
Abstract
Spleen tyrosine kinase (Syk) is a nonreceptor tyrosine kinase that is an important signaling enzyme downstream of immunoreceptors containing an intracellular immunoreceptor tyrosine activating motif (ITAM). These receptors encompass a wide variety of biological functions involved in autoimmune disease pathogenesis. There has been considerable interest in the development of inhibitors of the Syk pathway for the treatment of rheumatoid arthritis and systemic lupus erythematosus. We report that Syk inhibition mechanistically caused peri-islet hemorrhages and fibrin deposition in the rat pancreas and that this finding is due to a homeostatic functional defect in platelets. In more limited studies, similar lesions could not be induced in mice, dogs, and cynomolgus monkeys at similar or higher plasma drug concentrations. Irradiation-induced thrombocytopenia caused a phenotypically similar peri-islet pancreas lesion and the formation of this lesion could be prevented by platelet transfusion. In addition, Syk inhibitor-induced lesions were prevented by the coadministration of prednisone. A relatively greater sensitivity of rat platelets to Syk inhibition was supported by functional analyses demonstrating rat-specific differences in response to convulxin, a glycoprotein VI agonist that signals through Syk. These data demonstrate that the Syk pathway is critical in platelet–endothelial cell homeostasis in the peri-islet pancreatic microvasculature in rats.
Spleen tyrosine kinase (Syk) is a critical nonreceptor tyrosine kinase that signals downstream of immunoreceptors containing an intracellular immunoreceptor tyrosine activating motif (ITAM). These receptors encompass a wide variety of biological functions, including B-cell receptor activation, immune complex signaling through Fc receptors, and platelet activation through the glycoprotein VI (GPVI) and C-type lectin-like receptor 2 (CLEC-2) receptors (Mocsai, Ruland, and Tybulewicz 2010). Syk is functionally analogous to ZAP-70 in T cells and is predominantly expressed in cells of hematopoietic origin including B cells, mast cells, neutrophils, and platelets, with divergent functions in each (Turner et al. 2000). Because of its role in a wide array of pathogenic mechanisms in autoimmunity, there has been considerable effort to develop inhibitors of the Syk pathway for the treatment of diseases such as rheumatoid arthritis and systemic lupus erythematosus (Pamuk and Tsokos 2010).
Since the first description of Syk knockout animals in the late 1990s, there has been concern about the risk of bleeding associated with Syk inhibitors (Turner et al. 1995). Mice deficient in Syk exhibit petechial hemorrhage as early as embryonic day 11 as a result of defective vascular-lymphatic separation during development, followed by embryonic and perinatal lethality (Abtahian et al. 2003). Platelet CLEC-2 and lymphatic podoplanin interactions are critical for lymphangiogenesis, and these are dependent on Syk as well as Src family kinases (Pollitt et al. 2014; Bertozzi et al. 2010; Finney et al. 2012). It has also been shown that platelet ITAM signaling is important for maintaining vascular integrity in the context of inflammation, since pharmacologic inhibition of GPVI or genetic ablation of CLEC-2 in mice impairs the ability of platelets to prevent hemorrhage induced by inflammation (Boulaftali et al. 2013).
Platelets are critical for the maintenance of vascular integrity. Severe thrombocytopenia results in altered microvasculature leading to petechial hemorrhage and even fatal bleeding events (Slichter 2004). While it is unclear how exactly platelets maintain vascular integrity, it is clear that these mechanisms are distinct from the role of platelets in primary hemostasis (Ho-Tin-Noe et al. 2011). One proposed model is that platelets physically interact with the vascular endothelium and that this interaction is important for the health of the endothelium (Kitchens and Weiss 1975; Kitchens and Pendergast 1986; Gimbrone et al. 1969). Another hypothesis is that secreted factors have mitogenic activity on endothelial cells to promote integrity given that platelet releasate can increase the electrical resistance across pulmonary endothelial cells (Schaphorst et al. 2003). Released factors such as serotonin (5-HT), sphingosine-1-phosphate (S1P), lysophosphatidic acid, and others have also been suggested to maintain integrity of the vascular endothelial barrier (Schaphorst et al. 2003; Mineau-Hanschke et al. 1990; Alexander et al. 1998). Platelets deficient in GPVI are unable to rescue cutaneous bleeding after immune complex–mediated challenge via the reverse passive Arthus reaction (George et al. 2008; Boulaftali et al. 2013). In addition, a functional role of platelet GPVI has been implicated in activating neutrophils and subsequent vascular permeability and in the repair of vascular damage (Gros et al. 2015).
The pancreatic islet microvasculature is important for the transfer and dissemination of hormone signals involved in the regulation of blood glucose levels. To enable this process, the endothelium on the endocrine side of the peri-islet vasculature is fenestrated, while it is continuous on the exocrine side of this vascular bed (Henderson and Moss 1985). Maintenance of these vessels is of critical importance for the function of the islet. Both busulfan (Chia and Cattell 1985; Kaduk et al. 1987) and a compound with an undisclosed molecular target (Brenneman et al. 2014) have been reported to perturb the pancreatic microvasculature, including peri-islet hemorrhage with associated acinar fibroplasia. A phenotypically similar peri-islet lesion has also been observed in sublethally irradiated thrombocytopenic rats (Gude et al. 1974). The pancreatic lesion in both the thrombocytopenia experiments and the test article–induced lesion appeared to be rat-specific.
In the course of developing Syk inhibitors, we observed peri-islet hemorrhage and fibrin deposition in the rat. These findings were induced with multiple chemical series in the rat but were not observed in other preclinical species including mouse, dog, and cynomolgus monkey. Based on the lack of target expression in the pancreas, we hypothesized that the lesion was induced by inhibition of Syk in a nonendothelial cell type indirectly responsible for maintaining pancreatic islet vascular integrity. The investigations described were performed to understand whether the rat is unique from other preclinical species in the susceptibility to this toxicologic effect of Syk inhibition and to define the mechanisms responsible. This was performed in order to characterize the sensitivity of the rat to these effects and to evaluate the species as an appropriate model of potential Syk-related adverse changes. We describe the mechanisms leading to the pancreatic peri-islet hemorrhage and fibrin deposition in the rat and demonstrate that they are the result of selective inhibition of Syk within platelets. To inform species specificity and potential human risk, we evaluated functional responses in rat, mouse, cynomolgus monkey, dog, and human platelets in response to convulxin, a snake venom component that signals through the immunoreceptor GPVI. We identified inherent species differences in Syk-dependent platelet pathways.
Methods
Selective Syk Inhibitor
Compound 1 is a representative compound that was used to generate the data presented in these studies (Figure 1). The discovery and structure activity relationship around Syk in the furano[3,2-d]pyrimidine chemotype will be described in a future publication. The compound and related molecules are selective for Syk when compared with a panel of kinases (Supplemental Table 1) using a time-resolved fluorescence energy transfer competition-binding format.
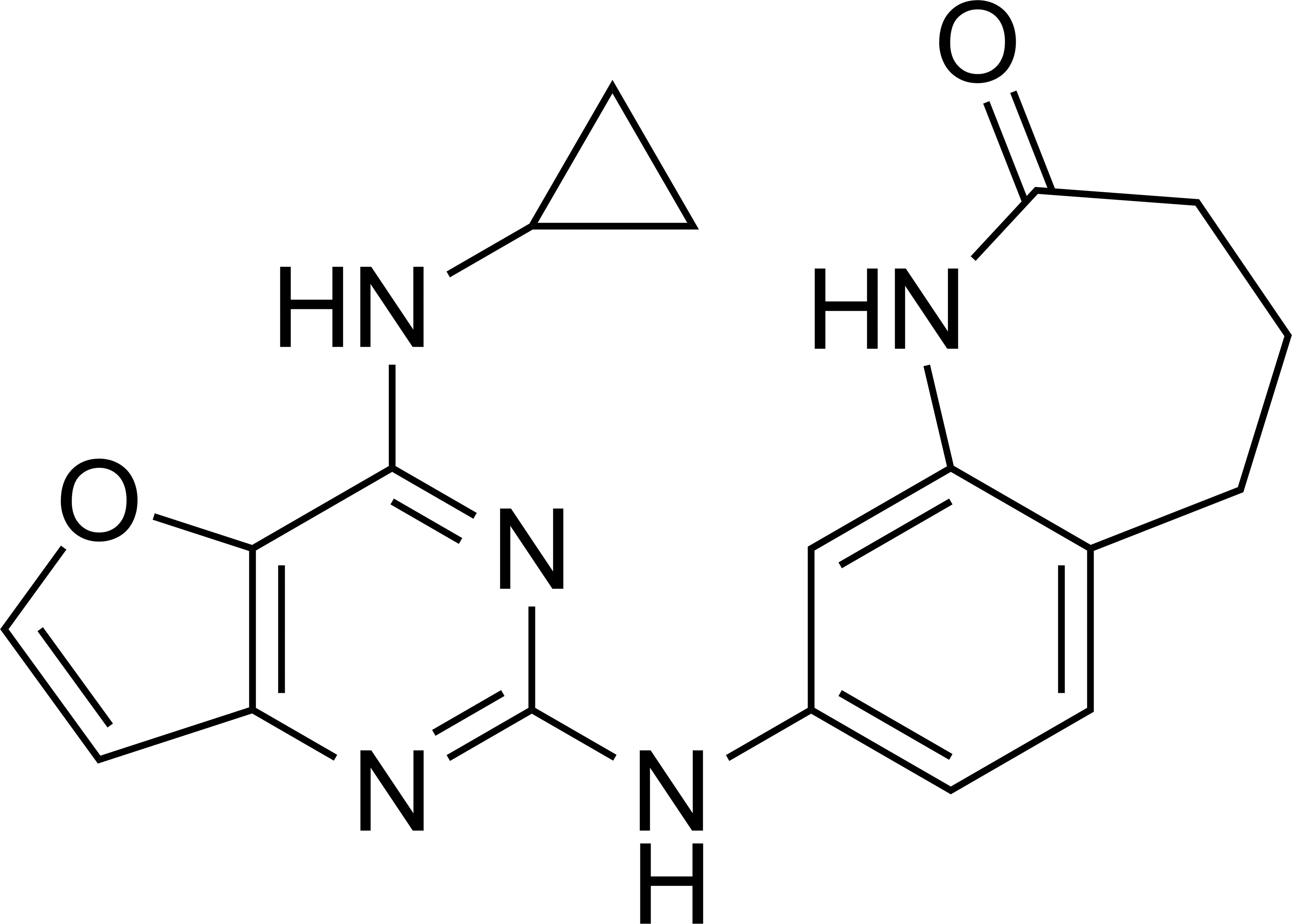
Chemical structure of a representative spleen tyrosine kinase inhibitor. Compound 1 (8-((4-(cyclopropylamino)furo[3,2-d]pyrimidin-2-yl)amino)-4,5-dihydro-1H-benzo[b]azepin-2(3H)-one) was used in both in vitro and in vivo studies.
