
Abstract
Small ubiquitin-like modifier (SUMO1–3) is a small group of proteins that are ligated to lysine residues in target proteins. SUMO conjugation is a highly dynamic process, as SUMOylated proteins are rapidly deconjugated by SUMO proteases. SUMO conjugation/deconjugation plays pivotal roles in major cellular pathways and is associated with a number of pathological conditions. It is therefore of significant clinical interest to develop new strategies to screen for compounds to specifically interfere with SUMO conjugation/deconjugation. Here, we describe a novel high-throughput screening (HTS)–compatible assay to identify inhibitors of SUMO proteases. The assay is based on AlphaScreen technology and uses His-tagged SUMO2 conjugated to Strep-tagged SUMO3 as a SUMO protease substrate. A bacterial SUMOylation system was used to generate this substrate. A three-step purification strategy was employed to yield substrate of high quality. Our data indicated that this unique substrate can be readily detected in the AlphaScreen assays in a dose-dependent manner. Cleavage reactions by SUMO protease with or without inhibitor were monitored based on AlphaScreen signals. Furthermore, the assay was adapted to a 384-well format, and the interplate and interday variability was evaluated in eight 384-well plates. The average Z′ factor was 0.83 ± 0.04, confirming the suitability for HTS applications.
Introduction
Small ubiquitin-like modifiers (SUMO1–3) are conjugated to lysine residues of target proteins and thereby modulate the stability, activity, and subcellular localization of these target proteins. The SUMO conjugation pathway plays pivotal roles in key cellular processes such as gene expression, DNA replication and DNA damage repair, and cell cycle control. 1 The SUMO conjugation pathway is associated with various disorders of high clinical significance, including cerebral ischemia/stroke, heart failure, diabetes, arthritis, cancer, and degenerative diseases.2,3 It is therefore of major clinical interest to develop new strategies to manipulate the SUMO conjugation pathway for preventive and therapeutic purposes.
SUMO is synthesized as a larger precursor that is processed by SUMO-specific proteases (endopeptidase activity) to expose two C-terminal glycine residues that are required for conjugation. SUMO is conjugated to lysine residues of target proteins in a complex process involving activating (E1), conjugating (E2), and ligating (E3) enzymes. SUMOylated proteins are rapidly deconjugated by SUMO proteases (isopeptidase activity). Both SUMO2 and SUMO3 have an internal SUMOylation site and can therefore form polymeric chains. SUMO proteases can perform three distinct functions: process SUMO precursors, remove SUMO from SUMOylated proteins, and depolymerize SUMO polymeric chains. So far, the most fully characterized SUMO proteases are sentrin-specific proteases (SENPs). 4
Recently, several new assays have been described to investigate different aspects of the SUMO conjugation pathway and to screen for small-molecule modifiers. These include Förster resonance energy transfer (FRET)–based assays and AlphaScreen technology-based assays to monitor SUMO protease activities and SUMO-modulated protein-protein interactions.5–7 AlphaScreen technology has been applied to detect SUMOylation, using His-tagged RanGAP1 as the SUMO target and glutathione S-transferase (GST)–tagged SUMO, and to detect the interaction between SUMO and the SUMO-binding motif in protein partners binding to SUMOylated proteins. FRET-based assays were developed to investigate all three functions of SENPs: processing of SUMO precursors, deconjugation of SUMOylated proteins, and depolymerization of SUMO polymeric chains conjugated to target proteins.
Here, we present a new, highly sensitive, high-throughput screening (HTS) compatible assay to search for small-molecule SUMO protease inhibitors. The assay is based on the AlphaScreen technology and uses SENP2 as the SUMO-specific protease and His-SUMO2 conjugated to Strep-SUMO3 as the SENP substrate.
Materials and Methods
DNA Constructs and Bacterial SUMOylation System
Two vectors were used to generate the SENP2 substrate His-SUMO2(K11R) conjugated to Strep-SUMO3ΔGG (SS3HS2). The vector pCOLA-E1/E2/His-SUMO2(K11R) was constructed by replacing the SUMO1 gene with the mouse SUMO2(K11R) sequence and inserting it into the pCOLA-E1/E2/6HisS1 vector (kindly provided by Dr. Verger, IRI USR 3078 CNRS, Parc de la Haute Borne, Villeneuve d’Ascq, Cedex, France). The K11R mutation was introduced using the QuikChange site-directed mutagenesis kit (Stratagene, Santa Clara, CA). The vector pET51b-Strep-SUMO3ΔGG was generated by inserting the mouse SUMO3ΔGG fragment into the bacterial expression vector pET51b (EMD Millipore, Billerica, MA). Bacterial production of His-SUMO2(K11R) conjugated to Strep-SUMO3ΔGG was performed as described elsewhere, 8 with minor modifications. Briefly, constructs pCOLA-E1/E2/His-SUMO2(K11R) and pET51b-Strep-SUMO3ΔGG were co-transformed into competent BL21 (DE3) Escherichia coli (New England Biolabs, Ipswich, MA), and colonies were selected with carbenicillin (50 µg/mL) and kanamycin (30 µg/mL). Three fresh colonies were picked and inoculated into 100 mL LB medium for overnight culture at 37 14;°C. This culture was then used as starter culture to inoculate 10 L LB medium. When the culture had reached an OD600 of 0.6, isopropyl β-D-1-thiogalactopyranoside (0.1 mM) was added to induce expression of proteins, and bacterial growth continued overnight at 20 14;°C. To enhance the SUMOylation reaction, the culture was grown for an additional 2 h at 25 14;°C. Bacteria were then harvested by centrifugation and stored at −80 14;°C until use.
