
Abstract
The protein-folding chaperone Hsp90 enables the maturation and stability of various oncogenic signaling proteins and is thus pursued as a cancer drug target. Folding in particular of protein kinases is assisted by the co-chaperone Cdc37. Several inhibitors against the Hsp90 ATP-binding site have been developed. However, they displayed significant toxicity in clinical trials. By contrast, the natural product conglobatin A has an exceptionally low toxicity in mice. It targets the protein–protein interface (PPI) of Hsp90 and Cdc37, suggesting that interface inhibitors have an interesting drug development potential.
In order to identify inhibitors of the Hsp90/Cdc37 PPI, we have established a mammalian cell lysate-based, medium-throughput amenable split Renilla luciferase assay. This assay employs N-terminal and C-terminal fragments of Renilla luciferase fused to full-length human Hsp90 and Cdc37, respectively. We expect that our assay will allow for the identification of novel Hsp90/Cdc37 interaction inhibitors. Such tool compounds will help to evaluate whether the toxicity profile of Hsp90/Cdc37 PPI inhibitors is in general more favorable than that of ATP-competitive Hsp90 inhibitors. Further development of such tool compounds may lead to new classes of Hsp90 inhibitors with applications in cancer and other diseases.
Keywords
Introduction
Hsp90 is a molecular chaperone that plays an essential role in the folding and maturation of more than 200 client proteins. These clients include protein kinases, growth factor receptors, and many enzymes. 1 Compared with normal cells, the expression of Hsp90 is 2- to 10-fold higher in a wide variety of human cancers, including breast, lung, prostate, colon, and skin cancers, which indicates a crucial function of Hsp90 in transformed cells.2,3 The higher expression of Hsp90 implies the increased stabilization of its client proteins, including mutant driver kinases, such as Raf, Her2, and Src in tumors.4,5 Thus, cancer cells rely on the Hsp90 chaperone machinery to protect mutated and overexpressed oncoproteins from degradation. Cancer cells can therefore be considered to be addicted to the Hsp90 chaperone machinery. 6
There are four different human Hsp90 paralogs: two cytoplasmic Hsp90 isoforms (Hsp90α and Hsp90β), Hsp90 of the endoplasmic reticulum (GRP94 [94 kDa glucose-regulated protein]), and mitochondrial Hsp90 (TRAP1 [tumor necrosis factor receptor-associated protein 1]). They display similar ATPase activity and conformational changes, but differ in their interactions with co-chaperones and due to their localization, serve as chaperones to different sets of protein. 7 Cytosolic Hsp90 contains a conserved C-terminal motif that interacts with numerous co-chaperones, which is not present in the other paralogs. 8
The Hsp90 chaperone machinery includes several co-chaperones and adaptor proteins such as Cdc37, Aha1, FKBP52, p23, STIP1, Hsp70, and Hsp40.9,10 Each of these proteins has its own function either associated with client selection or otherwise completing the Hsp90 chaperone machinery. Cdc37 plays a crucial role in loading kinase client proteins to the Hsp90 chaperone machinery.11–14 While various kinases, transcription factors, and E3 ligases interact with Hsp90, most of the interacting clients are kinases and their binding is mediated by Cdc37. Up to 60% of the human kinome interacts with Hsp90/Cdc37. 15 Cdc37 selectively recruits clients by challenging their conformational stability, as it locally unfolds them. 16 Thus, thermodynamically less stable kinases become clients for the Hsp90/Cdc37 complex. 17
According to a model proposed for the Hsp90-Cdc37-kinase cycle by Verba et al., an open-state kinase binds the N-terminal domain of Cdc37. Through the Cdc37 middle domain, this complex interacts with the N-terminus of an open-state Hsp90, which closes upon N-terminal ATP binding. Cdc37 then translocates to the middle domain of Hsp90. ATP hydrolysis results in Hsp90 opening and the kinase folding, while displacing Cdc37 from the complex. 18 Assistance from Hsp70/Hsp40 or other binding partners is probably required for the kinase cycle, as in the case of steroid hormone receptors. 19 However, the precise process of how these proteins are involved still remains unclear.