Spleen tyrosine kinase (Syk) inhibitor sensitivity in platelet functional assays.
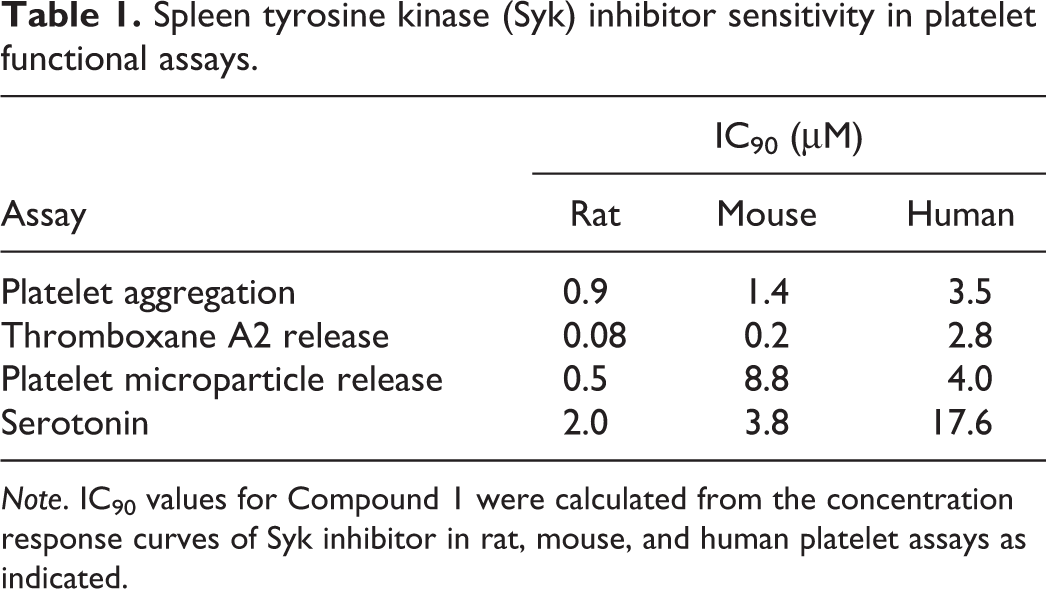
Note. IC90 values for Compound 1 were calculated from the concentration response curves of Syk inhibitor in rat, mouse, and human platelet assays as indicated.
Toxicity Studies with a Syk Inhibitor
All animal studies were performed in accordance with approved protocols from the AbbVie Institutional Animal Care and Use Committee. Compound 1 or vehicle (0.02% Tween 80, 0.5% hydroxypropylmethylcellulose) were administered at a dose of 100 mg/kg orally (PO [per oral]) by gavage once daily to 6- to 8-week-old Sprague-Dawley rats (n = 9/group). For the time-course studies, cohorts of animals dosed with either vehicle or Syk inhibitor were sacrificed after a single dose, and pancreata were analyzed for the presence of peri-islet hemorrhage. Pancreatic specimens were examined histologically, following a standard hematoxylin and eosin-staining protocol. Organs were embedded whole, sectioned, and analyzed longitudinally. Separate cohorts of vehicle and Syk inhibitor-treated animals were dosed daily for 28 days to follow the progression of the lesion. Standard toxicologic parameters were measured, including body weight, macroscopic and microscopic tissue evaluation, organ weights, hematology (CellDyn, Abbott Laboratories, North Chicago, IL), and plasma concentrations of the inhibitor tested. Standard clinical chemistries including markers of exocrine pancreatic injury (amylase and lipase) were evaluated on an Architect c4000 clinical chemistry analyzer (Abbott Laboratories).
For repeat dose studies conducted in Supplemental Figure 2, duration of studies is indicated in the table (either 7 or 14 days). Mouse studies were conducted for 7 days using male CD-1 mice (n = 5/group). Male Sprague-Dawley rats were used for rat studies (n = 9/group). Beagle dogs were used (n = 2/sex/group) in a 14-day study, and cynomolgus monkeys were used for nonhuman primate studies for either 7- or 14-day toxicity studies.
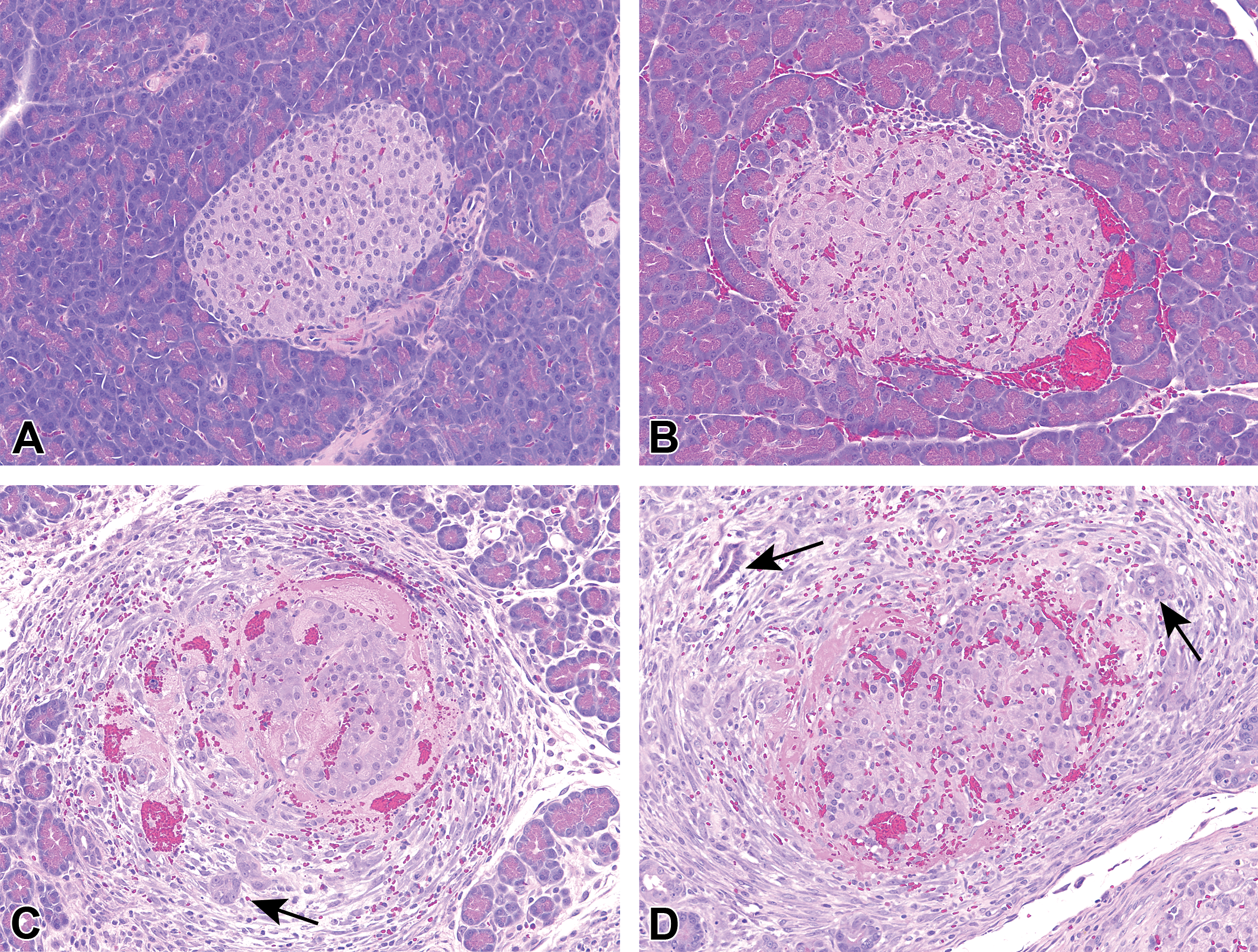
Peri-islet histologic changes in response to spleen tyrosine kinase inhibition. Compared with the normal islet (A), minimal fibrin deposition in the peri-islet capillaries and hemorrhage were the main features of the early stages of islet damage after a single dose of compound (B). After 7 days of dosing, fibrin deposition was still present along with duct hyperplasia of the exocrine pancreas (C and D, arrows) and fibroplasia (A–D, original magnification 20×; H&E).
Islet Immunohistochemistry for Glucagon and Insulin
Formalin-fixed paraffin-embedded pancreas tissue from Syk inhibitor–treated rats was cut at 5-μm sections and placed onto glass slides and dried. Sections were rehydrated with ethanol, and antigen was retrieved using Leica Bond epitope retrieval solution 1 for 30 min at 100°C on a Leica Bond RX instrument (Leica Biosystems, Melbourne, Australia). Primary antibody incubation with glucagon mouse monoclonal (0.25 μg/ml, Cat# ab10988; Abcam, Cambridge, MA) and insulin rabbit monoclonal (0.02 μg/ml, Cat# ab181547; Abcam) was 15 min. The isotype control antibodies were rabbit polyclonal sera (Cat # I-1000, Vector Labs, Burlingame, CA), mouse IgG1 (Cat# 550878; BD Pharmingen, San Jose, CA), and rabbit monoclonal IgG (Cat# ab125938; Abcam), respectively. All ancillary reagents were purchased from Leica. The Leica Refine detection kit (Cat# DS9800) includes peroxide block, postprimary rabbit antimouse, polymer goat antirabbit, 3,3'-Diaminobenzidine (DAB) Part 1, DAB Part B, and hematoxylin. Kit components were used per manufacturer’s instructions.
