
Abstract
Protein phosphatases (PP) are interesting drug targets. However, their ubiquitous presence and involvement in different, partially opposing signal pathways suggest that specificity may be achieved rather by targeting their interaction with subunits determining substrate specificity than the enzyme itself. An interesting subunit is phosphatase inhibitor-1 (I-1), which, in its protein kinase A–phosphorylated form (I-1P), inhibits the catalytic subunit of type 1 phosphatase (PP1c). In the current study, we established a colorimetric and a fluorescence-based assay system for the identification of compounds interfering with the inhibitory effect of I-1P on PP1c. The fluorescence assay exhibited 500-fold higher sensitivity toward PP1c. A nine-residue peptide containing the PP1c-binding motif (RVxF) of I-1 stimulated PP1c activity in the presence of I-1P (EC50 27 µM and 2.3 µM in the colorimetric and fluorescence assay, respectively). This suggests that the peptide interfered with the inhibitory effect of I-1P on PP1c and represents a proof-of-principle. The calculated Z′ factor for PP1c (0.84) and the PP1c–I-1P complex (0.73) confirmed the suitability of the fluorescence assay for high-throughput screenings (HTS). By testing several thousand small molecules, we suggest the advantages of kinetic measurements over single-point measurements using the fluorescence-based assay in an HTS format.
Introduction
Reversible protein phosphorylation is a major mechanism regulating the physiology of plant and animal cells. Steady-state phosphorylation reflects the balance between protein kinases and phosphatases. Protein phosphatase type-1 (PP1) is one of the major eukaryotic serine/threonine phosphatases, which regulates many biological processes, including synaptic plasticity, cell cycle, gene transcription, and metabolism. 1 The functional diversity of PP1 is accomplished by the association of its catalytic subunit with more than a hundred endogenous regulatory subunits. 2 One of the longest recognized regulators of PP1 is inhibitor-1 (I-1) that selectively and potently inhibits the PP1 catalytic subunit (PP1c, IC50 1–2 nM) only in its protein kinase A (PKA)–phosphorylated form (I-1-Thr35P). 3 I-1 and its neuronal homologue, dopamine- and cAMP-regulated phosphoprotein of 32 kDa (DARPP-32), belong to the family of intrinsically unstructured proteins (IUPs). 4 This could explain their extraordinary properties as acid- and heat-stable proteins. The cAMP-dependent activation of I-1 indicates its pivotal role in hormonal regulation of PP1. 5 In the heart, the involvement of I-1 in the β-adrenergic system was demonstrated by its hyperphosphorylation after stimulation with isoprenaline. 6 The interaction of phosphorylated I-1 (I-1P) with PP1c in the heart prevents dephosphorylation of several phosphoproteins, prolongs and amplifies β-adrenergic signaling, and subsequently increases cardiac output. 7 Out of several PP1-sensitive phosphoproteins, I-1 appears to specifically affect dephosphorylation of phospholamban (PLB) and ryanodine receptors (RyR) regulating sarcoplasmic reticulum Ca2+ uptake and release, respectively.7,8 I-1 expression and phosphorylation was shown to be decreased in heart failure similar to the downregulation of β1-adrenoceptors,8,9 suggesting that it contributes to the well-known desensitization phenomenon of β-adrenergic signaling in failing hearts and failing cardiomyocytes. Whereas some investigators provided evidence that restoration of high I-1 levels has beneficial effects under cardiac stress conditions, 10 we observed that genetic ablation of I-1 protected mice against acute and chronic catecholamine stimulation without affecting heart rate. 11 Conversely, overexpression of a truncated constitutively active form of I-1 improved cardiac output in the short term but exaggerated contractile dysfunction after chronic catecholamine infusion as well as spontaneously in aging mice. 12 These findings suggest that pharmacological I-1 blockade may represent a new strategy in treating heart failure.
Current assays for measuring PP1 activity, either as free catalytic subunit or as holoenzyme, are based on PP1c-mediated dephosphorylation of radioactively labeled phosphoproteins, particularly phosphorylase a.3,13 Those assays are relatively expensive, require facilities for radioactive work, and are not suitable for high-throughput drug screening. The general preference for nonradioactive high-throughput assays has provided an immense challenge for many investigators. Parker et al. 14 and Rodems et al. 15 presented a fluorescence polarization (FP)–based assay and a fluorescence resonance energy transfer (FRET)–based assay, respectively, for the detection of kinase and phosphatase activities. More recently, a high-throughput phosphatase assay was described using rhodamine 110–modified phosphopeptide substrates. 16 Although each of these systems is amenable for high-throughput screening (HTS), higher selectivity and reliability are still required, particularly for assessment of serine/threonine phosphatase activities. Moreover, the available phosphatase assays monitor the activity of the catalytic subunit of phosphatases alone, whereas serine/threonine phosphatases normally exist in a nonmonomeric form. Thus, assays are needed that are composed of both catalytic and regulatory subunits.
Here we developed a nonradioactive, reliable, and cost-efficient phosphatase-inhibitor assay and screened several thousand compounds for their effect on PP1c–I-1 interactions. By employing a chromogenic and a fluorogenic substrate we identified a small peptide that potently interfered with the inhibitory effect of I-1P on PP1c.
Materials and Methods
Colorimetric and Fluorescence Phosphatase Assays
The colorimetric assays were performed in transparent flat-bottom 96-well plates (Sarstedt AG, Nümbrecht, Germany) in a final volume of 220 µL. The hydrolysis of pNPP (Calbiochem, Darmstadt, Germany) generates a colored compound (pNP) that can be monitored by absorption at 405 nm. A stock solution of 50 mM pNPP was prepared freshly in the assay buffer containing 25 mM imidazole (pH 7.4), 1 mM dithiothreitol (DTT), 0.1 mg/mL bovine serum albumin (BSA), and 50 mM NaCl. The recombinant PP1c from rabbit skeletal muscle (specific activity 8.151 U/µg protein; Sigma-Aldrich, Steinheim, Germany) was applied in the assay. One unit hydrolyzed 1 nmol of pNPP per minute at pH 7.4 at 30 °C according to the manufacturer’s instruction. Enzyme (0.4–1.6 U/well) in a 40-µL enzyme dilution buffer (assay buffer plus 0.3 mM MnCl2) was added to the wells. The reactions were mixed by addition of 160 µL assay buffer and initiated by addition of 20 µL pNPP stock solution, yielding a final concentration of 4.5 mM. The substrate saturation curve was determined in the presence of 1 U PP1c at different substrate concentrations (0.5–50 mM). The absorptions were measured immediately for 30 min at 30 °C using a TECAN Safire 2 (TECAN, Männedorf, Switzerland) multiplate reader and analyzed with the Magellan software 5.03 (TECAN). The gradient of optical density (ΔOD/min) was considered as phosphatase activity for each sample.
