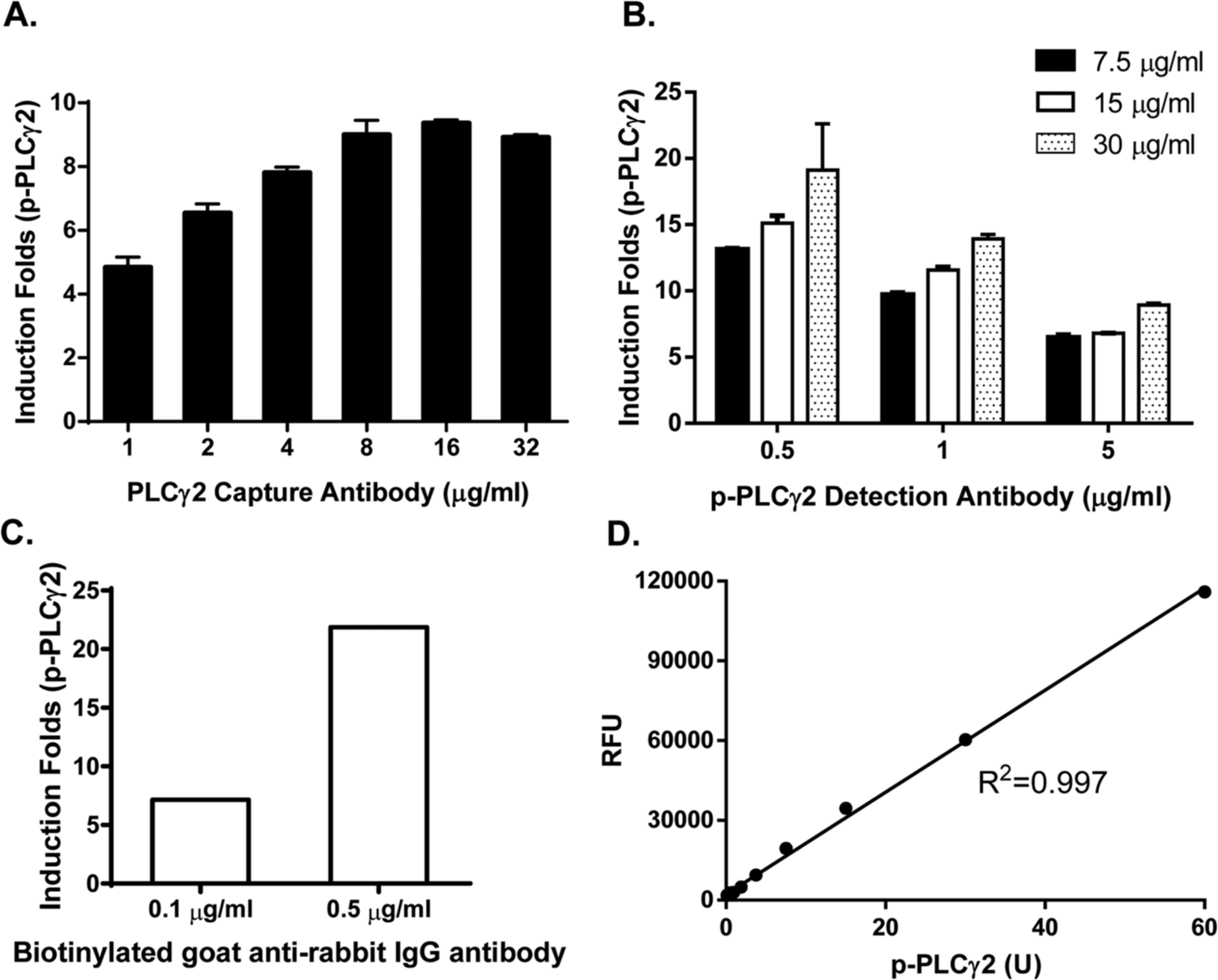
Development of the phospholipase C γ2 (PLCγ2) phosphorylation dissociation-enhanced lanthanide fluorescent immunoassay (DELFIA) assay. The phosphorylation of PLCγ2 was induced by stimulation of Ramos cells with anti-IgM for 5 min. The cell lysate was prepared and used for assay development and optimization. (
We next optimized the concentration of detection antibody against phosphorylated PLCγ2. p-PLCγ2 detection antibody (0.5, 1, or 5 µg/mL) was incubated with a different amount of stimulated Ramos cell lysate (7.5, 15, or 30 µg/mL). We found that 0.5 µg/mL of p-PLCγ2 detection antibody gave the best induction signals ( Fig. 2B ). Last, we repeated the above experiments with varying concentrations of biotin-labeled secondary anti–rabbit antibody and determined that the 0.5-µg/mL concentration yielded the best induction in PLCγ2 phosphorylation ( Fig. 2C ).