Oral Glucose Tolerance Test (OGTT)
Seven-week-old male Sprague-Dawley rats received vehicle (0.02% Tween 80, 0.5% hydroxypropylmethylcellulose) or Compound 1 (100 mg/kg) once daily by oral gavage for 7 weeks. The dose was selected to compare with previous studies, where the pancreatic lesion was observed. OGTT was conducted at baseline prior to initiating dosing and then weekly for 6 weeks. Animals were fasted 15 hr prior to OGTT. Thirty minutes after dosing with vehicle or Compound 1, the animals were given 3,000-mg/kg
Cell Culture for Syk Expression Studies
The rat basophilic leukemia cell line RBL-2H3 (ATCC CRL-2256™) was cultured in T150 flasks (Corning 430825) in minimal essential media (MEM) media (Cat# 11095-080; Gibco, Waltham, MA) supplemented with 10% heat-inactivated fetal bovine serum (FBS) (Cat# 10082147; Gibco), 2-mM
Syk Protein Expression by Western Blotting
Rat whole pancreas (nonperfused), lymph nodes, or dissected pancreatic islets (from perfused/digested pancreata) were homogenized in lysis buffer (Cat# 9803; Cell Signaling Technology, Beverly, MA) supplemented with protease inhibitor cocktail (Cat# 539134; Calbiochem, Billerica, MA). Heparinized rat whole blood was lysed for 15 min on ice using 10 volumes of cold 1× Pharm Lyse™ (Cat# 555899; Becton Dickinson, Franklin Lakes, NJ). White blood cells were then separated by centrifugation at 1,200 rpm for 5 min at 4°C. Cells were washed twice with cold 1× phosphate buffered saline (PBS) (Cat# 14190-144; Gibco) and then lysed on ice using lysis buffer with protease inhibitors. For all protein lysates, cell debris was centrifuged at 12,000 g for 10 min at 4°C. Total protein concentration was measured using the Pierce BCA protein assay kit (Cat# 23227; Thermo Scientific, Wilmington, DE). Ten μl of each lysate was resolved by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) for 2.5 hr at 125 V (constant voltage) using Novex 10 to 20% Tris–glycine gradient gels (EC61352). Resolved proteins were transferred to Immobilon polyvinylidene difluoride [PVDF] (IPV20200) for 90 min at 100 V (constant voltage). Membranes were then blocked for 1 hr at ambient temperature with PBS/0.5% Tween 20 (PBST)/3% gelatin from cold-water fish skin (Cat# G7765; Sigma). Membranes were then incubated at 4°C overnight with primary antibody (anti-Syk Santa Cruz sc-1077 or anti-GAPDH sc-25778) at 1:1,000 in PBST/1% fish gelatin. After 5 washes in PBST, membranes were incubated at room temperature for 1 hr on a shaker with goat antirabbit IgG-horseradish peroxidase antibody (Invitrogen G21234) at 1:5,000 in PBST/1% Tween 20. After 5 additional washes in PBST, the membranes were developed for 1 min in Amersham ECL mixture (RPN2106). Finally, Amersham Hyperfilm ECL (28906836) was exposed in the darkroom to the developed membranes.
Quantitative Polymerase Chain Reaction for Syk Messenger RNA (mRNA)
Rat whole pancreas (nonperfused), lymph nodes, or dissected pancreatic islets (from perfused/digested pancreata) were homogenized in QIAzol (Cat# 79306; QIAGEN, Germantown, MD). Heparinized rat whole blood was lysed for 15 min on ice using 10 volumes of cold 1× Pharm Lyse (Cat# 555899; Becton Dickinson). White blood cells were then centrifuged at 1,200 rpm for 5 min at 4°C, washed once with cold 1× PBS, and then lysed in QIAzol. RNA lysates were then processed to total RNA using the miRNeasy mini kit (Cat# 217004; QIAGEN) and quantified by Nanodrop ND-1000 (Thermo Scientific; absorption at 260 nm) and purity (260 nm/280 nm). Total RNA was converted to complementary DNA (cDNA) using the high-capacity cDNA Archive kit (Cat# 4322171; Applied Biosystems, Foster City, CA). cDNA was then diluted to 5 ng/µl and either rat Syk mRNA or rat 18S ribosomal RNA were specifically amplified using TaqMan® Gene expression assays (SYK = Rn00562684_m1 and 18S = Rn03928990_g1) and TaqMan Fast Universal Polymerase Chain reaction (PCR) Master Mix (Cat# 4352042; Applied Biosystems) on an Applied Biosystems 7500 fast real-time PCR system.
Irradiation and Platelet Transfusion Experiments in Rats
Healthy 8-week-old Sprague-Dawley rats (n = 9/group) with initial platelet counts ranging from 1,200 to 1,300 × 103/μl were irradiated in a cesium irradiator (Shepard and Associates, Mark I model 68A, San Fernando, CA) for 3.6 min at 250 rads/min for a total of 900 rads to induce thrombocytopenia (100–400 × 103/μl; Kassayova, Ahlersova, and Ahlers 1999). Irradiated rats (100–400 × 103 platelets/μl blood) received either 5-ml platelet-poor plasma (PPP), 5-ml platelet-rich plasma (PRP), or 5-ml PBS by slow infusion in the lateral tail vein daily, for either 4 or 5 consecutive days. Blood samples (250 μl) for platelet counts were taken every other day (CellDyn, Abbott Laboratories) for each animal. The platelet transfusion was given in the lateral tail vein as a slow infusion of 5 ml of PRP at 4.5 × 109 platelets/µl or the same volume of PBS or PPP. Animals were sacrificed on day 11, and pancreata were evaluated for peri-islet hemorrhage and/or fibroblasts.
PRP/PPP Preparation
Preparation of platelets was performed with a modification of methods previously described (Cazenave et al. 2004). Donor male Sprague-Dawley rats were euthanized via CO2 asphyxiation, and blood was collected via the vena cava using a 16-gauge needle and inverted in sodium citrate tubes. Blood was centrifuged at 4,200 rpm for 3 min to obtain a supernatant of PRP. Platelet counts were analyzed (CellDyn) and adjusted by addition of HBSS to yield 4.5 × 109 platelets/µl. To obtain PPP, the supernatant from above was centrifuged for an additional 8 min at 4,200 rpm, and the resulting supernatant was collected.
Prednisone Codosing with a Syk Inhibitor
Male Sprague-Dawley rats (n = 6/group) were dosed orally by gavage daily with either vehicle (0.02% Tween 80, 0.5% hydroxypropylmethylcellulose) or Syk inhibitor (100 mg/kg) with or without prednisone at doses of 0.3, 1, or 3 mg/kg for 7 days. Tissue was harvested after CO2 asphyxiation and processed as described. Numbers of pancreatic lesions were counted as defined by presence of peri-islet hemorrhage or fibroblasts.
Effects of Syk Inhibition on IN VITRO Platelet Function across Species
Platelet aggregometry
Whole blood samples from normal, healthy subjects from all species were collected into sodium citrate tubes (Becton Dickinson) and mixed by inverting. Blood from Sprague-Dawley rats (n = 2) or Swiss Webster mice (n = 10) was harvested via cardiac puncture after CO2 asphyxia and pooled. Beagle dog samples were obtained from the jugular vein under manual restraint, and cynomolgus monkey samples were obtained from the femoral vein during restraint with prior acclimation. Human whole blood was collected using a 21-gauge needle from 2 separate healthy individuals who had been free from non-steroidal anti-inflammatory drugs (NSAID) and anticoagulants for at least 48 hr. The 2 donors were tested individually. Human and rodent blood was stored at room temperature and warmed to 37°C approximately 30 min prior to incubation and aggregation assays were performed within 4 hr of blood draw. Dog and cynomolgus monkey samples were shipped at ambient temperature and warmed to 37°C approximately 30 min prior to incubation. Aggregation assays were performed 24-hr postblood draw.
Whole blood platelet aggregometry was evaluated using the Multiplate 5.0 Analyzer from DiaPharma (Franklin, OH; Defontis et al. 2013). Whole blood (300 μl) was diluted with 300-μl PBS in each test cell. Syk inhibitors were formulated in 100% dimethyl sulfoxide (DMSO) at a concentration of 20 mM and diluted in DMSO to maintain solvent concentrations across test groups and vehicle. Final assay concentration of DMSO was 0.25%. Final Syk inhibitor concentrations ranged from 0.1 to 50 μM. Inhibitor was added directly to the sample and allowed to incubate for 30 min at 37°C. Samples were then stimulated with convulxin and aggregation was allowed to progress for 6 min. All testing was performed at 37°C. For the 6-min aggregation plot, data were analyzed on an AUC basis. Experimental replication (a minimum of one repeat) was undertaken in order to ensure data consistency.
Platelet microparticle assay
Whole blood was collected as described and treated with Syk inhibitors at concentrations from 0.06 to 50 μM or DMSO vehicle (final concentration of 0.25%) for 30 min at 37°C in a sealed polypropylene microplate. Each treated sample was then stimulated for 15 min at 37°C with 0.5-µg/ml convulxin (Enzo, Farmingdale, NY) in the presence of 10-mM CaCl2 and 10-µg/ml hirudin (to inhibit thrombin and clot formation). After stimulation, antimouse CD41-FITC (MWReg30, BioLegend, San Diego, CA), antirat CD61-FITC (BD Biosciences, San Jose, CA, F11), and antihuman CD41-FITC (HIP8, BioLegend; Begamery et al. 2005) were added at room temperature for 15 min followed by fixation in formaldehyde (1% in HEPES saline) for 30 min prior to analysis on an Accuri C6 digital flow cytometer (Becton Dickinson). Platelet microparticles were identified by establishing a threshold on CD41- or CD61-FITC-positive events (depending on the species) and then gating out leukocyte–platelet aggregates based on size/granularity using forward scatter/side scatter. Platelet microparticle formation in the presence of convulxin was normalized to the vehicle.