In line with the significance of Hsp90 and kinases in tumorigenesis, Cdc37 itself is upregulated in many cancers. For example, in prostate cancer Cdc37 is overexpressed, whereas hepatocellular carcinoma cells overexpress both Cdc37 and Hsp90 compared with normal cells.20–23 Therefore, the collaboration of Hsp90 and Cdc37 likely plays an important role in the transformation and maintenance of cancer cells.
Several inhibitors of Hsp90 have been developed, but until now none have made it to the clinic. The first discovered Hsp90 inhibitor was geldanamycin. It is an N-terminal ATP-binding site inhibitor that blocks the ATPase cycle of Hsp90. Geldanamycin never entered clinical trials due to toxicity issues and poor pharmacological properties. Unfortunately, its derivatives, including 17-AAG (17-N-allyloamino-17-demethoxygeldanamycin), failed in the clinic partly due to high toxicity.24,25 Synthetic second-generation Hsp90 inhibitors, such as luminespib, have shown more favorable results.26,27
Three other classes of inhibitors interfere with the complex formation of Hsp90 and its co-chaperones. Compounds of the first class bind the C-terminal nucleotide-binding site (
Hsp90 Inhibitors Used in This Study and Their Activity in the Split Renilla Luciferase Assay.
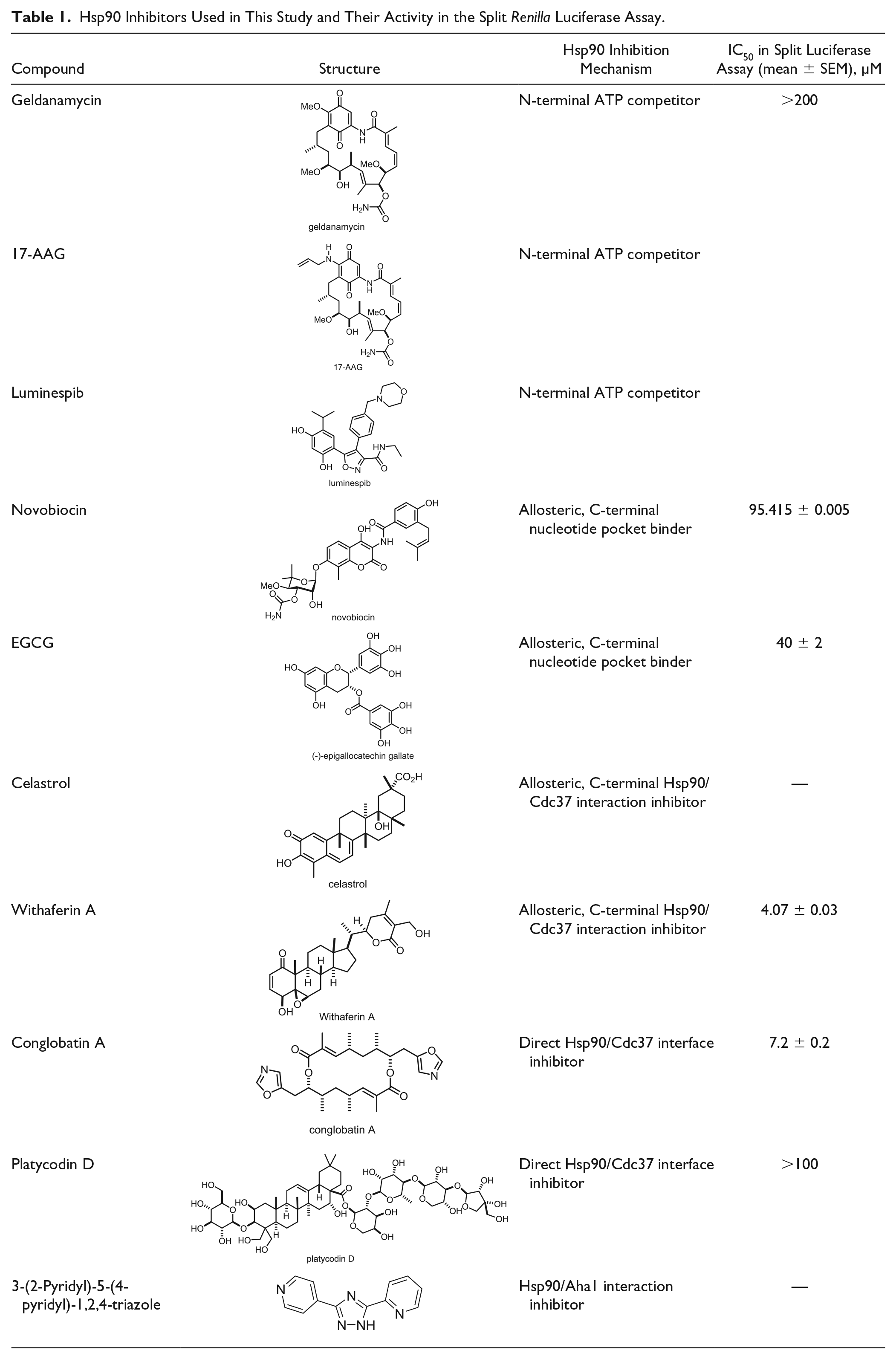
Additional C-terminal allosteric sites exist and can be engaged by a second class of inhibitors, such as celastrol and withaferin A, which disrupt Hsp90/Cdc37 complex formation.30–32 Finally, direct protein–protein interface (PPI) inhibitors of the Hsp90/Cdc37 complex are known. For example, conglobatin A and the recently discovered platycodin D fall into this category.33,34 Interestingly, conglobatin A selectively inhibits K-Ras membrane organization by an unknown mechanism and blocks stemness properties of cancer cells, while it exhibits an unusually low toxicity.35,36 This may suggest that Hsp90/Cdc37 inhibitors have an interesting potential against cancer stem cells. However, current Hsp90/Cdc37 inhibitors are complex natural products, which are typically difficult to synthesize. In order to discover chemo-synthetically more accessible compounds, we have developed a mammalian cell lysate-based assay that can be used for medium-throughput screening of Hsp90/Cdc37 interaction inhibitors.