The fluorescence assays were carried out in black flat-bottom 384-well plates (Greiner Bio-One, Frickenhausen, Germany) in a final volume of 100 µL. The fluorogenic substrate, DiFMUP (kind gift from Dr. Lars Kattner, Endotherm, Saarbruecken, Germany), was prepared in 50 mM Tris-HCl (pH 7.0) at a 10-mM stock solution and stored at −20 °C. Recombinant PP1c from rabbit skeletal muscle (specific activity 25 U/µg protein; New England Biolabs, Ipswich, MA; 1 unit definition is the amount of enzyme that hydrolyzes 1 nmol pNPP in 1 min at 30 °C) was prepared at 0.5 to 16 mU/well in 50 µL enzyme dilution buffer. Reactions were started by addition of 50 µL assay buffer containing DiFMUP (100 µM), resulting in a final concentration of 50 µM. Substrate saturation curve was obtained using a constant concentration of recombinant PP1c (2 mU) with different concentrations of DiFMUP (5–200 µM). The fluorescence intensities of hydrolyzed DiFMU, as a fluorogenic product (excitation 385, emission 490 nm), were monitored by the TECAN Safire 2 reader for 30 min at 30 °C. The change in relative fluorescence during the measurement time (ΔRFU/min) was considered phosphatase activity. Inhibition assays were performed with PP1c chemical inhibitors, including calyculin A (Enzo Life Science, Farmingdale, NY), okadaic acid (OA; Enzo Life Science), and cantharadin (Sigma-Aldrich), and PP1 protein inhibitors, including inhibitor-2 (I-2; New England Biolabs) and I-1, after preincubation with PP1c for 10 min at room temperature. Preparation and dilution of the inhibitors were carried out in DMSO or enzyme dilution buffer. I-1–derived peptides with the sequences 6PRKIQFTV 14 and 19PHLDPEAAEQI 29 (PANATecs, Heilbronn, Germany) were applied as positive and negative controls for PP1c–I-1P inhibition, respectively. The lyophilized acetate salt of the peptides was solved in H2O in a stock concentration of 10 mM, and aliquots were stored at −80 °C.
PKA Phosphorylation/Thiophosphorylation of Recombinant I-1 Protein
I-1 complementary DNA (cDNA) of mouse heart subcloned in pGEX-2T expression vector, provided by Dr. Katrin Wittköpper, was used to express I-1 as a fusion protein with glutathione-S-transferase (GST) as described earlier. 7 The recombinant I-1 protein was in vitro phosphorylated at Thr35 using the purified catalytic subunit of PKA (PKAc, specific activity ~10 U/µg protein; Sigma-Aldrich) to become activated. Phosphorylation/thiophosphorylation of I-1 protein was performed using a protocol described by Endo et al. 17 The phosphorylation reaction was carried out in phosphate-buffered saline (PBS) at pH 7.4, containing 6 mM MgCl2 (Merck, Darmstadt, Germany) and 0.3 mM adenosine triphosphate (ATP; Sigma-Aldrich) with 1 U PKAc per 1 µg I-1 protein, for 90 min at 30 °C. A control reaction was performed in parallel, containing I-1 and PKAc, without ATP and MgCl2. The reactions were terminated by incubating at 95 °C for 5 to 10 min and desalted in the enzyme dilution buffer using Amicon, 10K devices (Millipore, Billerica, MA) according to the manufacturer’s guideline. Thiophosphorylation of the I-1 protein was performed essentially as described above except that ATP was substituted with 0.3 mM ATP-γ-S (adenosine 5′-[3-thiotriphosphate]), which is a nonhydrolyzable ATP analogue. Instead of 6 mM, 2 mM MgCl2 was used and the reaction was incubated at 30 °C for 24 h. The phosphorylated/thiophosphorylated proteins were stored at −80 °C.
Western Blot Analysis
Recombinant I-1 proteins before and after phosphorylation/thiophosphorylation were prepared with the buffer containing 10 mM Tris-HCl (pH 6.8), 2% sodium dodecyl sulfate (SDS), 10% glycerol, 100 mM DTT, and 0.01% bromophenol blue; heated at 95 °C for 5 min; and loaded on a 12% SDS-containing polyacrylamide gel. The SDS extracts were blotted on nitrocellulose membranes (Protran, Schleicher & Schüll, Dassel, Germany) for 90 min on ice with a constant current of 300 mA. The membranes were incubated with polyclonal antibodies raised against I-1 (1:1000, produced in house as described previously 7 ) and phospho–DARPP-32 (1:1000; Cell Signaling Technology, Danvers, MA) for detection of I-1 protein or phosphorylated/thiophosphorylated level, respectively.
Radioactive Phosphatase Assay
The activities of recombinant PP1c (Sigma-Aldrich) in the presence of I-1P and I-1SPRKIQFTV or I-1PHLDPEAAEQI were determined using [32P]-phosphorylase a as substrate as described previously. 7 After preincubation of PP1c–I-1P complex (0.1 U PP1c + 3 nM I-1P) with the peptide for 20 min at 30 °C, 5 mg/mL [32P]-phosphorylase a was added and the reaction was carried out for 20 min and then terminated by the addition of 50% trichloroacetic acid (TCA). After centrifugation, aliquots of the supernatant were counted in a liquid scintillation counter (Canberra Packard, Meriden, CT).