5-HT assay
Whole blood was collected as described and aliquoted in 96-, deep-well plates and incubated for 30 min at 37°C with Syk inhibitors at concentrations from 0.01 to 25 μM or DMSO vehicle. The final concentration of DMSO in the assay was 0.5%. Ten µm of indomethacin (Cat# I8280; Sigma, St. Louis, MO) and 1-mM ethylene glycol tetraacetic acid (Cat# E4378; Sigma) were then added to each well immediately prior to stimulation with 1-µg/ml convulxin for 5 min at 37°C. Plates were then centrifuged at 2,000 rpm for 10 min at room temperature; 50 µl of plasma was assayed using the 5-HT enzyme-linked immunosorbent assay (ELISA; Rocky Mountain Diagnostics, Colorado Springs, CO, BAE-8900) per the manufacturer’s instructions. The manufacturer indicates assay is compatible with multiple species (Song et al. 2005).
Thromboxane B2 assay
Whole blood from healthy human donors and normal animals was collected as described. Blood was centrifuged at 200 g for 2 min to prepare PRP. Platelets were further isolated by centrifugation of enriched plasma at 900 g for 15 min at room temperature in HBSS without calcium chloride (CaCl2) and supplemented with 1-mM prostaglandin E1 (Cat# P5515; Sigma). After removal of the supernatant, platelets were resuspended in 6 ml of HBSS to adjust the platelet count to 4 × 108 platelets/ml; 50 μl of platelets were combined with 50-μl Syk inhibitor or vehicle in 0.5% DMSO, followed by a 10-min preincubation. Samples were then stimulated with 50 μl of 0.25 mg/ml convulxin for 10 min at 37°C and quenched by addition of 100 μl of a solution containing 5-mM EDTA (Cat# E6758, Sigma) and 10-mM indomethacin (Cat# I8280; Sigma). Assay plates were centrifuged at 1,000 g for 15 min, and supernatants were analyzed for thromboxane B2 concentration by ELISA (Cat# 519031; Cayman Chemical, Ann Arbor, MI) as per manufacturer’s instructions.
Statistical Analysis
Comparison of results from the vehicle and Syk inhibitor groups on days 7, 14, 28, 35, and 42 were performed using Students paired t-test. Significant differences of glucose tolerance test over time (AUC) were performed using 2-way analysis of variance (ANOVA) with Bonferroni’s multiple comparison. Significance of prednisone codose experiments was determined using 1-way ANOVA with Bonferroni’s multiple comparison.
Results
Development of Pancreatic Lesion
In rat toxicology studies with Syk inhibitors (Figure 1), we consistently identified a pancreatic peri-islet hemorrhage that leads to fibroblast proliferation, secondary degeneration, and necrosis in the exocrine pancreas and, in severe cases, lobular atrophy (Figure 2). The histomorphologic findings were characterized through single-dose, time-course, and dose-range studies. After a single 100-mg/kg dose, focal hemorrhage and fibrin deposition (Figure 2B) were evident, suggesting vascular leak as compared to normal islets (Figure 2A). After 3 to 7 days of continued dosing, peri-islet injury progressed with mixed cellular infiltrates, including fibroblasts, and involved the exocrine pancreas, including hyperplasia of the exocrine pancreatic ducts (Figure 2C and D, arrows). The histomorphologic findings initially involved either a single islet or a small number of islets, suggesting that not all islets were equally sensitive to the effects of Syk inhibition at any given time. The incidence and severity of the lesion increased in a dose-responsive manner, and the number of islets affected increased with longer dosing duration (data not shown).
The lesions shown in Figure 2 occurred with Compound 1 which is selective for Syk inhibition when tested in a panel of 84 kinases (Supplemental Table 1) as well as a panel of 75 nonkinase biological targets. More than 10 Syk inhibitors represented by 5 structurally distinct chemical series, each with different kinase selectivity profiles, caused similar lesions in toxicology studies in rats (data not shown). Therefore, it is unlikely that the lesion is due to a common metabolite, aberrant kinase activity, or off-target effect. Compound 1 did not accumulate in the pancreatic tissue (data not shown). In multiple toxicology studies with Syk inhibitors in rat, mouse, dog, and cynomolgus monkey, the lesion was observed only in the rat, suggesting that the rat pancreas is uniquely sensitive to Syk inhibition. Four compounds were tested in mice, and a single compound was tested in monkey and dog (Supplemental Table 2). These findings were morphologically identical to those reported in thrombocytopenic rats and in rats administered either busulfan or compounds against an undisclosed target (Gude et al. 1974; Brenneman et al. 2014; Kaduk et al. 1987). The nature of the lesion was consistent with a primary vascular toxicity affecting the microvasculature directly surrounding the pancreatic islets. Serum amylase and lipase were elevated in occasional animals but did not correlate with incidence or severity of the lesion at any time point (data not shown).
Functional Consequence of Pancreatic Lesion
We sought to determine if long-term administration of Syk inhibitors would have deleterious effects on islet function. To test this, we treated rats with a Syk inhibitor at 100 mg/kg once per day for six weeks and challenged the rats with an OGTT once per week. Glucose levels were normal after challenge for both vehicle- and compound-treated animals at day 7, but by day 14 the inhibitor-treated animals began to show statistically significant elevation in blood glucose concentrations over time (Figure 3A). Relative to vehicle controls, blood glucose concentrations continued to increase through day 42, consistent with progressive decrease in insulin production subsequent to beta cell loss (circulating insulin levels were not measured; Figure 3B). To confirm pancreatic endocrine perturbation, we evaluated islets for the presence of insulin and glucagon by immunohistochemistry (Figure 4). The typical peripheral distribution of glucagon-positive alpha cells (Figure 4, right panels), and the central distribution of insulin-positive beta cells (Figure 4, left panels) was lost in rats administered the Syk inhibitor as early as 72 hr of treatment (Figure 4, middle panels). The presence of individual glucagon- and insulin-positive cells and small cellular aggregates in the exocrine pancreas at 6 weeks was interpreted as a compensatory response to the loss in endocrine function stemming from damage of the islets (Figure 4, bottom panels).
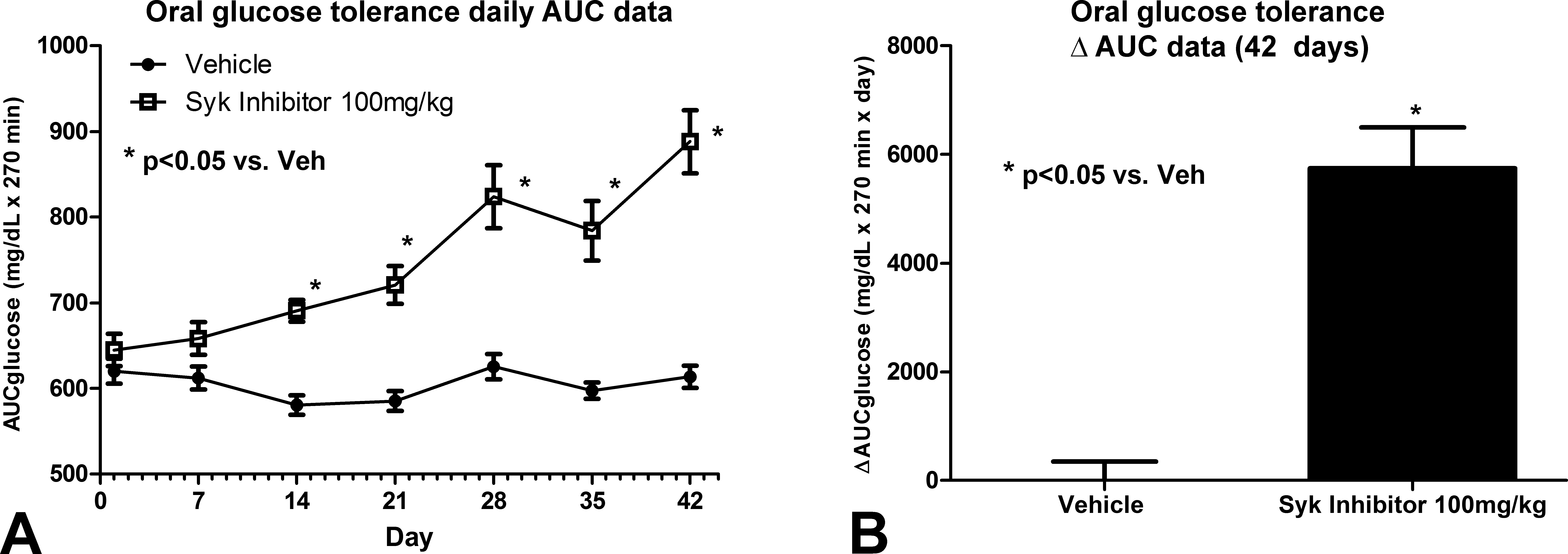
Consequence of long-term spleen tyrosine kinase inhibition on islet function. Sprague-Dawley rats were administered Compound 1 for 6 weeks. (A) Weekly oral glucose tolerance measurements are represented as the area under the curve (AUC) for the 4-hr period following glucose challenge (n = 9 rats/group; *p < .05, paired t-test). (B) Data from panel (A) were integrated as AUC over for the 6-week observation period (*p < .05, 2-way analysis of variance with Bonferroni’s multiple comparisons).