Using these assay conditions, we next generated a standard curve using serially diluted Ramos cell lysate stimulated with anti–human IgM. An arbitrary unit (U) was assigned for p-PLCγ2, where 1 µg/mL of total Ramos cell lysate equals 1 U of p-PLCγ2. As shown in Figure 2D , the assay linearity ranged between 0.2 and 60 U.
Evaluation of DELFIA Phospho-PLCγ2 Assay Using SYK and BTK Inhibitors
We next used the optimized DELFIA assay to evaluate GPVI-induced PLCγ2 phosphorylation in human platelet samples in the presence or absence of the BTK inhibitor, RN486. Human platelets were isolated from 25 healthy human volunteers and pretreated with RN486 prior to convulxin stimulation. The DELFIA p-PLCγ2 assay was performed in parallel with a second DELFIA assay to measure t-PLCγ2 as an internal reference control for sample normalization by determining the ratio of quantified p-PLCγ2 to t-PLCγ2. As shown in Figure 3A , the mean IC50 for RN486 was 71 ± 26 nM, with each inhibition curve representing an individual donor. The standard deviation of ±26 nM among the individuals is within the normal range observed in other previously whole-blood–based assays for assessing SYK/BTK activity.
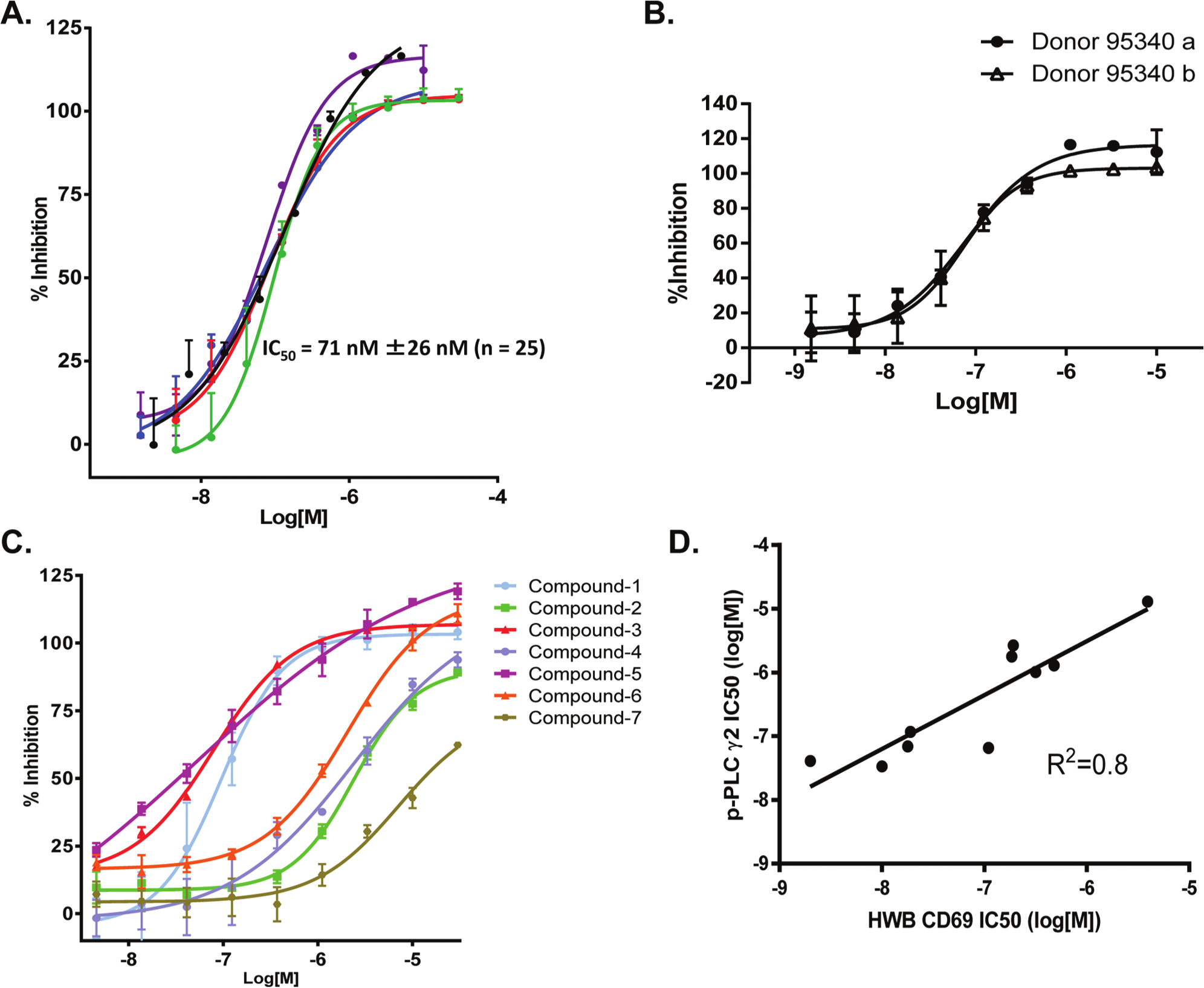