PP1c–I-1P High-Throughput Screening
Screening of a library of drug and lead-like small molecules from the ChemBionet library 18 in a high-throughput format was performed by the facilities of European ScreeningPort, GmbH (Hamburg, Germany). The reactions were carried out in 384-well plates in a final volume of 10 µL. The PP1c–I-1P mixture was prepared freshly. The components (PP1c and I-1P) at 43.2 pM and 60 nM, respectively, were preincubated for 10 min. A PP1c mixture containing only PP1c was prepared as a control for phosphatase activity in the absence of I-1P. “Assay-ready” plates were produced from compound library stocks (10 mM in 100% DMSO) using acoustic dispensing (Echo 550; Labcyte, Sunnyvale, CA) at 4 to 6 h prior to screening. To each well, 5 µL of the PP1c–I-1P mixture was added and compounds preincubated with the mixture for 15 min at 30 °C. The PP1c activity was initiated by addition of 5 µL assay buffer containing DiFMUP (10 µM stock concentration). Final reaction concentrations were 21.6 pM (2 mU/well) PP1c, 30 nM I-1P, 5 µM DiFMUP, 20 µM test compounds, and 0.5% DMSO. The lidded plates were then incubated at 30 min incubation at 30 °C. The fluorescence intensity (excitation 385 nm, emission 490 nm) was then measured for a further 30 min in kinetic mode (1 read per minute) on an EnVision multimode reader (PerkinElmer, Hamburg, Germany). Time-series data were subject to linear regression analysis (EnVision software) and plate-level statistics (Z′) calculated in the IctivityBase software (IDBS, Guildford, UK). Plates with a Z′ value lower than 0.3 were excluded.
Results
Colorimetric PP1c–I-1P Assay System
In a first step, pNPP was employed as a chromogenic substrate. Originally, pNPP had been used in conjunction with alkaline phosphatases due to its intense hydrolysis in basic milieu, but it is also feasible for the determination of acidic phosphatases, tyrosine phosphatases, and serine/threonine phosphatases. 19 The activity of recombinant PP1c (0.4–1.6 U/well) was monitored in the presence of 4.5 mM pNPP ( Fig. 1A ). The optical density (OD) increased linearly with increasing phosphatase concentrations. PP1c at 1 U/well was used for further experiments. We observed a linear increase in OD between 5 and 30 min ( Fig. 1B ). Substrate concentration-dependency was measured with 1 U PP1c and a 30-min incubation and showed a typical Michaelis-Menten saturation curve with a calculated Km value of 3 mM (GraphPad Prism 5.02; GraphPad Software, La Jolla, CA) ( Fig. 1C ).
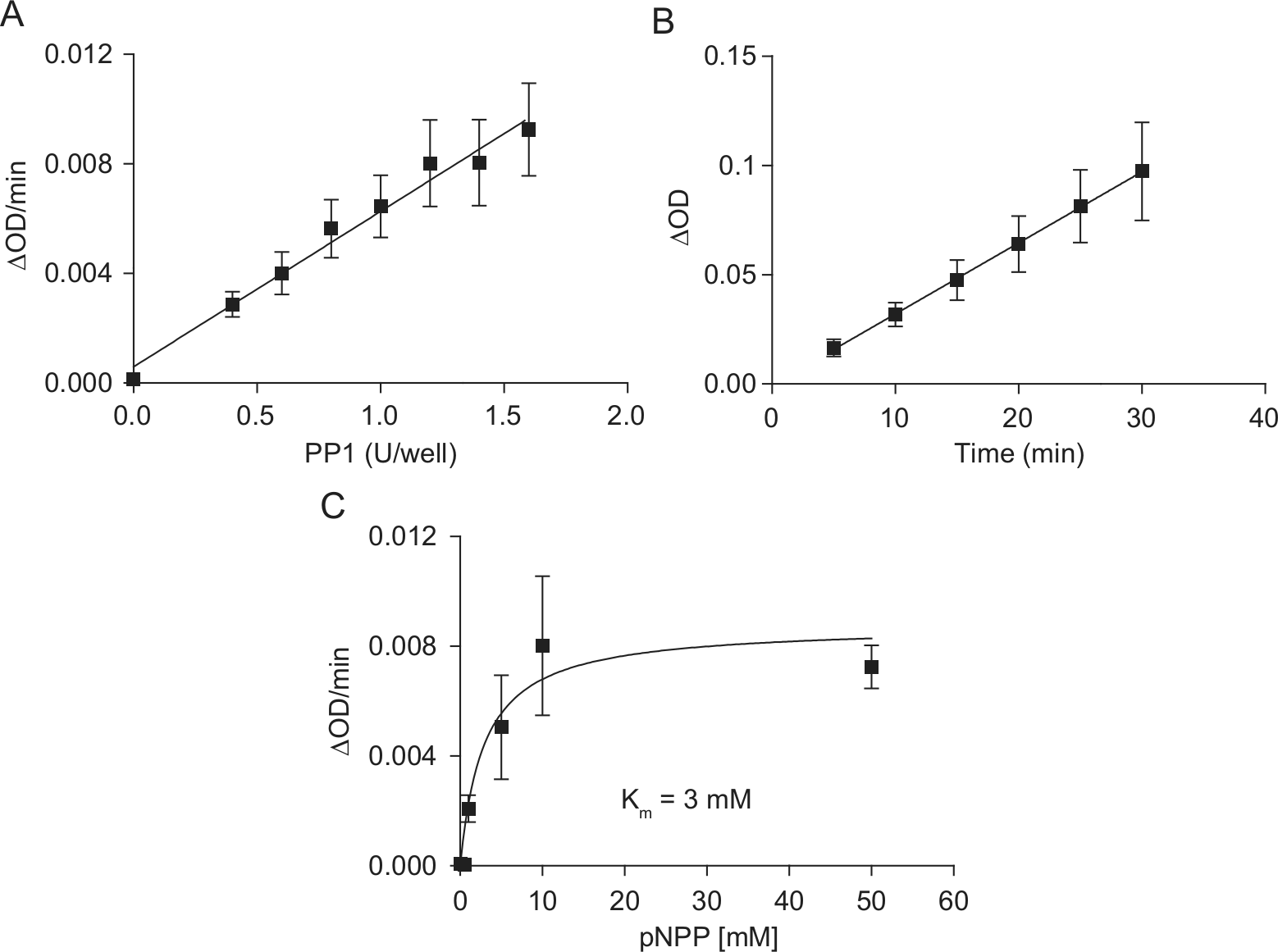
pNPP as a substrate for PP1. (
Next, we compared the inhibitory effects of various PP1c inhibitors to determine the assay pharmacology. I-2 was used as a selective protein inhibitor of PP1c; calyculin A, OA, and cantharidin were used as nonselective chemical inhibitors of PP1c. Concentration-inhibition curves in the presence of 1 U PP1c are shown in Figure 2A – D . The IC50 values obtained were consistent with earlier reports. 20 These data indicated the principal feasibility of the colorimetric assay.