Protein Purification
The strategy for large-scale purification of substrate SS3HS2 is illustrated in Figure 1A . The bacterial pellet from the 10-L culture was resuspended in 400 mL of binding buffer (20 mM Tris-HCl [pH 8.0], 500 mM NaCl, 20 mM imidazole, 1 mM phenylmethanesulfonyl fluoride). Cells were lysed using a cell cracker (Microfluidics, Newton, MA), and lysates were cleared by centrifugation at 10 14;000 g for 1 h. The cleared lysates were incubated with 6 mL Ni-NTA agarose (Qiagen, Valencia, CA) for 2 h at 4 14;°C with gentle shaking on a platform shaker and subsequently transferred to a gravity-flow column. Beads were washed intensively with 400 mL binding buffer, and bound proteins were eluted with 40 mL binding buffer supplemented with 500 mM imidazole.
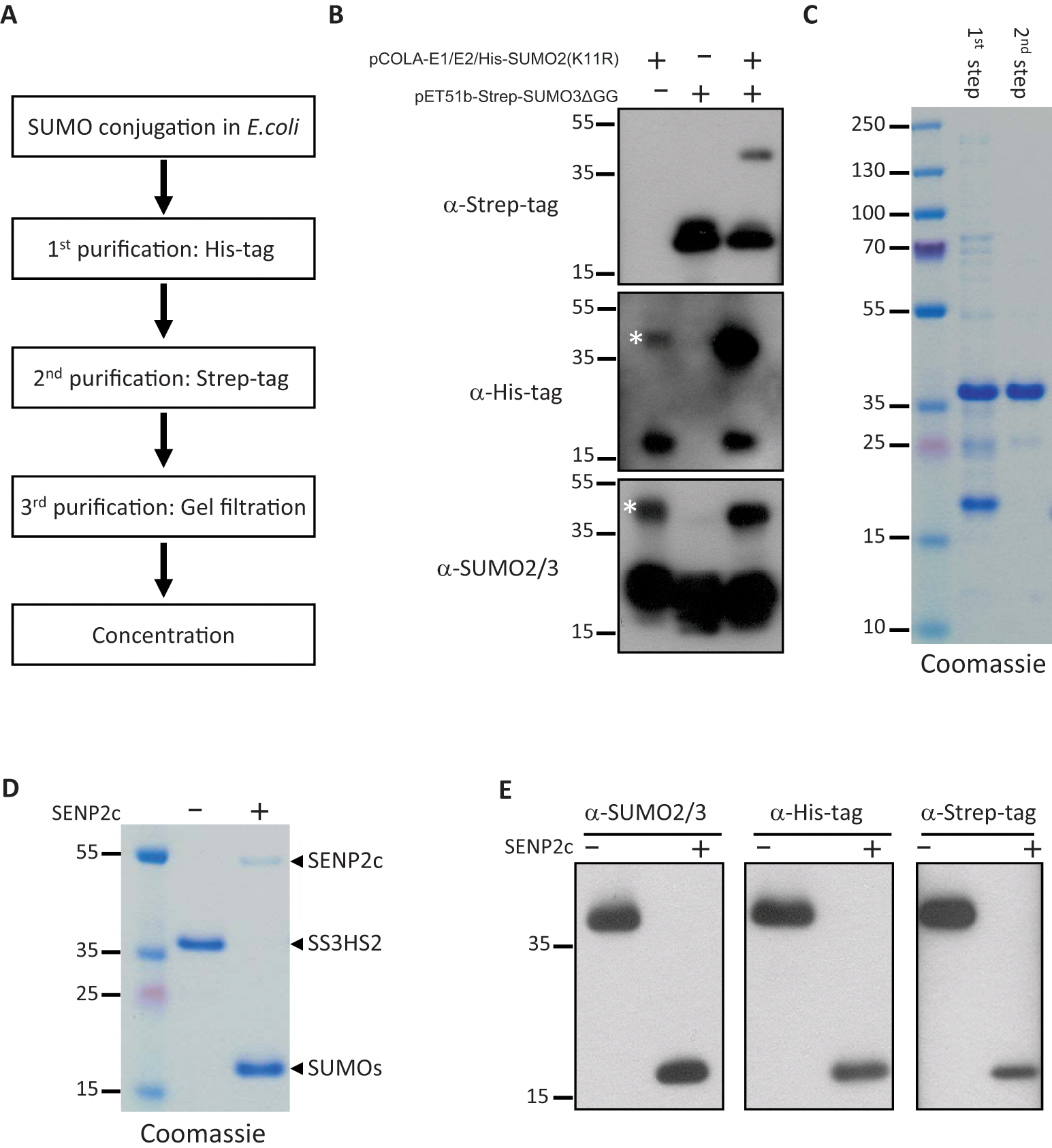
Production of the highly purified substrate SS3HS2. (
For the second purification step, fractions containing most of the substrate protein were combined and applied to a hand-packed gravity column with 10 mL Strep-Tactin superflow resin (IBA, Olivette, MO) at room temperature. After washing with buffer W containing 100 mM Tris-HCl (pH 8.0), 150 mM NaCl, and 1 mM EDTA, protein SS3HS2, featuring both His tag and Strep tag, was eluted in buffer W supplemented with 10 mM desthiobiotin (IBA). Fractions containing SS3HS2, as confirmed by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) analysis, were pooled and concentrated using a 10-kDa cutoff Vivaspin column (Vivascience, St. Louis, MO).