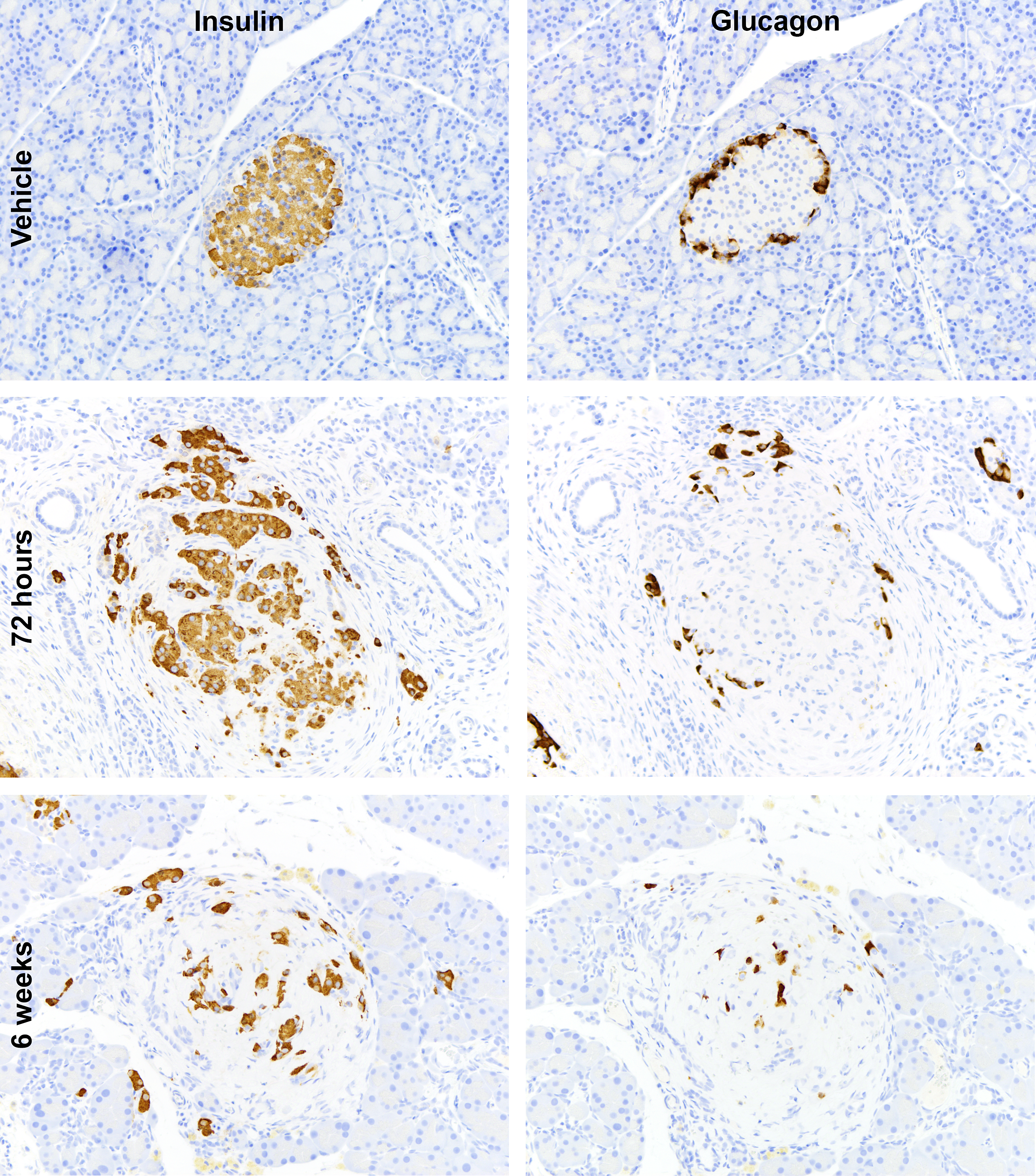
Consequences of long-term spleen tyrosine kinase (Syk) inhibition on islet architecture. Sections of rat pancreas were stained by immunohistochemistry with antibodies for insulin to stain the beta cells (left panels) or glucagon to stain the alpha cells (right panels). Sections from vehicle animals (top panels) were compared to Syk inhibitor–treated animals after 72 hr of dosing (middle panels) or after 6 weeks of dosing (lower panels) (original magnification 20×; insulin stain, right panels; glucagon, left panels).
Syk Is Not Expressed in the Pancreatic Islet
The hypothesis that Syk inhibitors have a direct effect on Syk in the islet was tested by examining Syk expression in the pancreas. We determined by immunohistochemistry (IHC) that Syk was not expressed in the islet (Figure 5A), whereas the same antibody did stain cells in the lymph node, serving as a positive control (Figure 5B). Western blot analyses showed that Syk is highly expressed in lymph node, peripheral blood mononuclear cells and RBL-2H3 cells, but it was below the limit of detection in islet preparations (Figure 5C). Using a third method to measure expression, quantitative PCR was used on the same tissues as the Western blot (Figure 5D). Compared with control lymph node and blood samples, Syk expression in the islet was not detectable (Figure 5D). Given the lack of islet Syk expression and the highly selective nature of the molecules, the toxicity cannot be attributed to direct effects on the islet. When taken together, the data indicate that the pancreatic lesions are likely mediated through inhibition of Syk in a target cell outside of the pancreas.
Spleen tyrosine kinase (Syk) is not expressed in the pancreatic islets. (A) Syk was not expressed in the pancreatic islet of the rat by immunohistochemistry. (B) As a positive control, lymph nodes were stained with the same antibody as in panel (A). Syk is highly expressed in the lymphoid follicles of rat lymph nodes, which corresponds to the B-cell areas (A and B, original magnification 40×; Syk IHC, hematoxylin counterstain). (C) Western blot analysis of protein lysates from tissues as indicated. (D) Quantitative PCR from the same samples as in panel (C). Data indicate no expression compared with PBMC, lymph node, or cell line (RBL-293) positive controls.
Platelets Are an Important Target Cell for Lesion Induction
Gude et al. (1974) reported a phenotypically similar pancreatic lesion in the rat after either antibody or irradiation-induced thrombocytopenia. To test the hypothesis that thrombocytopenia may induce the pancreatic lesion, we reproduced the Gude study in rats by irradiation-induced bone marrow collapse, which resulted in severe thrombocytopenia by day 10 (Figure 6A). When the platelet number dropped below 50,000/μl, the rats exhibited a pancreatic lesion phenotypically similar to that induced by compound (Figure 6B and C). To confirm the platelet dependence of the lesion and to exclude a broad effect of irradiation via bone marrow collapse, we reconstituted irradiated rats with PRP or PPP. The animals transfused with PRP, which achieved platelet counts of at least 200,000/μl, did not develop the pancreatic lesion (Figure 7A). The rats that received PPP developed the lesion similar to irradiated rats, further supporting the specific role of platelets in the formation of the lesion and suggesting that the specific factor involved is not a long-lived soluble factor (Figure 7A). The protective effect of the PRP reconstitution could be reversed with systemic administration of a Syk inhibitor at the time of reconstitution suggesting that Syk pathway in platelets is important for maintaining the microvasculature integrity of the pancreatic islet (Figure 7B). Since Syk inhibitors do not cause thrombocytopenia (data not shown), we hypothesized that Syk inhibition contributes to a functional deficit in platelets that disrupts peri-islet microvasculature homeostasis.
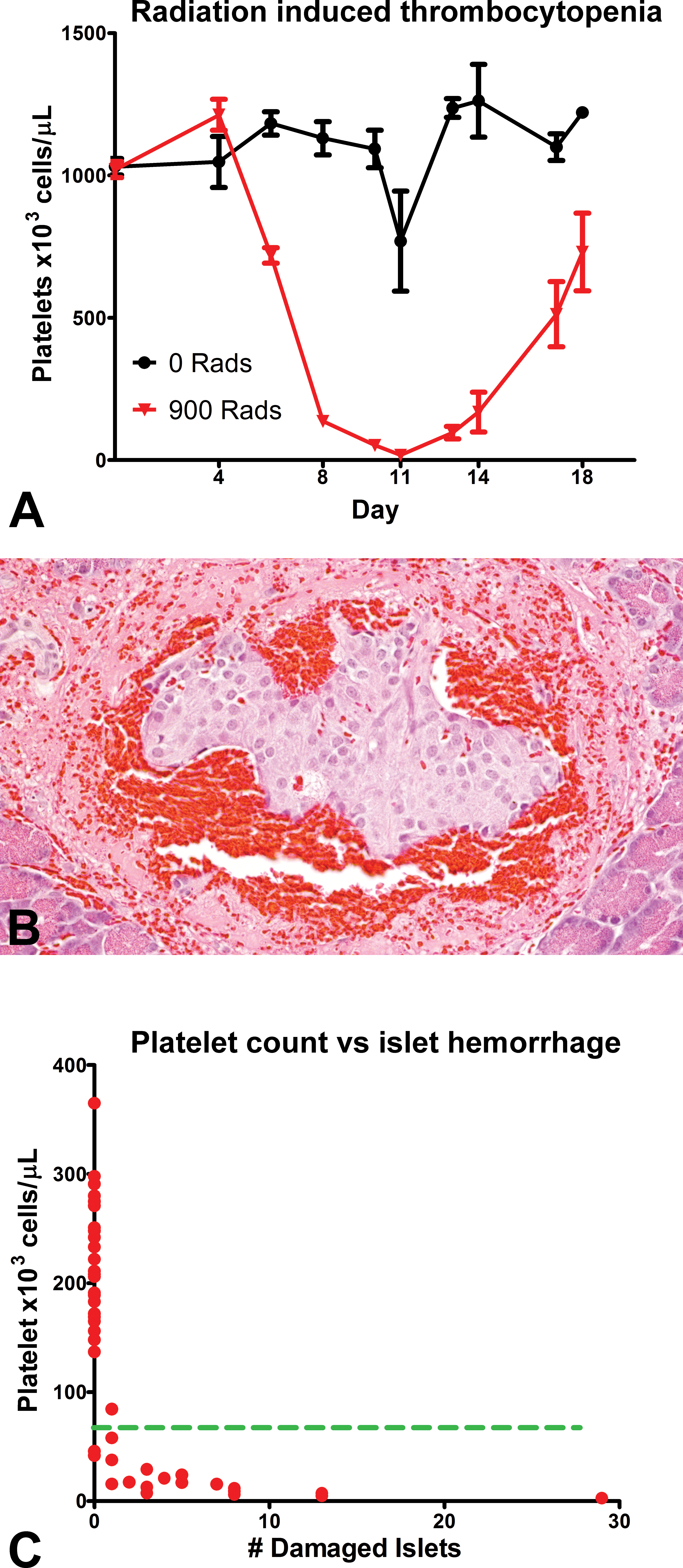
Sublethal irradiation in rats recapitulates the phenotype induced by spleen tyrosine kinase inhibitors. (A) Sublethal irradiation (900 rad) of Sprague-Dawley rats (n = 9) results in progressive thrombocytopenia with a nadir between days 10 and 12 (red line) compared with nonirradiated control rats (black line). Platelet counts were measured for each rat over time. (B) Histologic sections of pancreas from animals at day 10. The most acute stage of damage consisted of congestion of the peri-islet capillary with fibrin deposition (original magnification 40×; H&E). (C) Incidence of damaged islets in relation to platelet count after irradiation. The pancreatic lesions occurred only in rats with less than 50 × 103 platelets (green dashed line).