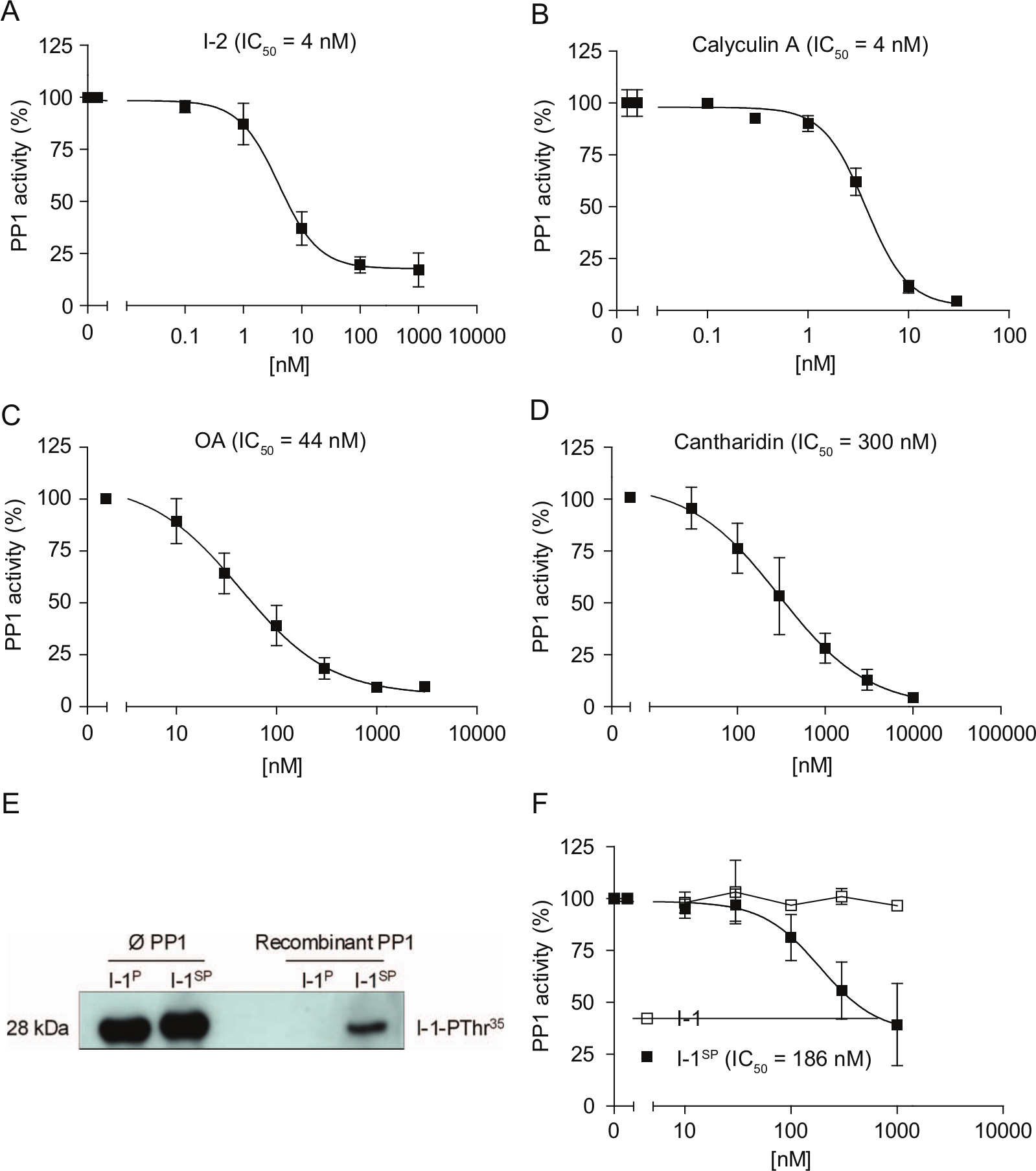
PP1 inhibitors in the colorimetric assay. Concentration-inhibition curves of (
To establish a PP1c–I-1P assay system, we generated recombinant I-1 protein. Since I-1 phosphorylation on Thr35 is essential for its activity against PP1c, I-1 was phosphorylated by PKAc in the presence of ATP. Robust phosphorylation was verified by Western blot with phospho-specific antibodies ( Fig. 2E ). However, in the presence of PP1c, the I-1 phosphorylation state completely disappeared during the 5-min incubation period with PP1c. This indicates that recombinant PP1c at the high concentrations used herein was able to dephosphorylate I-1P. By using the nonhydrolyzable ATP analogue ATP-γ-S instead of ATP (I-1SP), we achieved partial resistance against PP1c-mediated dephosphorylation ( Fig. 2E ). I-1SP inhibited PP1c activity with an IC50 value of 186 nM, whereas nonphosphorylated I-1 was without effect ( Fig. 2F ). The observed IC50 value was ~100-fold higher than values reported for native PP1c and natural substrates (1–2 nM).3,21
Fluorescence PP1c–I-1P Assay System
To circumvent the problem of I-1P-dephosphorylation at the high PP1 concentrations, DiFMUP was used as a fluorogenic substrate. DiFMUP is a derivative of the 4-methylumbelliferyl phosphate (4-MUP) with superior spectral properties to 4-MUP at pH 7.0. 22 At 50 µM DiFMUP and a 30-min incubation, the fluorescence signal increased linearly between 0.5 and 16 mU PP1c/well ( Fig. 3A ). At 2 mU PP1c and 50 µM DiFMUP, the assay was linear between 5 and 30 min ( Fig. 3B ) and showed a substrate concentration-dependency according to a Michaelis-Menten equation with a Km value of 91 µM ( Fig. 3C ).