For the third purification step, concentrated SS3HS2 was loaded onto a HiLoad 16/60 Superdex 200 prep grade (GE Healthcare, Pittsburgh, PA) in buffer containing 100 mM Tris-HCl (pH 8.0) and 500 mM NaCl. Fractions containing SS3HS2, as verified by SDS-PAGE analysis, were pooled and concentrated using a 10-kDa cutoff Vivaspin column. Protein concentration of the final SS3HS2 stock was determined by the BCA protein assay (Pierce, Rockford, IL). Glycerol was added to a final concentration of 20%, and the stock was snap-frozen in liquid nitrogen and stored at −80 14;°C.
SDS-PAGE and Western Blot Analysis
To characterize the substrate SS3HS2 in SDS-PAGE gels, SENP inhibitors were preincubated with 10 nM SENP2c (GST-tagged SENP2 catalytic domain; ENZO Life Sciences, Farmingdale, NY) for 30 min at room temperature followed by incubation with 1.5 µM substrate SS3HS2 for 30 min in cleavage buffer containing 100 mM sodium phosphate, 150 mM NaCl (pH 7.2). For titration of SENP2c in the AlphaScreen assay buffer, various concentrations of SENP2c were incubated with 1.5 µM substrate SS3HS2 for 30 min at room temperature in buffer containing 25 mM HEPES (pH 7.4), 100 mM NaCl, 0.1% bovine serum albumin (BSA), and 0.05% Tween-20. Reactions were then stopped by adding sample loading buffer and proteins were separated in 4% to 12% NuPAGE gels and stained with SimplyBlue (Invitrogen, Carlsbad, CA).
For Western blot analysis, after electrophoresis, proteins were transferred to PVDF membranes (Bio-Rad, Hercules, CA). Membranes were then blocked for 1 h in Tris-buffered saline solution supplemented with 0.1% Tween-20 and 5% skim milk powder before incubation with the primary antibodies for 16 h at 4 14;°C. The following antibodies were used: rabbit polyclonal anti-SUMO2/3 (Covance, Denver, PA), mouse monoclonal anti–His tag (Cell Signaling, Danvers, MA), and rabbit polyclonal anti–Strep tag (Genscript, Piscataway, NJ). After extensive washing, blots were incubated with the secondary antibody: goat anti-rabbit or anti-mouse horseradish peroxidase conjugates (Santa Cruz Biotechnology, Santa Cruz, CA) for 1 h at room temperature. Protein bands were visualized using the ECL Western Blotting detection reagents (GE Healthcare).
AlphaScreen Assay Development
AlphaScreen experiments were carried out in white 96-well (PerkinElmer, Santa Clara, CA) or 384-well (Corning, Tewksbury, MA) plates in a total reaction volume of 50 µL (96-well plates) or 20 µL (384-well plates) under subdued light at room temperature. All dilutions and reactions were performed in assay buffer containing 25 mM HEPES (pH 7.4), 100 mM NaCl, 0.1% BSA, and 0.05% Tween-20. AlphaScreen detection reagents, Strep-Tactin donor beads, and Nickel-Chelate acceptor beads (PerkinElmer) were used at a final concentration of 20 µg/mL. The 96-well and 384-well plates were read on EnSpire (PerkinElmer) or PHERAstar HTS (BMG Labtech, Ortenberg, Germany) plate readers, respectively, using standard AlphaScreen settings.
A half-log dilution series of substrate SS3HS2 was used for titration in the 96-well plates to determine the substrate concentration that generated optimal AlphaScreen signals. To each well, 20 µL assay buffer, 10 µL SS3HS2 (0.08 nM–2.528 µM final concentration), and 10 µL acceptor beads were sequentially added. After incubating for 30 min at room temperature, 10 µL donor beads was added to generate the AlphaScreen signal, which was recorded 30 min later.