Evaluation of the dissociation-enhanced lanthanide fluorescent immunoassay (DELFIA) phospho–phospholipase C γ2 (p-PLCγ2) assay using spleen tyrosine kinase (SYK) and Bruton’s tyrosine kinase (BTK) inhibitors. (
To assess the day-to-day assay performance, we drew blood samples from the same donor at different days (30 days apart), treated them with RN486, and ran the assay as described above. As shown in Figure 3B , the assay remained robust with comparable IC50 values at 53 nM and 58 nM for samples prepared and assayed on different days.
We next tested the performance of the assay by using it to rank the potency of a number of BTK and SYK inhibitors, selected based on their varying potencies in blocking BCR-mediated B-cell activation in a human whole-blood CD69 assay.19,20 The results showed that the DELFIA p-PLCγ2 assay effectively ranked compounds from the most potent to the least, with IC50 values determined at 0.03, 0.04, 0.71, 1.01, 1.04, 2.77, and 13.01 µM ( Fig. 3C ). IC50 values of each individual compound from both the human whole-blood CD69 B-cell activation assay and the DELFIA p-PLCγ2 assay were plotted on a curve showing a linear R2 value of 0.8 ( Fig. 3D ). Importantly, despite a shift in compound potency between the two assays, the relative compound potency in each assay remained the same.