Materials and Methods
Materials
Conglobatin A was purchased from BioAustralis (cat. BIA-C1022, Smithfield, NSW, Australia). 17-AAG was purchased from Santa Cruz Biotechnology Inc. (cat. sc-200641, Santa Cruz, CA). 3-(2-Pyridyl)-5-(4-pyridyl)-1,2,4-triazole was purchased from Alfa Aesar (cat. H50665, Karlsruhe, Germany). Celastrol (cat. 70950) and (–)-epigallocatechin-3-gallate (EGCG; cat. 70935) were purchased from Cayman Chemical (Ann Arbor, MI). Geldanamycin (cat. T6343, TargetMol, Boston, MA), luminespib (cat. HY-10215, MedChemTronica, Sollentuna, Sweden), novobiocin (cat. N825320, Toronto Research Chemicals, Toronto, ON, Canada), platycodin D (cat. P578200, Toronto Research Chemicals), and withaferin A (cat. NP-007425, AnalytiCon Discovery, Potsdam, Germany) were purchased through MolPort (Riga, Latvia). Stock solutions of these compounds (1 mM for measuring effects on Renilla luciferase activity at 20 μM, and 10 mM for IC50 determination) were prepared in DMSO, which was purchased from Santa Cruz Biotechnology Inc. (cat. sc-358801). JetPRIME transfection reagent was purchased from Polyplus-transfection (cat. 114-07, Illkirch, France) and the Renilla Luciferase Reporter Assay System was purchased from Promega (cat. E2820, Nacka, Sweden).
Plasmid Constructs
The plasmids used in the study were described previously. 37 The full-length human Hsp90 and Cdc37 cDNAs were genetically fused to the N-terminal fragment (NRL, residues 1–229) and C-terminal fragment (CRL, residues 230–311) of Renilla luciferase in a pcDNA3.1(+) vector backbone to produce pcDNA3.1(+)-NRL-Hsp90 and pcDNA3.1(+)-Cdc37-CRL, respectively. The NRL or CRL fragments cloned into the pcDNA3.1(+) vector backbone were used as controls. pGL4.74 expressing full-length Renilla luciferase was used for counterassay experiments.