For titration of SUMO protease SENP2c, 10 µL of the substrate SS3HS2 (400 nM final concentration) was mixed with 10 µL assay buffer and 10 µL SENP2c (0.125–16 nM final concentration) and incubated at room temperature for 30 min. Then, 10 µL acceptor beads was added, plates were incubated for 30 min, 10 µL donor beads was added, and plates were incubated for an additional 30 min before recording the AlphaScreen signal. The same strategy was used to evaluate the efficacy of N-ethylmaleimide (NEM, 0.32 µM–5 mM; Sigma-Aldrich, St. Louis, MO) to inhibit SENP2c and to evaluate DMSO (0.25%–5%) tolerance. NEM was preincubated with SENP2c for 30 min before adding the substrate.
To investigate the robustness and quality of our new assay system, the assay was adapted for the 384-well format and the Z′ factor was calculated from eight plates performed on 3 different days. To the wells, 5 µL of the substrate SS3HS2 (400 nM final concentration) was added followed by 5 µL SENP2c (4 nM final concentration) or 5 µL assay buffer. After incubating for 30 min at room temperature, the AlphaScreen signal was recorded, as described above, with 5 µL each of donor and acceptor beads. These assays were run in the presence of 0.1% DMSO. The Z′ factor was calculated using the equation described by Zhang et al. 9 : Z′ factor = 1 – 3 × (σH + σL)/|µH – µL|, where σH and σL represent the standard deviations of the AlphaScreen signal of the substrate in the absence (high signal, H) and presence (low signal, L) of the protease, and µH and µL represent the mean values of high signal and low signal, respectively.
Data Analysis
All data analyses were performed with Prism 6 (GraphPad Software, La Jolla, CA). All data are presented as mean ± SD.
Results and Discussion
To take advantage of the high sensitivity and low background interference of AlphaScreen technology, we designed an assay to identify inhibitors of SUMO proteases ( Fig. 2 ). First, we decided to use a physiological substrate (i.e., a SUMO-conjugated protein). Compared with some fluorogenic substrates such as SUMO-AMC, a SUMO-conjugated protein as substrate is more physiologically relevant and offers great potential for identifying specific inhibitors of the isopeptidase activity of a SUMO protease.
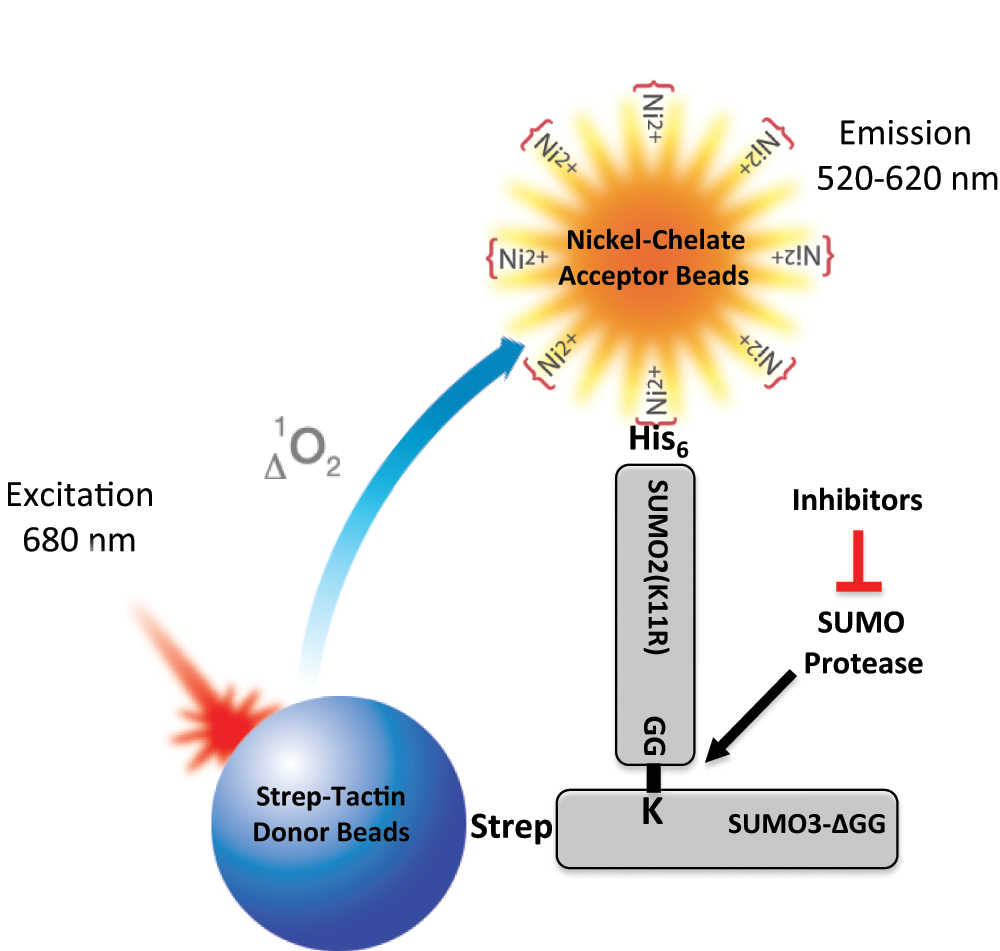
Principle of the AlphaScreen-based small ubiquitin-like modifier (SUMO) protease inhibitor assay. In this assay format, His-SUMO2(K11R) conjugated to Strep-SUMO3ΔGG (SS3HS2) is used as the SUMO protease substrate. When substrate SS3HS2 is mixed with Strep-Tactin donor beads and Nickel-Chelate acceptor beads, donor beads and acceptor beads bind to Strep-SUMO3ΔGG and His-SUMO2(K11R), respectively. Upon laser excitation, donor beads generate singlet oxygen that diffuses to and activates acceptor beads when in close proximity, resulting in signal emission. When the substrate is cleaved by SUMO protease, the distance between His tag and Strep tag is increased, resulting in a marked decrease in signal intensity due to a short lifetime of singlet oxygen in aqueous solutions. SUMO protease inhibitors block de-SUMOylation and thereby maintain signal intensity in the presence of SUMO protease.