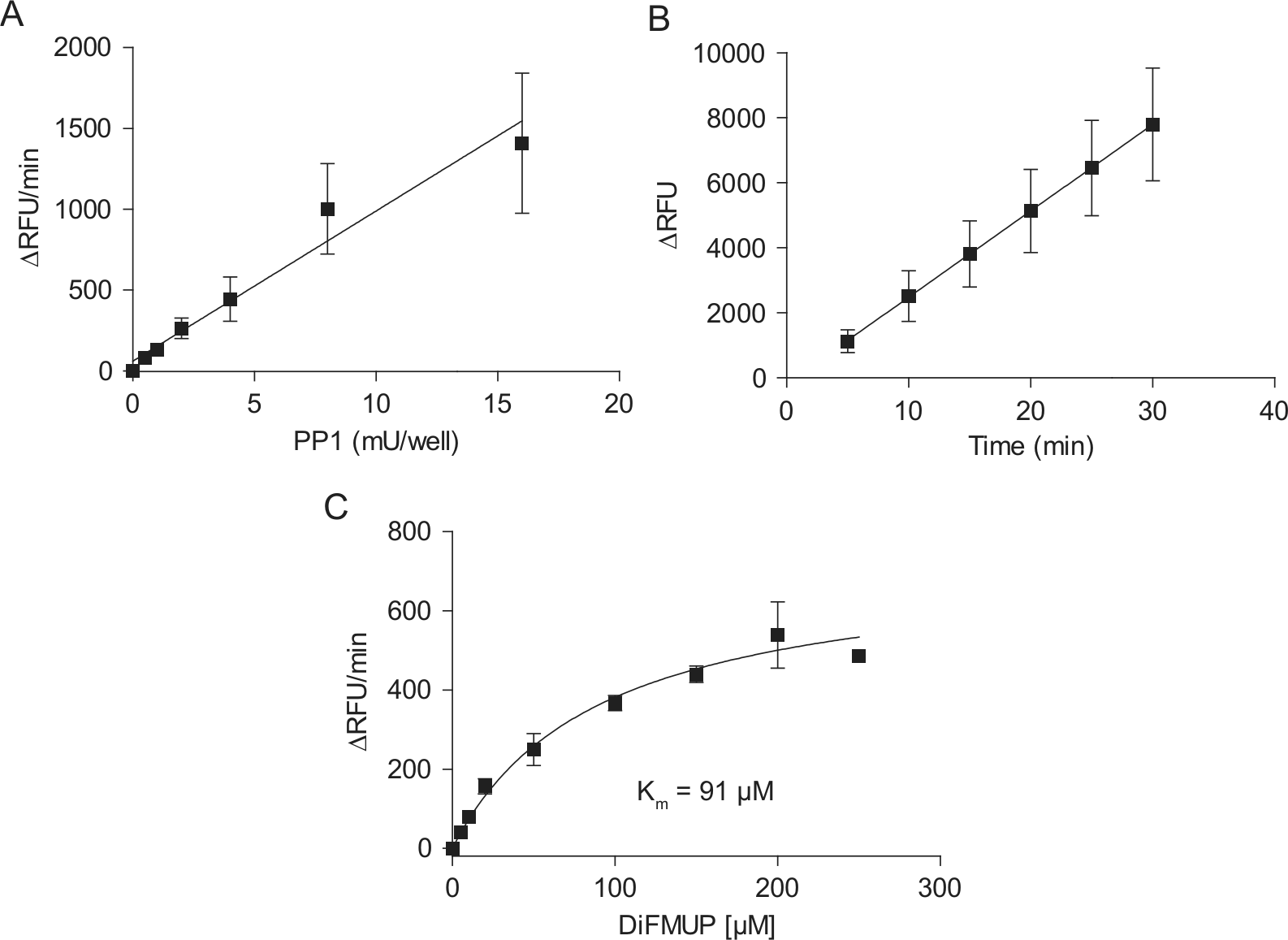
DiFMUP as a substrate for PP1. (
The high activity of PP1c against DiFMUP allowed us to use a 500-fold lower amount of PP1c than in the colorimetric assay (2 mU vs. 1U). Under these conditions, the IC50 values of I-2, calyculin A, and cantharadin were ~30-fold, 8-fold, and ~2-fold, respectively, lower than in the colorimetric assay (
Fig. 4A
,
B
,
D
), whereas that of OA was similar (
Fig. 4C
). I-1P inhibited the low concentration of recombinant PP1c in the fluorescent assay with an IC50 value of 14 nM (
Fig. 4E
). This value was relatively close to the value obtained with native PP1c using phosphorylase a as substrate (1–2 nM). Comparison between 4 mU PP1c from mammalian tissue (native PP1c) and recombinant PP1c exhibited identical inhibition by I-1P (IC50 16 nM vs. 23 nM;
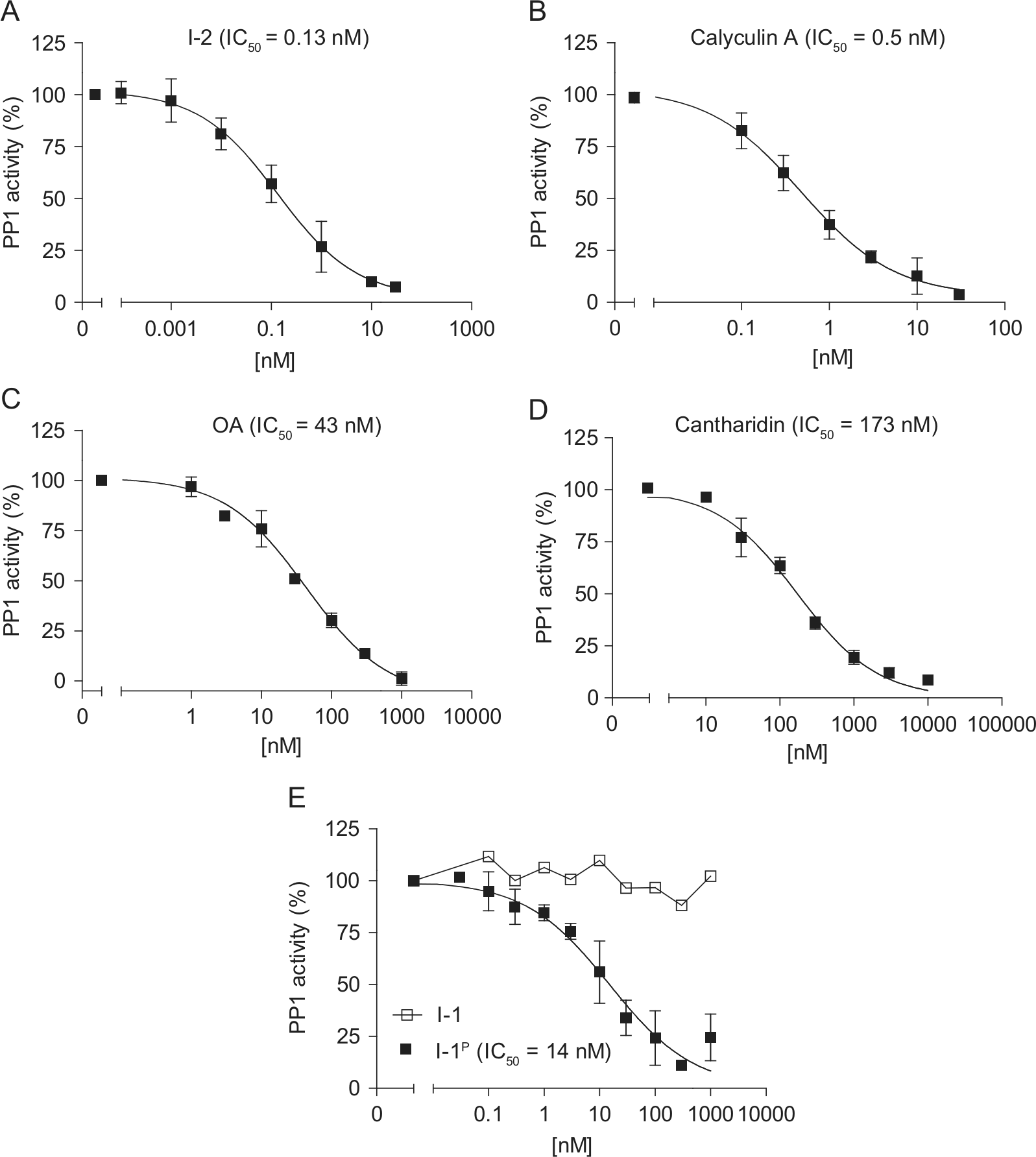
PP1 inhibitors in the fluorescence assay. Concentration-inhibition curves of (
Identification of an I-1 Nonapeptide Interfering with the Effect of I-1P on PP1c