Stability of the p-PLCγ2 DELFIA Assay
The use of blood from patients may require a 24-h window for blood drawing in a clinic and transportation to laboratory; hence, it is critical to assess the stability and activity of human platelets after 24-h storage/shipment at room temperature or at 4 °C. We observed a significant decrease in PLCγ2 phosphorylation (almost 70% of p-PLCγ2) in convulxin-induced human platelets when the blood was stored for 24 h at 4 °C compared with samples experimented on the same day ( Fig. 4A ). Although the levels of PLCγ2 phosphorylation were also reduced when the blood samples were stored for 24 h at room temperature, the induction fold of p-PLCγ2 after stimulation with convulxin remained sufficient for assay performance ( Fig. 4A ). To understand the significant loss of the level of p-PLCγ2 in platelets at 4 °C, we counted the number of platelets in PRP and compared blood samples stored for 24 h at 4 °C versus at room temperature. As shown in Figure 4B , we noted a significantly lower platelet count (about 75% decrease) when the blood was stored at 4 °C compared with samples stored at room temperature, which likely explains the loss of a measured level of p-PLCγ2. Consistent with our observation, literature reports indicate that platelets do not tolerate refrigeration. Hypothermic (4 °C) storage conditions cause deep modifications in platelet shape and functionality, which compromise viability of cold stored platelets.24,25
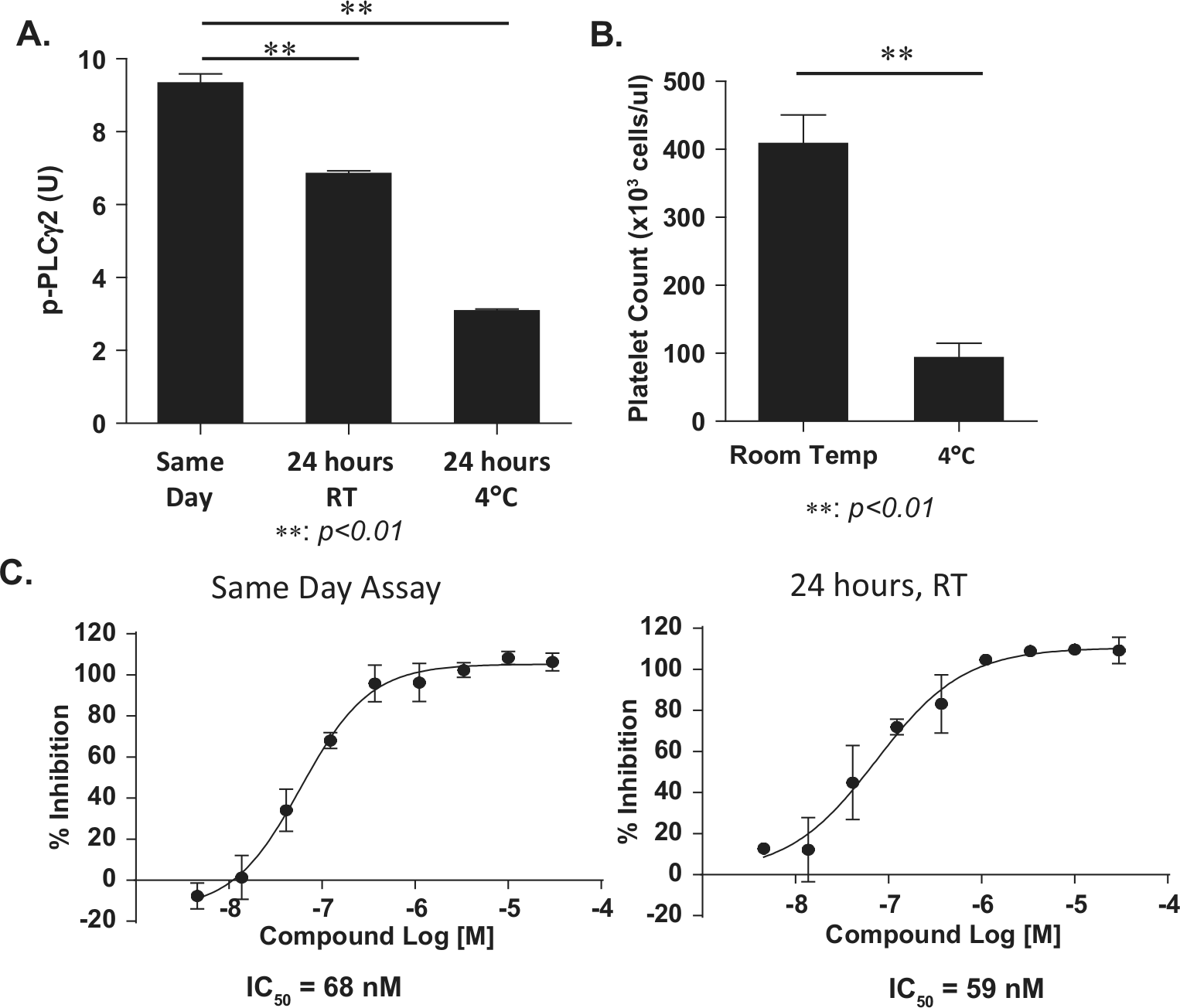
Stability of the phospho–phospholipase C γ2 (p-PLCγ2) dissociation-enhanced lanthanide fluorescent immunoassay (DELFIA) assay. (