The bacterial in vivo SUMOylation system developed by Uchimura et al. 8 makes it possible to produce large quantities of SUMO-conjugated proteins. Initially, we sought to generate polySUMO2/3 chains in bacteria by using GST-SUMO3 and His-SUMO2. However, we experienced two problems with this design. First, GST tag dimer formation rendered it difficult to obtain pure polySUMO2/3 due to co-purifying GST-SUMO3. Second, owing to the nature of the bacterial SUMOylation system, it is almost impossible to control the distribution of different polySUMO2/3 chains. To overcome these problems, we replaced the GST tag with the Strep tag for SUMO3, mutated the internal SUMOylation site of SUMO2 to SUMO2(K11R), and deleted the C-terminal GG of SUMO3. In this way, we were able to obtain only His-SUMO2(K11R) conjugated to Strep-SUMO3ΔGG, a substrate we named SS3HS2. Accordingly, Strep-Tactin donor beads and Nickel-Chelate acceptor beads were used to generate AlphaScreen signals.
The strategy for large-scale production and purification of substrate SS3HS2 is illustrated in Figure 1A . It involves conjugation of His-SUMO2(K11R) to Strep-SUMO3ΔGG in E. coli co-transformed with vectors to express tagged SUMO paralogues together with the heterodimeric activating enzyme SAE1/SAE2 (E1) and the conjugating enzyme Ubc9 (E2). To evaluate our vector system, E. coli were transformed with pCOLA-E1/E2/His-SUMO2(K11R), pET51b-Strep-SUMO3ΔGG, or both vectors. Substrate SS3HS2 formation was verified by Western blot analysis using antibodies against Strep tag, His tag, or SUMO2/3 ( Fig. 1B ). Co-transformation with both constructs resulted in formation of substrate, as indicated by the band above 35 kDa on Strep tag, His tag, and SUMO2/3 Western blots. We also observed a faint band indicated by an asterisk, on His tag and SUMO2/3 Western blots in protein extract from bacteria transformed only with pCOLA-E1/E2/His-SUMO2(K11R). We confirmed by Western blot analysis with Ubc9 antibody that this band results from Ubc9 conjugated with His-SUMO2. After the second purification step, this faint band disappeared (data not shown), indicating that His-SUMO2-Ubc9 did not interfere with substrate production.
A highly purified substrate was generated from the cleared E. coli lysate using a three-step purification process, as outlined in Figure 1A . At each step, the substrate was assessed for purity by SDS-PAGE followed by Coomassie staining of the gel and Western blot analysis using antibodies against SUMO2/3, His tag, and Strep tag, respectively ( Fig. 1C – E ). Overall, the bacterial SUMOylation system combined with the three-step purification resulted in high yield of the pure product. The purity was estimated to be >95%, as assessed by SDS-PAGE gel stained with Coomassie blue, and the yield of the purified substrate was estimated to be 0.94 mg from 1 L of bacterial culture, which is comparable to a previous report. 10
We assume our substrate SS3HS2 can be cleaved by most SUMO proteases. In this study, SENP2 was selected due to its strong isopeptidase activity but much weaker endopeptidase activity. 11 We used GST-tagged SENP2c (catalytic domain) to test our substrate. The purified substrate SS3HS2 was cleaved by SENP2c into His-SUMO2 and Strep-SUMO3, as expected ( Fig. 1D , E ).