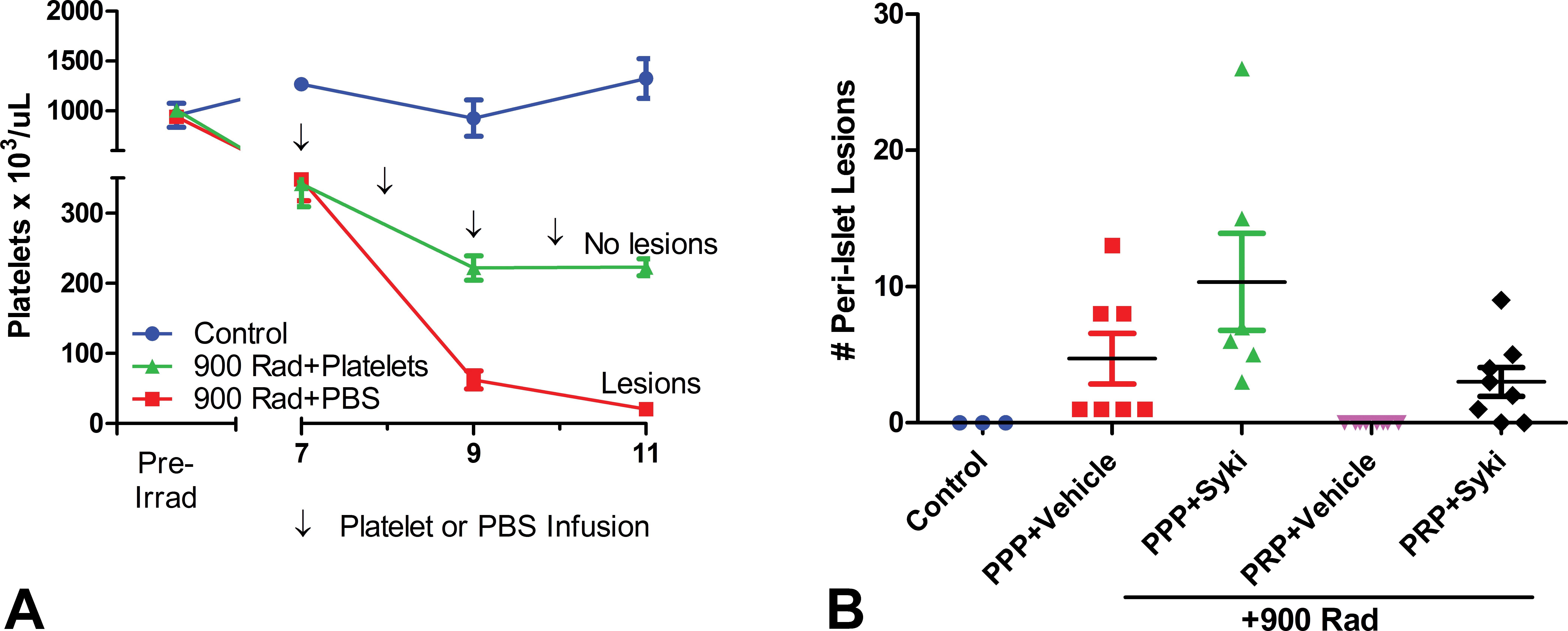
Platelet transfusion protects irradiated rats protects from peri-islet hemorrhage. (A) Platelet counts of nonirradiated rats (blue line), irradiated rats with platelet-poor plasma infusion (red line), and irradiated rats with platelet-rich plasma infusion (green line) were measured on days 7, 9, and 11. Arrows indicate infusion times. (B) The number of peri-islet lesions was counted on day 11 after irradiation, platelet transfer, and systemic administration of 100 mg/kg spleen tyrosine kinase inhibitor.
Kitchens and Pendergast (1986) reported that thrombocytopenia in humans resulted in thinning and fenestration of the endothelium. These related changes could be reversed with the administration of glucocorticoid, indicating a direct effect on endothelium given the lack of platelets. To test whether glucocorticoid could protect rats against Syk inhibitor-induced lesion formation, we codosed rats with prednisone and Compound 1. Administration of prednisone significantly reduced the number of islet lesions in a dose-dependent manner (Figure 8). Rats dosed with Syk inhibitor alone developed a mean number of 8 damaged islets. Administration of prednisone significantly reduced the number of impacted islets in a dose-dependent manner.
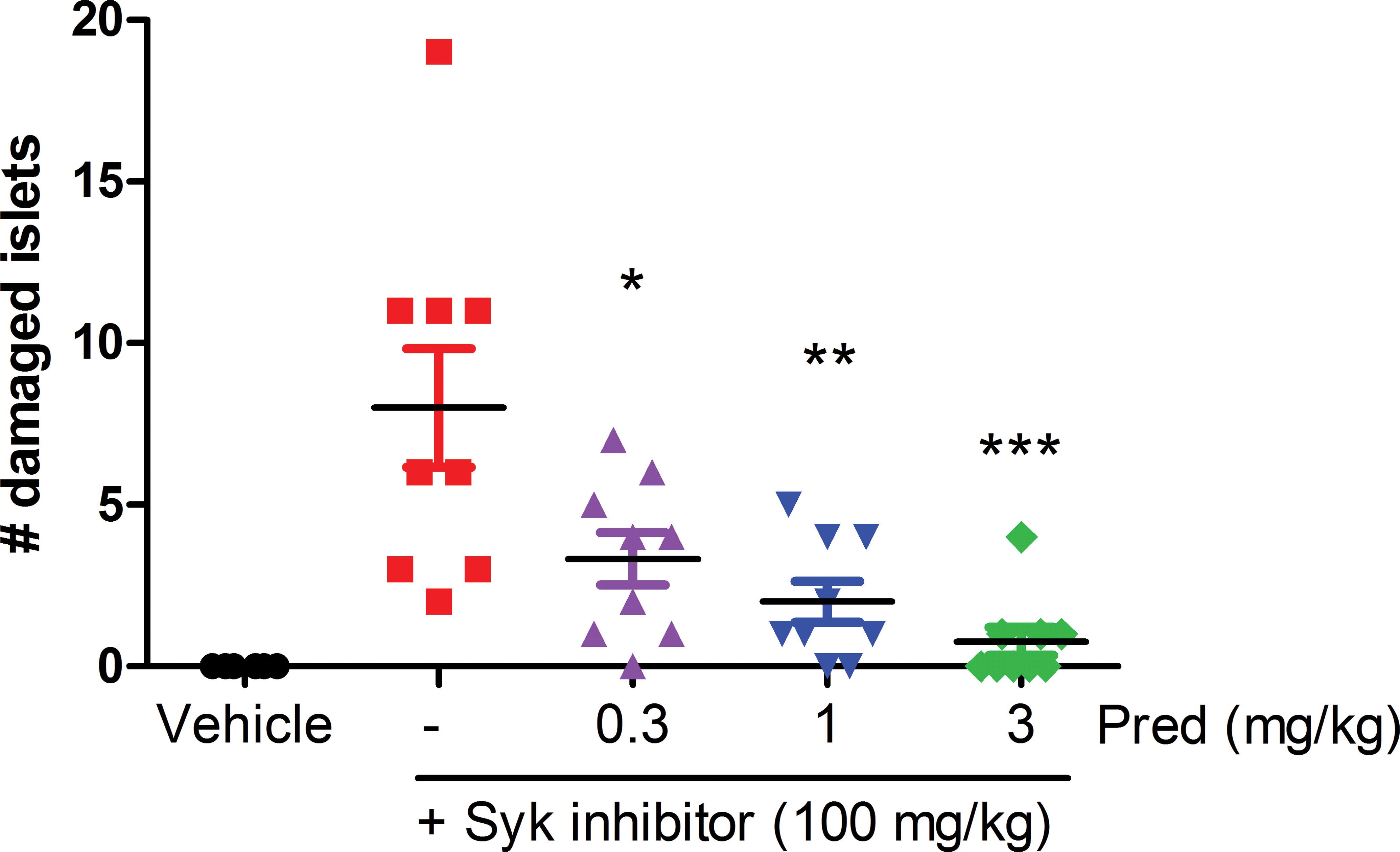
Coadministration of prednisone with spleen tyrosine kinase (Syk) inhibitor dose dependently protects against formation of the pancreatic lesion. Syk inhibitor was dosed at 100 mg/kg with or without prednisone (0.3, 1, or 3 mg/kg) concurrently for 7 days (n = 6). The number of damaged islets was counted on histologic sections by the number of lesions across the entire pancreas. Lesions were reduced in all prednisone treated groups (*p < .05, **p < .01, ***p < .001, one-way analysis of variance with Bonferroni’s multiple comparison). Data are representative of 2 independent experiments.
Cross-species Analysis of Syk Expression and Function in Platelets
Because the development of the pancreatic lesion appeared to only occur in the rat, we next evaluated Syk expression and Syk-dependent functions in platelets across species. Syk expression was assessed by Western blot in human, mouse, rat, dog, and cynomolgus monkey platelets. Syk is highly expressed in platelets across species (Figure 9A). The lack of expression of Syk in pancreatic islets was also confirmed in the same experiment. Given that platelet Syk expression did not appear different across species, we examined the effect of Syk inhibition on select assays of platelet function. Platelet aggregation was measured in the whole blood of mouse, rat, dog, cynomolgus monkey, and human using convulxin, a specific GPVI agonist whose downstream signaling is Syk-dependent. Convulxin stimulation of platelet aggregation was concentration-dependent in all species tested (Figure 9B). Human platelets were most sensitive (EC50 = 0.04 nM), while EC50 values ranged from 0.12 nM (mice) to 0.45 nM (cynomolgus monkey). The rat EC50 was 0.56 nM. The magnitude of the aggregation response across species was different as well. At 10-nM convulxin, rat platelet responses were consistently lower compared to the robust induction observed in dog, mouse, and human blood (Figure 9C). There were more modest species differences in other platelet functional responses such as release of platelet microparticles, 5-HT, and thromboxane B2 (data not shown). Although it is unclear whether GPVI-mediated aggregation is the mechanism responsible for the maintenance of peri-islet microvasculature, these data support inherent differences in Syk-dependent platelet function.