Next, we determined the assay performance by using blood samples stored at room temperature for 24 h. Platelets from the same donor were prepared and pretreated either on the same day of blood drawing or after a 24-h storage, at room temperature, with RN486 before stimulation with convulxin. The calculated IC50 values for both methods were similar ( Fig. 4C ), suggesting assay performance remains stable for 24 h at room temperature. The 24-h time frame should increase the flexibility of the assay management, especially in the clinical setting.
Detecting PLCγ2 Phosphorylation in Platelets from Samples of RA Patients
Finally, we used the assay to monitor PLCγ2 phosphorylation in platelets prepared from RA patients. Blood samples from six RA patients were acquired from Bioreclamation, LLC (Westbury, NY) and shipped to our laboratory in 24 h at room temperature. Platelets were prepared and treated with RN486 as described above. A representative concentration-dependent inhibition curve of RN486 for one RA donor is shown in Figure 5A . When the mean IC50, IC75, and IC90 values of RN486 calculated from RA patient samples were compared with those from healthy volunteers, there were no significant differences between the IC75 and IC90 values, as shown in Figure 5B . However, the IC50 values were slightly higher in RA patients compared with those in healthy volunteers (p < .05). This is probably due to high basal systemic inflammation in RA patients, who may need a higher concentration of compound to attenuate the p-PLCγ2 signal. Indeed, a comparison showed that platelets from RA patients expressed a higher baseline of p-PLCγ2 compared with those from healthy subjects, as shown in Figure 5C (p < .05). If these results can be reproduced using more RA patient samples, the awareness of a higher basal p-PLCγ2 in RA patients could guide better dosage projection for clinical development of SYK or BTK inhibitors.