Next, we used SDS-PAGE and Coomassie staining to evaluate substrate cleavage by SENP2c in the absence and presence of known SENP inhibitors and in the AlphaScreen assay buffer ( Fig. 3A – C ). We tested three different SENP inhibitors: the general cysteine protease inhibitor NEM, the specific SENP inhibitor SUMO2-aldehyde, and SDS. Various concentrations of these inhibitors were preincubated with 10 nM SENP2c for 30 min at room temperature followed by incubation with 1.5 µM substrate for 30 min ( Fig. 3A , B ). At concentrations >1.25 mM and 100 nM, respectively, NEM and SUMO2-aldehyde appeared to completely block the cleavage of SENP2c. In agreement with a previous study, SDS efficiently inhibited SENP activity at a very low concentration (0.01%). 12
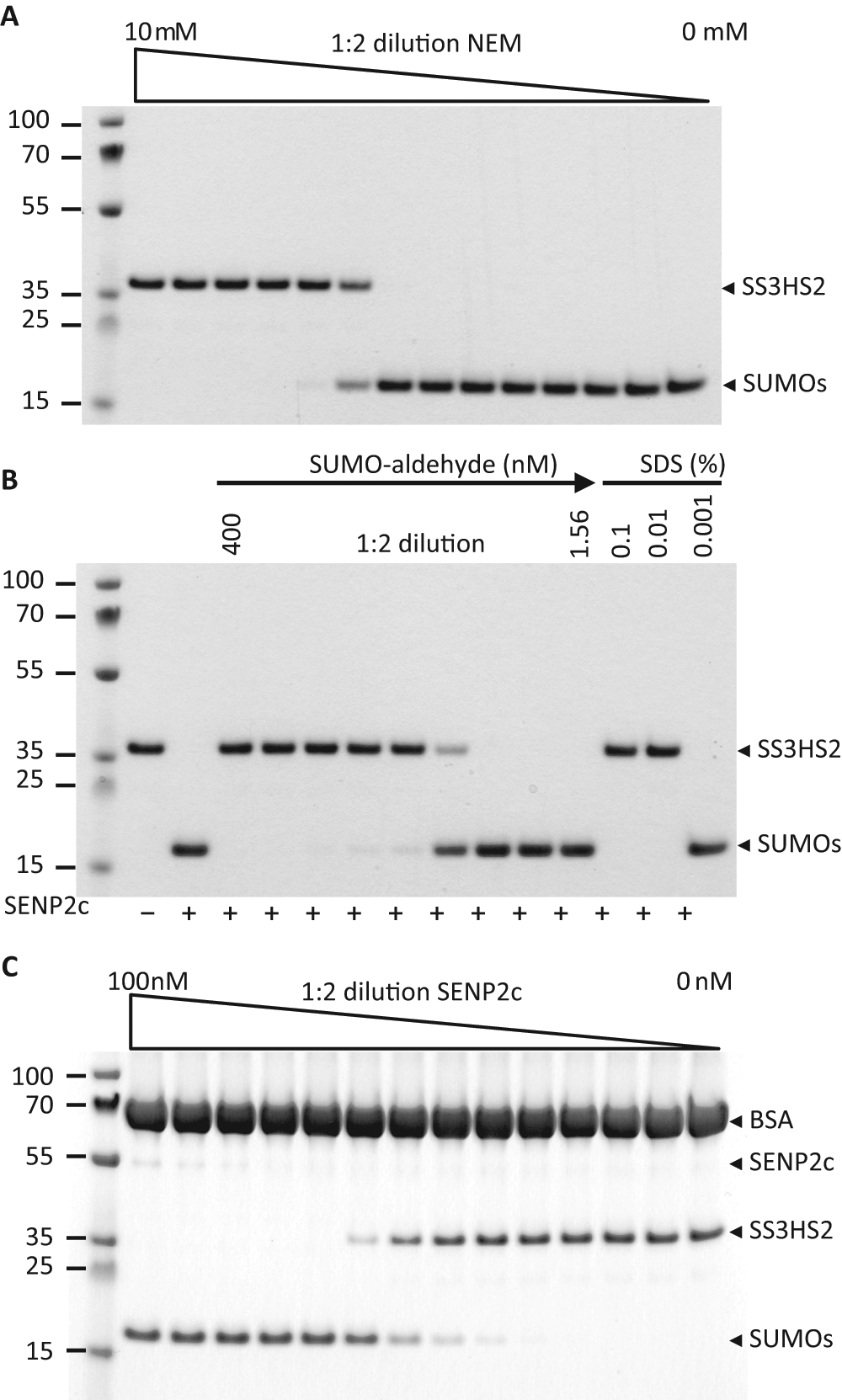
Gel-based analysis of substrate cleavage by SENP2c. (
We then determined the effectiveness of SENP2c in cleaving the substrate by titrating SENP2c in the AlphaScreen assay buffer (25 mM HEPES [pH 7.4], 100 mM NaCl, 0.1% BSA, and 0.05% Tween-20) with 1.5 µM substrate. The substrate was almost completely cleaved into His-SUMO2 and Strep-SUMO3 when the SENP2c concentration was above 6.15 nM ( Fig. 3C ). This indicates that cleavage by SENP2c in the AlphaScreen assay buffer is as efficient as in the cleavage buffer (data not shown). Together, these data demonstrated that our substrate can be used to characterize SUMO proteases and their inhibitors.