Cell Culture
Human Embryonic Kidney 293 EBNA (HEK 293) cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM; cat. D6171, Sigma-Aldrich, Helsinki, Finland) supplemented with 10% fetal bovine serum (FBS; cat. S1810, Biowest, Nuaille, France) and 2 mM
DNA Transfections
One million HEK 293 cells were seeded in 10 cm culture dishes and incubated at 37 °C for 24 h. The next day, cells were transfected with 10 μg of either pcDNA3.1(+)-NRL-HSP90 or pcDNA3.1(+)-Cdc37-CRL using 20 μL of jetPRIME transfection reagent according to the manufacturer’s instructions. For some control experiments, pcDNA3.1(+)-NRL or pcDNA3.1(+)-CRL was used. After 48 h of incubation, cells were harvested by trypsinization. Cell pellets were washed thoroughly with phosphate-buffered saline (PBS) and used for cell lysate preparation or directly stored at −80 °C for up to 5 months.
Cell Lysate Preparation
Cells were lysed for 10 min on ice in 1 mL of 1× lysis buffer provided with the Renilla Luciferase Reporter Assay System kit. Lysates were cleared by centrifugation at 13,000g for 1 min at 4 °C. Lysates were kept on ice throughout the assay procedure. One milliliter of lysate prepared from one 10 cm culture dish was sufficient for 200 reactions or wells. Before performing the assay, the quality of lysates, reagents, and positive control was checked by performing the assay in 3 to 5 wells of a 96-well plate as described in the standard split Renilla luciferase assay protocol below. If the relative luminescence unit (RLU) was between 40,000 and 50,000 after 40 s, lysates and other reagents were considered to meet the requirements to perform further experiments.
Split Renilla Luciferase Assay
Twenty microliters of assay buffer without Renilla luciferase substrate was added to each well of a white, solid, flat-bottom, opaque 96-well plate (cat. 3917, Costar, Corning Inc., Corning, NY). Test compound (20 μM; typically 1 μL of a 1 mM stock solution) or 2% DMSO was added as negative control. The DMSO concentration was always adjusted to 2% at all compound concentrations. Next, 5 μL of NRL-Hsp90-lysate was added in one row (12 wells) of the plate. The plate was agitated for a few seconds to mix reagents and then incubated for 5 min at room temperature (RT). This readily allowed the compounds to bind to Hsp90. Then 5 μL of Cdc37-CRL-lysate was added to the NRL-Hsp90-lysate-containing wells and incubated at RT for 2 min. In the next step, 20 μL of buffer containing 2× Renilla luciferase substrate was added to the same wells using an electronic pipette. The resulting 50 μL reaction mix was incubated for 30 s at RT and then shaken for 10 s. Finally, the row was read with a synergy H1 Hybrid Multi-Mode reader (BioTek, Winooski, VT) in the luminescence detection mode with a gain value of 250 and integration time of 1 s.
Data Analysis
The Z factor (Z′) was determined as
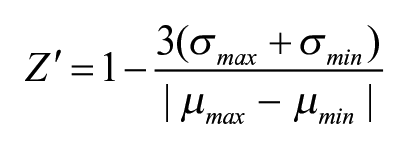
where µ is the mean value and σ is the standard deviation. 38
The statistical significance of differences between samples was examined using one-way analysis of variance (ANOVA) followed by Tukey’s multiple comparison tests using GraphPad Prism version 7 (GraphPad Software, La Jolla, CA).