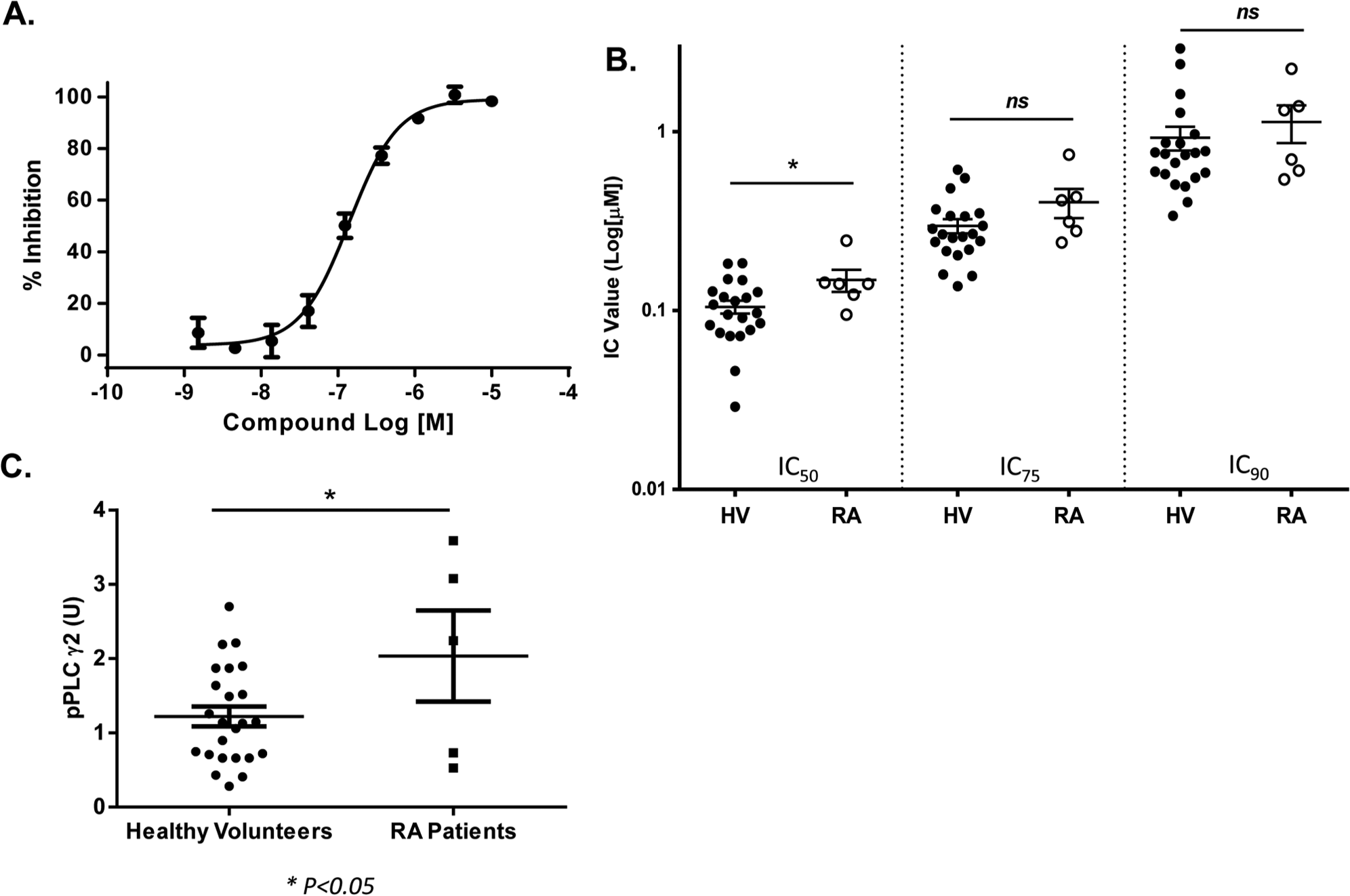
Detecting phospholipase C γ2 (PLCγ2) phosphorylation in platelets from samples of patients with rheumatoid arthritis (RA). (
In summary, there is tremendous pharmaceutical interest in the development of agents that target the SYK-BTK axis for the treatment of inflammatory diseases and hematological malignancies. 1 We have developed and established a quantitatively reliable and robust PD assay that uses GPVI-mediated phosphorylation of PLCγ2, which is the proximal target of the SYK-BTK axis. The assay showed good stability and consistency across different donors (healthy volunteer or RA patients) over 24 h at room temperature. Measuring PLCγ2 phosphorylation in human platelets allows for excellent flexibility in terms of fast processing of samples, ease of storage, and batch analysis. Furthermore, platelets are abundant in the blood and can be easily isolated by simple centrifugation, providing convenience as a whole-blood assay. Thus, our assay could be easily manipulated to fit in various laboratory settings and should facilitate the development and clinical evaluation of pharmacological inhibitors targeting the SYK-BTK axis.
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors were employees of Hoffmann-La Roche during the preparation for the manuscript.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.