After characterizing the substrate, we set out to develop an AlphaScreen assay based on this substrate and to optimize assay conditions for future HTS applications. First, substrate titration experiments were performed in 96-well plates, and a typical bell-shaped curve was obtained, showing the hook effect when the concentration of the substrate was greater than 800 nM ( Fig. 4A ). Considering both optimal signal and cost-effectiveness of protein consumption for future studies, 400 nM of the substrate was chosen for subsequent experiments. Next, serial dilutions of SENP2c were used for titration ( Fig. 4B ). According to the recorded AlphaScreen signals, the substrate was completely cleaved in the presence of 4 nM SENP2c; thus, this concentration was used for all subsequent experiments. The low signals in the presence of 4, 8, and 16 nM SENP2c are comparable to the baseline signal in the absence of substrate, suggesting that once cleaved, almost no AlphaScreen signal is produced by His-SUMO2 and Strep-SUMO3. These data confirmed that we have developed a sensitive assay to monitor the isopeptidase activity of SUMO proteases.
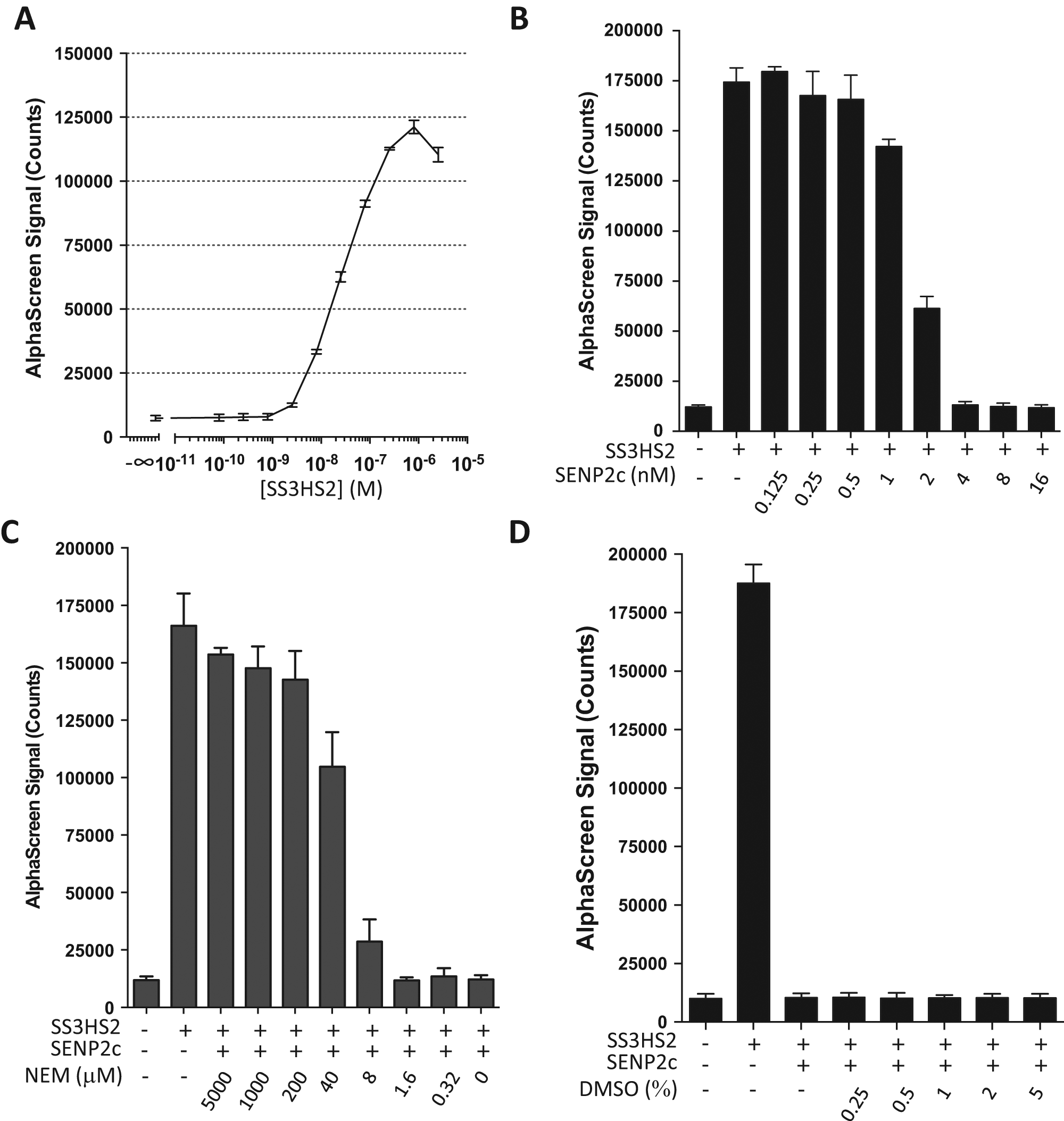
AlphaScreen assay development. (
To determine whether this assay could be used to evaluate the inhibitors of SENPs, we titrated NEM (0.32–5000 µM final concentration) and evaluated its efficacy for blocking SENP2c-induced substrate cleavage ( Fig. 4C ). At a concentration of 200 µM, NEM almost completely blocked SENP2c-induced substrate cleavage, as indicated by the high AlphaScreen signal intensity. These data are comparable to the gel-based analysis ( Fig. 3A ), demonstrating that this assay accurately detects cleavage reactions. Next, given that the compounds in most screening libraries are dissolved in DMSO, we assessed the effect of DMSO on our assay reaction. We verified that up to 5% DMSO in the assay did not interfere with SENP2c-induced cleavage and the corresponding sharp drop in signal intensity ( Fig. 4D ).