We hypothesized that a peptide that mimics the region of I-1 that binds to PP1c should interfere with the effect of I-1P on PP1c and should exert stimulatory effects in our PP1c-I-1P assay. We therefore designed a nonapeptide derived from the N-terminal domain of I-1 ( 6 SPRKIQFTV 14 ). The peptide contains the KIQF motif, which is essential for I-1 binding to PP1c.3,17 An 11-residue peptide, containing amino acids following the KIQF motif of I-1 ( 19 PHLDPEAAEQI 29 ), was applied as a control peptide. Both peptides were preincubated at different concentrations with PP1c–I-1SP or PP1c–I-1P complexes in the presence of pNPP or DiFMUP, respectively. As expected, I-1SPRKIQFTV concentration-dependently restored PP1c activity with an EC50 value of 27 µM and 2.3 µM in the colorimetric and fluorescence assays, respectively ( Fig. 5A , B ). In contrast, I-1PHLDPEAAEQI did not affect the inhibitory effect of I-1 on PP1c activity. The effect of the peptides was further examined in a radioactive assay using [32P]-phosphorylase a as the substrate. Incubation of the PP1c–I-1P complex (0.1 U PP1c + 3 nM I-1P) with 10 µM of I-1SPRKIQFTV abolished the inhibitory effect of I-1P on PP1c activity, whereas 10 µM I-1PHLDPEAAEQI was not effective ( Fig. 5C ).