Results
Assay Principle
The assay we describe here specifically detects Hsp90/Cdc37 interaction inhibitors, but not ATP-competitive Hsp90 inhibitors that bind the N-terminal ATP pocket of the chaperone. Three classes of Hsp90/Cdc37 interaction inhibitors exist. The first two classes are allosteric inhibitors that bind the C-terminus of Hsp90, at either the nucleotide-binding pocket or another allosteric site (
In order to measure the Hsp90/Cdc37 interaction, we employed a previously described split Renilla luciferase assay, where the N-terminal fragment of the luciferase was fused to the N-terminus of full-length human Hsp90 and the C-terminal fragment to the C-terminus of human Cdc37
39
(NRL-Hsp90 and Cdc37-CRL, respectively;
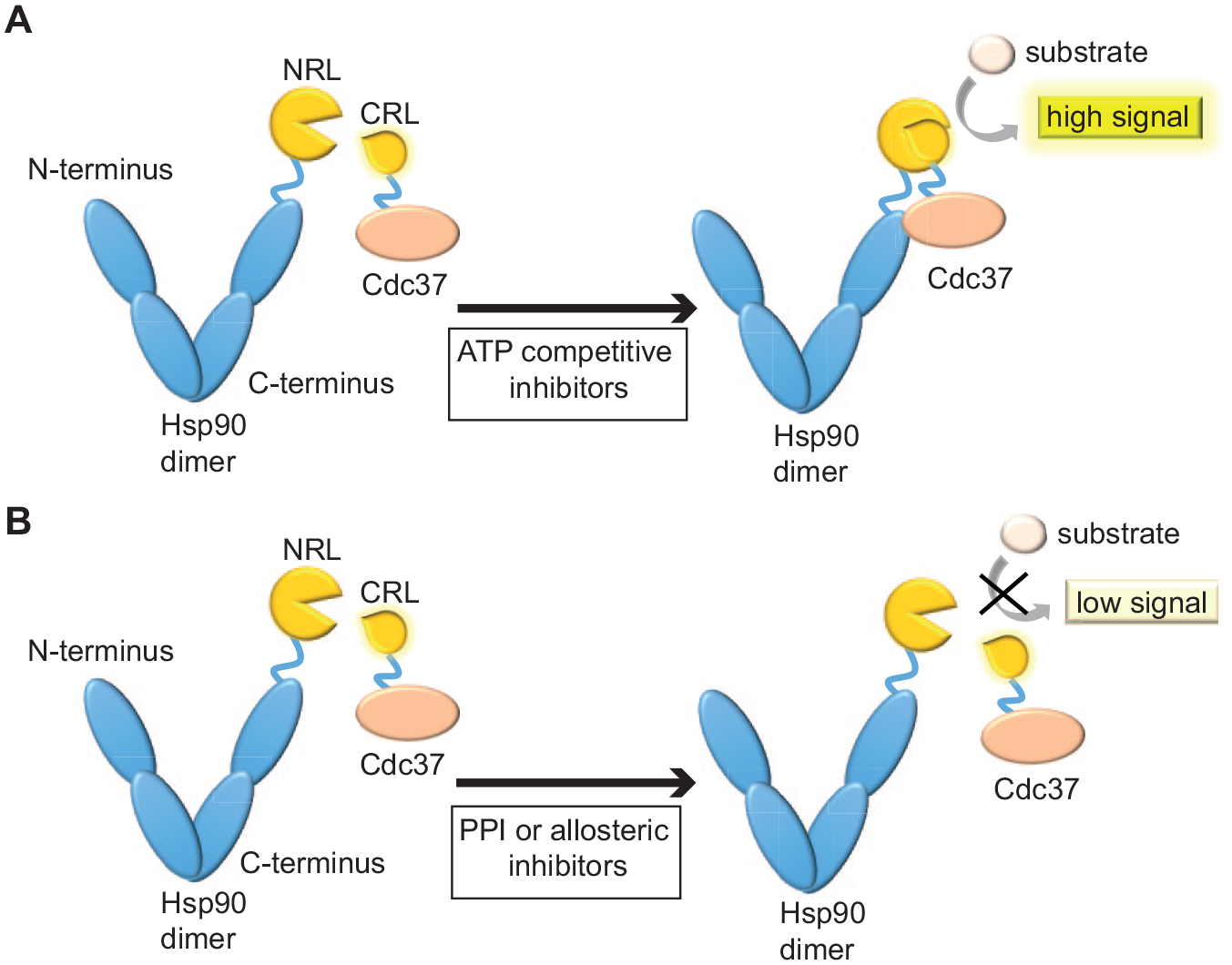
Schematic representation of the split Renilla luciferase assay to detect inhibitors of the Hsp90/Cdc37 interaction. Hsp90 and Cdc37 are overexpressed in HEK 293 cells as fusion proteins with the NRL and CRL fragments of Renilla luciferase, respectively. (