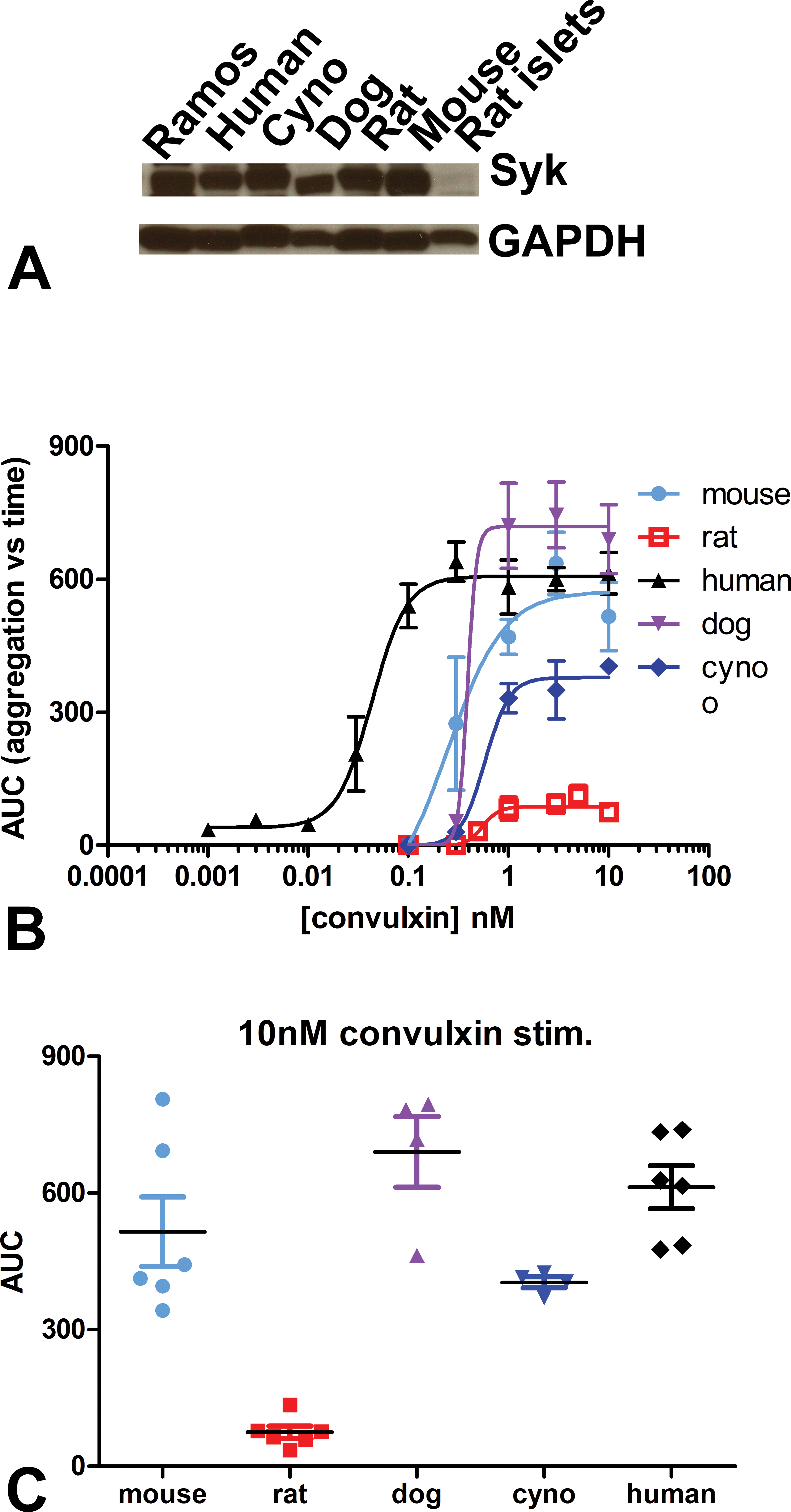
Cross-species comparison of spleen tyrosine kinase (Syk) expression and platelet function. Platelet protein extracts from multiple species (human, cynomolgus monkey, dog, rat, and mouse) were resolved by SDS-PAGE and immunoblotted for Syk expression. The Ramos B-cell line was used as a positive control, while rat pancreatic islets served as a negative control (A). Platelet aggregation was measured in response to convulxin (B). Platelet aggregation responses at high concentration of convulxin (10 nM) across species highlight the difference in magnitude of response (C). Each point represents an individual replicate (n = 6).
Another explanation for the rat-specific susceptibility to the pancreas lesion was differential sensitivity of platelets to inhibitor across species. Syk inhibitors were tested in multiple platelet functional assays in rat, mouse, and human (Figure 10), assessing aggregation, microparticle release, and secretory responses. In all cases, rat platelets were more sensitive to Syk inhibition than mouse and human (Figure 10). The greatest difference in IC90 values of the Syk inhibitor between species was in the thromboxane B2 and 5-HT release assays showing a 10- to 35-fold difference between rat and human (Table 1). The differences in the aggregation and microparticle release responses between species were more modest (Figure 10A and D).
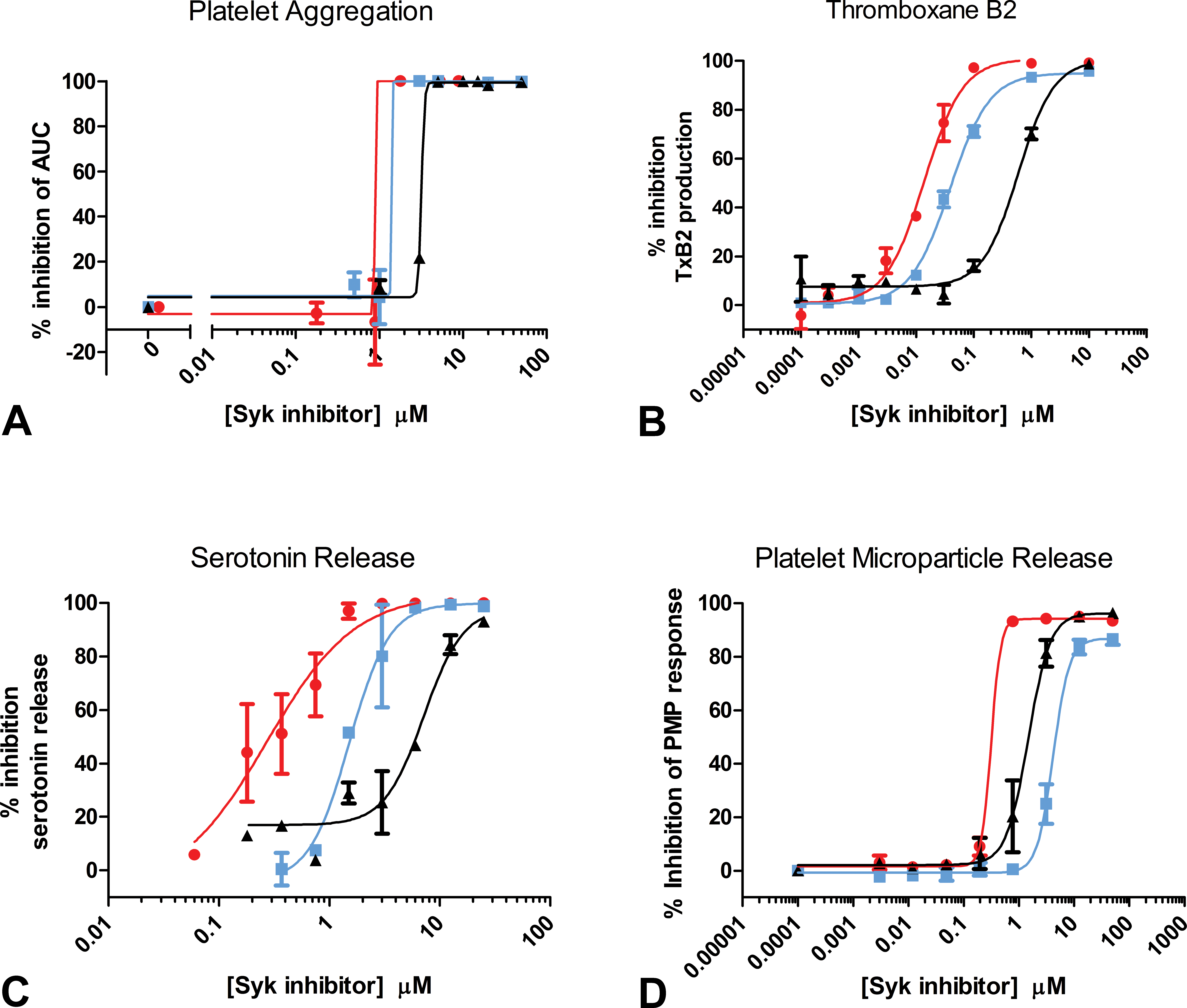
Spleen tyrosine kinase inhibitor responses in multiple platelet functional assays. The effects of Compound 1 were tested in whole blood platelet aggregation (A), thromboxane B2 release (B), serotonin release (C), and micro particle release (D) in response to 0.5 ug/ml convulxin. Assays tested in rat (red), mouse (blue), and human (black). Data presented are from combined independent experiments (n = 2). Percentage inhibition was calculated, comparing blockade of Syk inhibitor to baseline (no stimulation). Syk inhibitors were able to completely inhibit convulxin-induced responses.