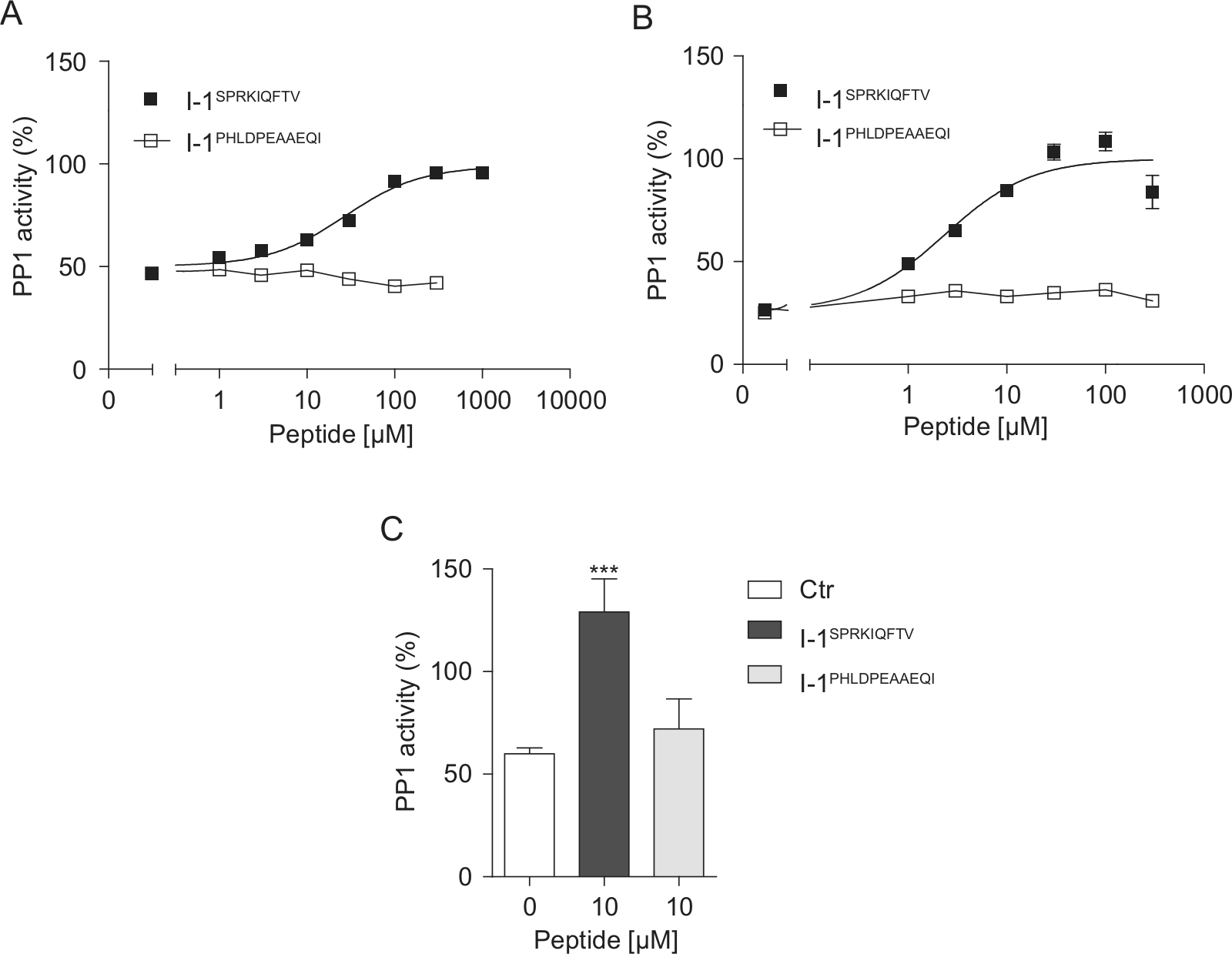
The effect of I-1 peptides on the PP1c–I-1 interaction. Phosphatase activities were monitored after preincubation of various concentrations of I-1SPRKIQFTV or I-1PHLDPEAAEQI with PP1c–I-1 complex in the presence of (
Characterization of the PP1c–I-1P Assay for HTS
The data above indicated the suitability of both colorimetric and fluorescence assays for the identification of PP1c–I-1P inhibitors. However, the higher sensitivity of the DiFMUP assay favored its utilization for HTS. We therefore determined the Z′ factor as a measure of the quality and reliability of assays, particularly for HTS. Determination of Z′ factor was performed by monitoring the fluorescence signals of wells containing buffer without enzyme (blank), PP1c (4 mU/well), or PP1c–I-1P complex (4 mU/well PP1c + 50 nM I-1P) for ~60 min. The assay window demonstrated an appropriate signal dynamic between blank, PP1c, and PP1c–I-1P, which was essential for the detection of compounds with I-1P– or PP1c-inhibitory effect (
A library of 4576 compounds was tested for inhibition of the PP1c–I-1P interaction by adding them to the PP1c–I-1P solution. Kinetic measurements were carried out between 30 and 60 min after reaction start (
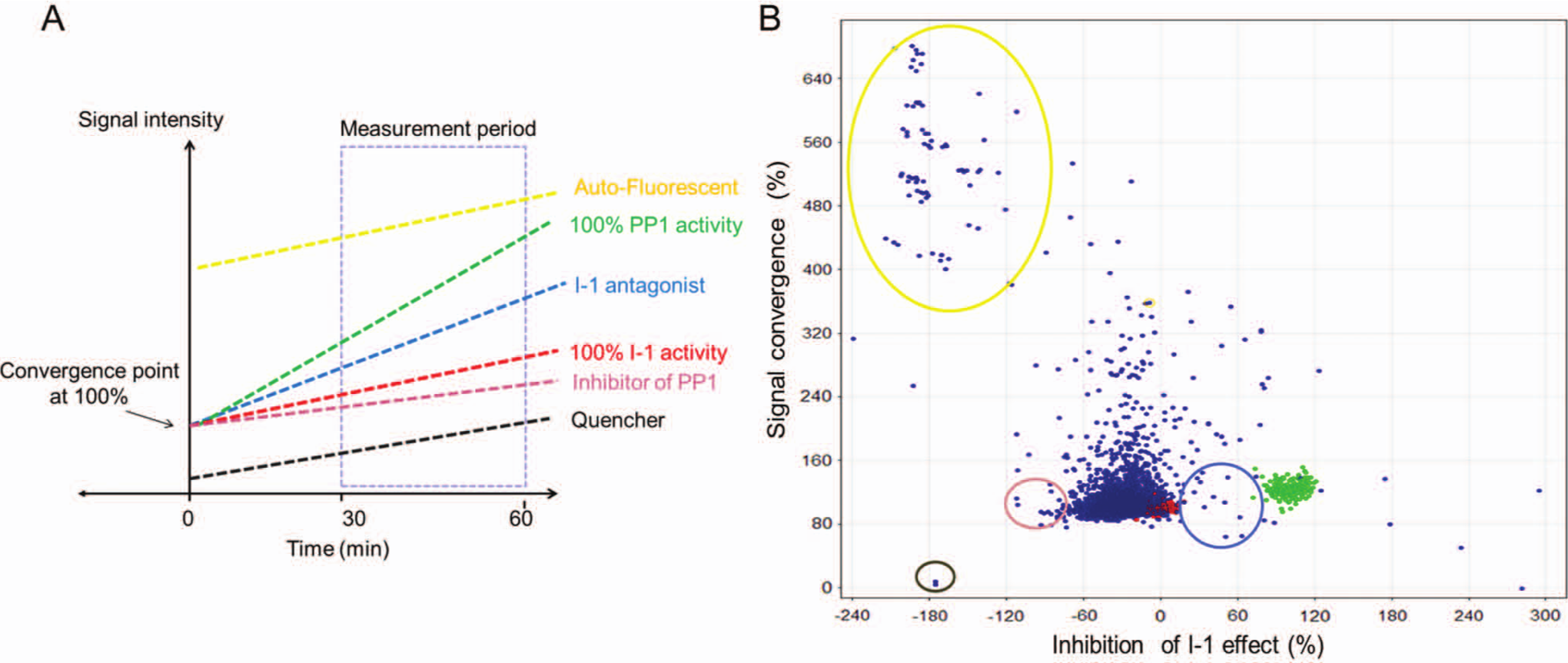
Kinetic measurements of the high-throughput screening (HTS). (
Discussion
Protein phosphatases are notoriously difficult drug targets. 2 Reasons include their involvement in essentially all signaling processes and the observation that specificity is not primarily determined by phosphatase catalytic isoforms but rather their targeting to specific compartments via regulatory subunits. Modulation of PP1 regulatory subunits has become a substantial aim in many disease areas such as diabetes (PP1/hepatic glycogen-targeting subunit [GL]), hypertension (PP1/smooth muscle regulatory subunit [M110]), and Parkinson disease and drug addiction (PP1/DARPP-32).24–26 Recently, salubrinal and guanabenz have been identified as relatively potent inhibitors of the PP1/growth arrest and DNA damage-inducible protein (GADD34) complex.27,28 These molecules target pathways involved in endoplasmic reticulum stress by disruption of the stress-induced dephosphorylation of the α-subunit of translation initiation factor 2 (eIF2α).