Finally, we adapted the assay for 384-well plates and evaluated the Z′ factors of the assay in eight plates on 3 days. The Z′ factor is commonly used to check the robustness and quality of the assay. Assays were run in the presence of 0.1% DMSO and a final concentration of 400 nM substrate in the absence (high signal) or presence (low signal) of SENP2c (4 nM). Overall, variation in the average high signal and low signal from eight plates was small (
Several assays have been developed to study the isopeptidase activity of SUMO proteases, including an assay based on FRET with potential for HTS applications.5,6 These assays use in vitro SUMOylation systems to generate a substrate composed of a SUMO-conjugated protein tagged with fluorescent proteins, normally cyan fluorescent protein and yellow fluorescent protein. In the presence of SUMO proteases, the decrease in the FRET signal can be converted to measure the concentrations of cleaved substrate. However, the feasibility of these assays for HTS applications has not yet been verified.
The AlphaScreen-based platform used for the HTS-compatible assay described here offers the great advantages of high sensitivity and straightforward data analysis. Since the excitation wavelength is higher than the emission wavelength, this assay platform avoids interferences caused by auto-fluorescence, resulting in an excellent signal-to-noise ratio. The AlphaScreen signal is recorded in a time-resolved manner, thereby greatly increasing specificity and sensitivity. Finally, the Z′ factor was above 0.8 when multiple experiments were performed on different days, demonstrating the excellent reproducibility of this assay platform (
Here we present a new HTS-compatible assay platform to screen for SUMO protease inhibitors. The assay could easily be reconfigured, depending on the specific application interests. We have optimized the reactions and used 4 nM SENP2c, 400 nM substrate, and a fixed reaction time for verification and calculating the Z′ factor (
SUMO conjugation is activated after transient cerebral ischemia and is believed to be a protective stress response shielding neurons from damage induced by a period of insufficient blood supply.3,14 Various pediatric and adult cardiovascular procedures involve cardiopulmonary bypass that requires a period of circulatory arrest. To protect organs from damage, these operations are usually performed during deep hypothermia. Moderate to deep hypothermia activates SUMO conjugation, and this activation is believed to contribute to the protective effect provided by hypothermia. 15 If an increase in levels of SUMO-conjugated proteins could enhance cell protection from ischemic damage, it would be of major clinical interest to identify small molecules that activate this protective pathway in normothermic patients, thus avoiding the potentially adverse effects associated with deep hypothermia.
Two different therapeutic strategies can be envisioned that result in an increase in levels of SUMO-conjugated proteins: activate SUMO conjugation or block deconjugation. Any pharmacologic approach to activate SUMO conjugation has the potential for both beneficial and adverse effects by conjugating SUMO to proteins that are otherwise not physiological targets. Blocking SUMO deconjugation, on the other hand, will result in accumulation of SUMO-conjugated proteins that were SUMOylated by the cellular SUMO conjugation machinery. We therefore believe that blocking SUMO deconjugation is a powerful strategy to modulate the SUMO conjugation pathway for protective and therapeutic purposes in the clinical setting, such as protecting organs against damage triggered by ischemia.
In summary, we presented here a novel and robust HTS-compatible assay for screening SUMO protease inhibitors. This assay is also a powerful tool to identify new SUMO proteases and study kinetics of the isopeptidase activity of SUMO proteases. The combination of the bacterial SUMOylation system and the AlphaScreen technology guarantees low cost, high sensitivity, and excellent signal-to-noise ratio, thus making this assay particularly suitable for HTS projects to screen large small-molecule libraries. Identifying compounds that inhibit the isopeptidase activity of SUMO proteases could have a broad spectrum of clinical applications from cancer treatment to increasing the resistance of organs to the potentially damaging effects of a transient ischemic episode.
Footnotes
Acknowledgements
The authors thank Professor Li-An Yeh, Director of the Biomanufacturing Research Institute and Technology Enterprise (BRITE), North Carolina Central University, for helpful discussions; Mark Hughes at BRITE for helping to set up the analysis for calculating the Z′ factor; Dr. Arun Kumar Shukla at the Department of Medicine, Duke University Medical Center, for his help in substrate purification; Dr. Joern Coers, Department of Molecular Genetics and Microbiology, Duke University Medical Center, for permitting access to an EnSpire plate reader; and Kathy Gage, Department of Anesthesiology, Duke University Medical Center, for her editorial contribution in the preparation of this manuscript.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by funds provided by the Department of Anesthesiology, Duke University Medical Center, and in part by NIH RO1 grants HL095552 and NS081299 to W.P. and a Scientist Development Grant from the American Heart Association (12SDG11950003) to W.Y.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.