Discussion
We characterized a unique peri-islet microvascular lesion in the pancreas of rats that was most likely caused by inhibition of the nonreceptor tyrosine kinase Syk. Inhibition was associated with peri-islet hemorrhage and fibrin deposition. Chronic exposure to Syk inhibition resulted in exocrine pancreatic fibroplasia, loss of alpha, beta, and delta cells, and attenuated islet function. The lesion is likely the result of perturbation of the microvascular endothelium leading to fibrin deposition, fibroplasia, and loss of islet function. We have shown that the effect of Syk inhibitors is not a direct effect on the islet, given the absence of Syk expression in the islet or surrounding endothelium but rather an indirect effect through inhibition of Syk signaling in platelets. We recapitulated data from Gude et al. showing that thrombocytopenia induced via irradiation resulted in the formation of a phenotypically similar lesion and that platelet reconstitution with PRP was able to prevent formation of the lesion. This protection could be reversed by the systemic administration of a Syk inhibitor, thus implicating Syk in platelets as an important factor in maintaining microvascular homeostasis in the pancreas of rats.
Platelets are known to play a critical role in vascular integrity and endothelial permeability. Induced thrombocytopenia has been shown to result in disruption of microvascular permeability (Lo et al. 1988), with thinning and fenestration of the endothelium (Kitchens and Pendergast 1986). Signaling through receptors upstream of Syk in platelets has been implicated in vascular integrity as well. Individuals that are deficient in GPVI have a mild bleeding phenotype (Kojima et al. 2006). This is consistent with recent reports that demonstrate that platelet GPVI is important for modulation of vascular permeability and repair in the context of inflammatory challenge (Gros et al. 2015). Mice deficient in Syk have a platelet defect that results in developmental perturbations of the vascular/lymphatic interface, petechial hemorrhage, and perinatal mortality (Finney et al. 2012). Administration of Syk inhibitors, however, does not result in a decrease in platelet count or a change in bleeding time, suggesting that platelet functional changes occur independent of an effect on the clotting cascade or thrombopoiesis.
Platelets store numerous proteins, lipids, and other vasoactive mediators in α-granules, dense granules, and lysosomes that are released upon adhesion receptor activation (Smyth et al. 2009). Via these mediators, platelets influence endothelial cell proliferation, survival, and barrier function (Ho-Tin-Noe et al. 2011). A recent example is the demonstration that interaction between platelet CLEC-2 and lymphatic endothelial podoplanin maintains high endothelial venule (HEV) microvascular integrity (Herzog et al. 2013). Podoplanin was expressed in the fibroblastic reticular cells surrounding the HEV. This interaction resulted in the release of S1P, which was required for the expression of vascular endothelial cadherin (VE-cadherin) in the HEV adherens junctions. Platelets deficient in S1P were unable to maintain the integrity of the microvasculature in the HEV. The contribution of CLEC2/podoplanin and GPVI in platelets implicates a Syk-dependent pathway in the maintenance of microvascular integrity. The data presented in this report suggest that the factor responsible for endothelial integrity is not a long-lived soluble factor, given that thrombocytopenic rats reconstituted with PPP could not protect the islets from the hemorrhage while PRP could (Figure 6A). This does not, however, preclude the release of a local or short-lived soluble mediator.
In addition to the role of platelets in the lesion formation, another question that arises from these studies is why the rat appears to be the most sensitive species for the formation of the peri-islet lesion. This is of particular importance in the development of inhibitors for clinical use, given that the most sensitive toxicological species is used to assess human risk. In toxicological studies performed in rat, mouse, dog, and cynomolgus monkeys, the rat is the only species in which the pancreatic lesion was observed, similar to what has been previously described for an undisclosed test article (Brenneman et al. 2014). While we found no major Syk expression differences in platelets from different species, there were functional differences in the Syk-dependent GPVI pathway. As mentioned, platelets and platelet lysates have been shown to modulate the permeability of endothelial cultures. Many factors derived from platelets have been shown to exert these effects. Multiple platelet function assays were used to determine if specific components or activities of platelets were responsible for the species sensitivity. The GPVI agonist convulxin was used to elicit aggregation responses, 5-HT release (dense granule release), thromboxane B2 release (positive feedback for primary hemostasis), and microparticle release. Microparticles are small cellular fragments containing bioactive molecules that are released from platelets after activation that have direct effects on endothelium (Ho-Tin-Noe et al. 2011). There was a striking species difference in the platelet aggregation response with rats (Figure 9B and C) showing inherent differences in platelet function in a pathway dependent on Syk signaling. These differences support the hypothesis that the primary hemostatic defect with Syk inhibition in rats involves aberrant interactions between platelets and endothelial cells. In addition, the Syk inhibitors were more potent in rat platelet functional assays compared with other species. This alone is unlikely to explain the rat sensitivity, because the magnitude of the potency difference in the in vitro platelet assay is much less than the difference in circulating drug concentrations observed in the in vivo toxicology studies. This is especially true in the mouse, where the drug concentrations were over 100-fold higher than the rat. The difference in species sensitivity is likely due to a combination of the inherent functional differences and the sensitivity of rat platelets to inhibitor.
Species differences in peri-islet microvasculature may also be a reason for the rat sensitivity to platelet perturbation. We have shown that rats can be protected from Syk inhibitor-induced lesion with the coadministration of prednisone (Figure 7). Kitchens and Pendergast (1986) demonstrated that endothelial structural changes observed with thrombocytopenia in humans could be reversed after treatment of glucocorticoid. This is most likely a direct effect on the endothelium, given the lack of platelets. Glucocorticoids have been shown to increase the expression of endothelial tight junction proteins such as occludin and cadherins (Salvador, Shityakov, and Forster 2014), suggesting a potential mechanism. The protective effect of steroids in the current study suggests that prednisone is sufficient to compensate for the lack of a Syk-dependent factor derived from platelets and that this may be through a direct effect on the endothelium. All vasculature structures are not the same. The peri-islet microvasculature may have distinct properties (e.g., receptor expression, adhesion molecules) that confer sensitivity to platelet-derived factors or interactions. Differences in phenotypes of macro- and microvasculature have been demonstrated particularly in cardiac vascular beds (Edelberg, Christie, and Rosenberg 2001). Studying platelet interactions with endothelium through adhesion under flow, platelet spreading, or endothelial permeability assays may enhance the understanding of the specific relationship. Including the pancreatic microvasculature endothelium in these types of studies will be critical, given that this vascular bed appears to be most sensitive to perturbation with Syk inhibition.
In addition to induced thrombocytopenia, similar lesions in the rat have been induced by an undefined test article (Brenneman et al. 2014) and by busulfan, an alkylating antineoplastic agent (Kaduk et al. 1987). The mechanism of the busulfan-induced lesion had been hypothesized to be local accumulation of the drug in pericapillary pericytes (Brenneman et al. 2014; Kaduk et al. 1987). Induction of thrombocytopenia is a known effect and a common experimental use of busulfan (Kitchens and Weiss 1975). It is possible that the lesion is secondary to the thrombocytopenia induced by the drug rather than a direct effect of the drug on the islet. Kitchens et al. administered busulfan to rabbits and showed structural changes in the endothelium that were secondary to thrombocytopenia. Brenneman et al. (2014) also described a test article–related pancreatic lesion specific to the rat that followed a similar time course of induction. In that study, 4 to 7 days of test article treatment did not result in pancreatic dysfunction as measured by OGTT. The results shown here are consistent with that observation with no significant functional deficit after 7 days of dosing with a Syk inhibitor. When a Syk inhibitor was dosed beyond 7 days, however, significant changes in blood glucose after OGTT were observed at 14 days and progressed through day 42. This suggests cumulative damage leading to compromised pancreatic function as a result of Syk inhibition. Brenneman et al. hypothesized that the lesion was most likely due to off-target effects, based on a lack of (undisclosed) target expression in the pancreas. Our studies (a) report that Syk is a molecular target whose inhibition results in the pancreatic lesion, (b) confirm the lack of target (Syk) expression in the pancreatic islet and accompanying endothelium, and (c) indicate the effect occurs through a functional defect in platelets.
Although the mechanism by which Syk blockade in platelets results in peri-islet toxicity remains to be fully elucidated, our findings are limited to rats. The key question in the development of Syk inhibitors for use in human is the relative risk of developing the pancreatic lesion. There is evidence to suggest that the pancreatic lesion may not be relevant to humans. Humans that are chronically thrombocytopenic with idiopathic thrombocytopenia purpura, for example, do not exhibit any signs of pancreatic dysfunction or induced diabetes. Additionally, busulfan has been used clinically for decades without any reported incidences of diabetes or pancreatic insufficiency. In the absence of a translational biomarker that provides a means to monitor the formation of the lesion in humans, further study will be needed to identify the exact mechanism responsible for the maintenance of the pancreatic microvasculature. The identification of the platelet as an important component of the mechanism enables future translational studies. Additional understanding of whether the rat is uniquely susceptible to this mechanistic pancreatic lesion may enable a path forward for the appropriate assessment of human risk with Syk inhibitors.
Footnotes
Authors’ Note
All authors are employees of AbbVie. AbbVie participated in the interpretation of data, review, and approval of the publication.
Acknowledgments
The authors would like to dedicate this article to the memory of Marc Barnard who passed away during its preparation. Marc’s enthusiasm and dedication to science will be missed. The authors are grateful for the assistance of Karin Orsi, Sarah Heighton, Christine Nelson, and Stephanie Gaudette for the immunohistochemistry and histology, David J. Desmond for the clinical pathology assays, and Tatiana Sharapova and Rita Ciurlionis for biomarker exploration.
Author Contributions
Authors contributed to conception or design (AL, ES, RM, CH, MB, DS, DC, RC, DH, NW, MH, LD, DF, CS, EO, IM, AS); data acquisition, analysis, or interpretation (AL, ES, RM, CH, MB, DS, DC, RC, DH, NW, MH, LD, DF, CS, EO, IM, AS); drafting the manuscript (AL, ES, RM, CH, MB, DS, RC, IM, AS); and critically revising the manuscript (AL, ES, RM, CH, MB, DS, DC, RC, DH, NW, MH, LD, DF, CS, EO, IM, AS). All authors gave final approval and agreed to be accountable for all aspects of work in ensuring that questions relating to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The design, study conduct, and financial support for this research were provided by AbbVie.