Our previous studies with genetically engineered mice indicated that dampening of the intracellular β-adrenergic signaling in the heart by genetic deletion of I-1 might be beneficial for chronic heart disease.8,11,12 The present study established the first assay feasible for HTS for small molecules interfering with the inhibitory effect of I-1P on PP1c. We used PKA-phosphorylated recombinant mouse I-1, recombinant rabbit PP1c, and either pNPP or DiFMUP as substrates. Both are universal phosphatase substrates and commonly used for the determination of phosphatase activities and the effect of natural toxins with the PP1c and PP2Ac inhibitory effect such as microcystin.22,29 The DiFMUP-based fluorescence assay was shown to be clearly superior to the pNPP assay, 500-fold more sensitive, cheap, and robust with Z′ factors (0.84 and 0.73), well above currently accepted limits. Moreover, it compared well with the gold standard radioactive assay with recombinant PP1c and [32P]-labeled phosphorylase a as substrate (
Fig. 5C
). The sensitivity of PP1c to inhibition by I-1P in the DiFMUP assay was similar to values measured previously with the radioactive assay (14 nM vs. 1–2 nM3,21). The small difference may reflect normal variability or could indicate a true difference between recombinant PP1c and native PP1c used in previous studies.6,21 A lower sensitivity of recombinant PP1c against I-1P (compared with fresh native PP1c) was previously described by Endo et al.
17
and is likely due to lack of metals such as Mn2+ in the active site of the enzyme. Despite this difference, recombinant and native PP1c behaved very similar when directly compared in the DiFMUP assay, both in terms of negligible I-1P dephosphorylation when used at a concentration of 4 mU/100 µL and sensitivity to inhibition by I-1P (
Importantly, both nonradioactive assay formats proved useful in characterizing an I-1 nonapeptide as a novel inhibitor of PP1c–I-1P interactions. The peptide was derived from an N-terminal region of I-1 that had been proposed previously to be involved in its interaction with PP1c.3,17 In contrast to a control peptide, the peptide containing the KIQF motif stimulated PP1c activity concentration-dependently when assayed together with the PP1c–I-1P/I-1SP complex (in both assays) and showed reasonable potency (2–30 µM depending on the assay format). The effect of I-1SPRKIQFTV on PP1c activity was further confirmed in the radioactive phosphorylase a assay ( Fig. 5c ). However, the peptides showed no significant effect on PP1c alone (not shown). These results do not formally prove but suggest that the peptide interrupted the protein-protein interaction of I-1P with PP1c. Thus, we regard the peptide as a proof-of-principle for the validity of the assay.
The DiFMUP-based assay was shown to be suitable for an HTS format, but approximately 10% of the investigated compounds showed auto-fluorescence at a magnitude that obviated evaluation. This is a well-known problem of assays based on short-wavelength fluorescence substrates. A much lower fraction of compounds interfered by signal-quenching effects. To circumvent the interfering signals, we performed large-scaled kinetic measurements similar to our original assay format. Based on two calculated factors, gradient and signal convergence at time 0 (T = 0; Fig. 6A ), the compounds could be categorized into neutral, interferer, potential I-1, and PP1c inhibitors in one assay system. Moreover, effective compounds with simultaneous auto-fluorescence activity also could be recognized. Direct effects on PP1c were evaluated in a counterscreen. Beside these merits, a critical point of the kinetic measurements could be a relatively long read time (30 min) per plate compared with the single-point measurements. This may restrict the throughput.
In conclusion, we present a robust, sensitive, and simple phosphatase-inhibitor kinetic assay system suitable for drug discovery purposes. Putative small-molecule antagonists were identified in the primary screen phase using the methods described. However hit confirmation during secondary screens was not successful, reflecting the inherent difficulty in obtaining unequivocal data at the primary HTS. This is associated with the nature of the protein-protein interactions, which mean very few compounds would be expected to be specific hits. Nevertheless, the methodology was sound in that peptide-based compounds produced consistent results. In future studies, we intend to apply the same screening method with larger libraries, including protein-protein interaction compounds and/or in silico screenings to improve the chances of identifying and confirming hits.
Footnotes
Acknowledgements
We thank Jutta Starbatty and Thomas Schulze for excellent technical assistance. DiFMUP was a kind gift from Dr. Lars Kattner, Endotherm, Saarbruecken, Germany.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by the Deutsche Forschungsgemeinschaft (DFG, FOR 604 to A.E.-A. and T.E. and SFB1002 TPA02 to A.E.-A.) and the European Union (EUGene Heart to A.E.-A. and T.E.).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.