Assay Workflow
In order to obtain a high expression of the split luciferase fusion proteins NRL-Hsp90 and Cdc37-CRL in HEK 293 cell lysates, they were separately expressed for 2 days. After harvesting and cell lysis, lysates were cleared by centrifugation (
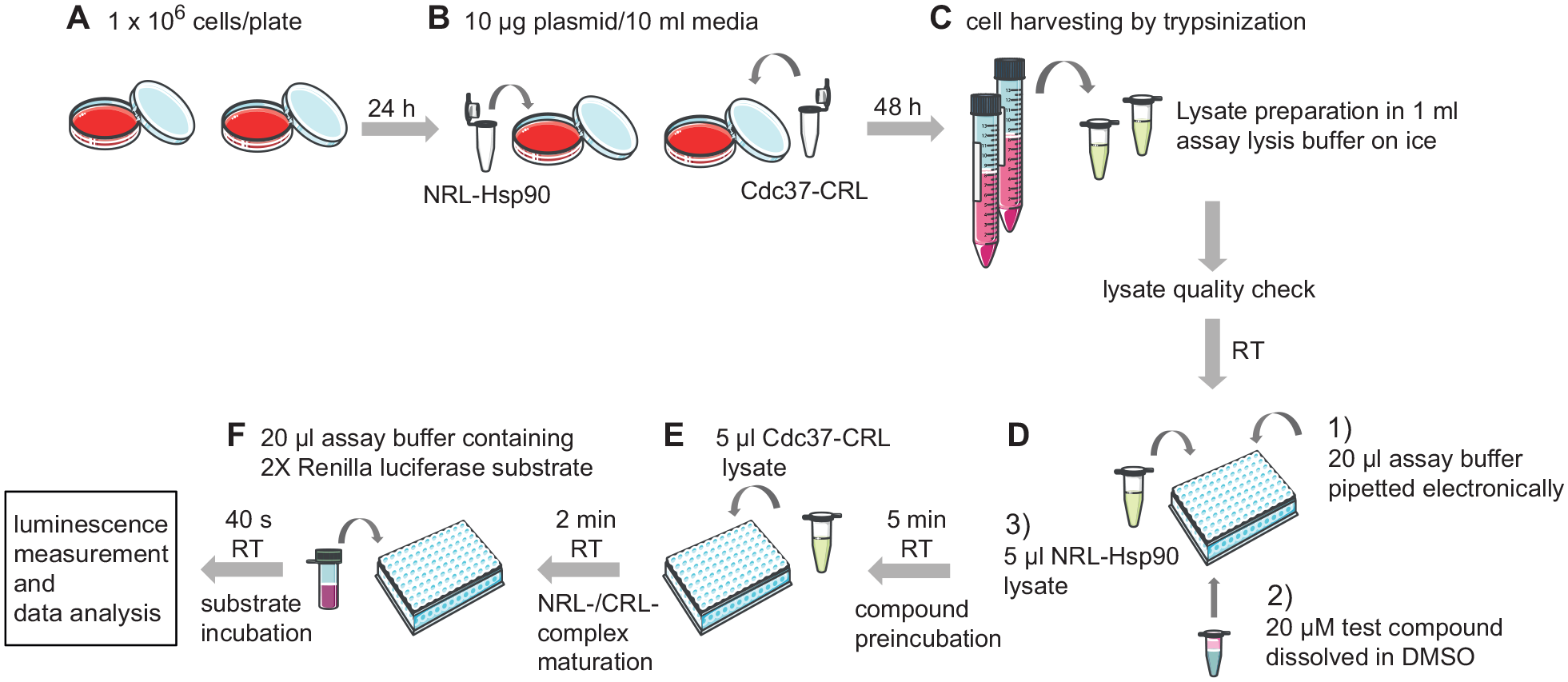
Standard assay workflow. (
Cell Pellets, but Not Lysates, Can Be Stored Frozen
In order to determine whether cell lysates or cell pellets can be stored frozen for later use, we compared the signal of freshly prepared lysates with those of frozen and then thawed lysates. As compared with the fresh lysate, storage of lysates at −20 °C for 1 or 3 days caused a 35% or 60% decrease in luminescence signal, respectively (
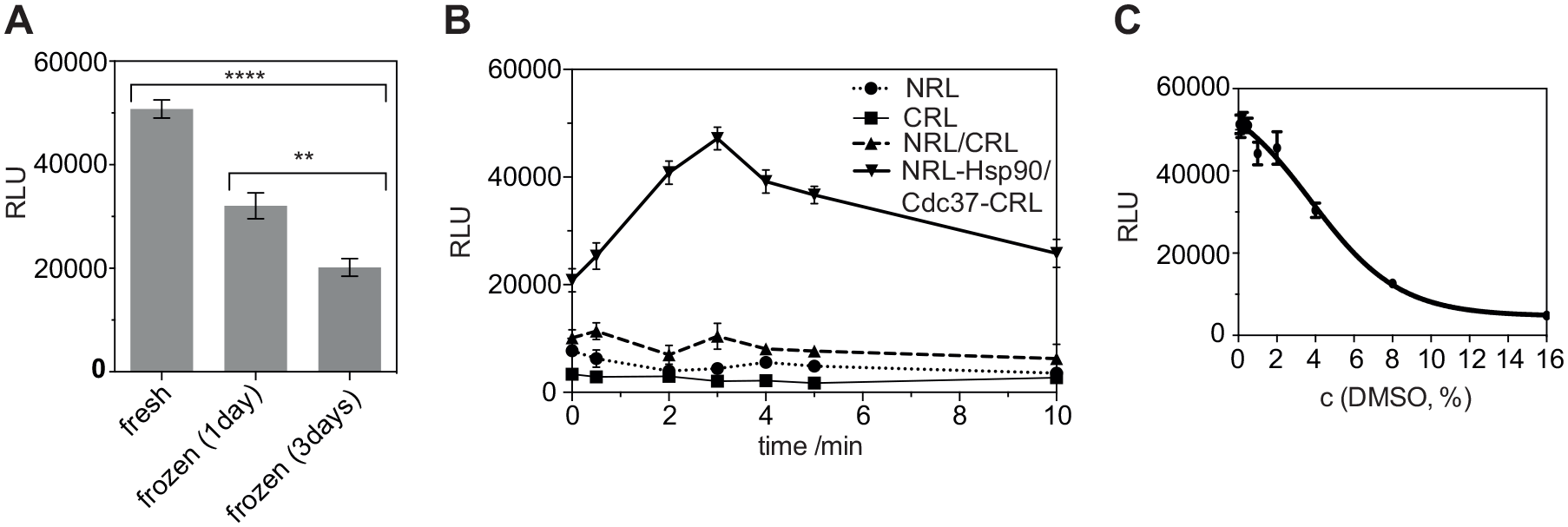
Lysate storage, background recombination, and DMSO tolerance. (
Background Recombination of Split Luciferase Fragments Is Negligible
The split luciferase fragments could spontaneously recombine and thus produce an unspecific luminescence signal that does not report on the Hsp90 and Cdc37 interaction. We therefore tested whether only the NRL and CRL fragments of Renilla luciferase without the Hsp90 and Cdc37 fusion could produce a signal in our assay. The specific luminescence signal resulting from the interaction of NRL-Hsp90 and Cdc37-CRL was 4.5-fold higher at our readout time in the standard protocol than the signal produced by the NRL and CRL fragment mix (
The Split Luciferase Assay Tolerates 2% DMSO
Next, we tested for the DMSO tolerance of the assay. For compound screening in biochemical assays, a 1%–5% DMSO concentration can be acceptable. When titrating increasing concentrations of DMSO in the split luciferase reaction mixture, we found that there was no significant effect of DMSO on the split luciferase Hsp90/Cdc37–fusion protein complex formation up to approximately 2% DMSO (IC50 = 5.6 ± 0.9%) (
Compounds Can Be Preincubated with Either NRL-Hsp90 or Cdc37-CRL
Before combining the N- and C-terminal fragments of the luciferase fusion proteins, we implemented a compound preincubation step (
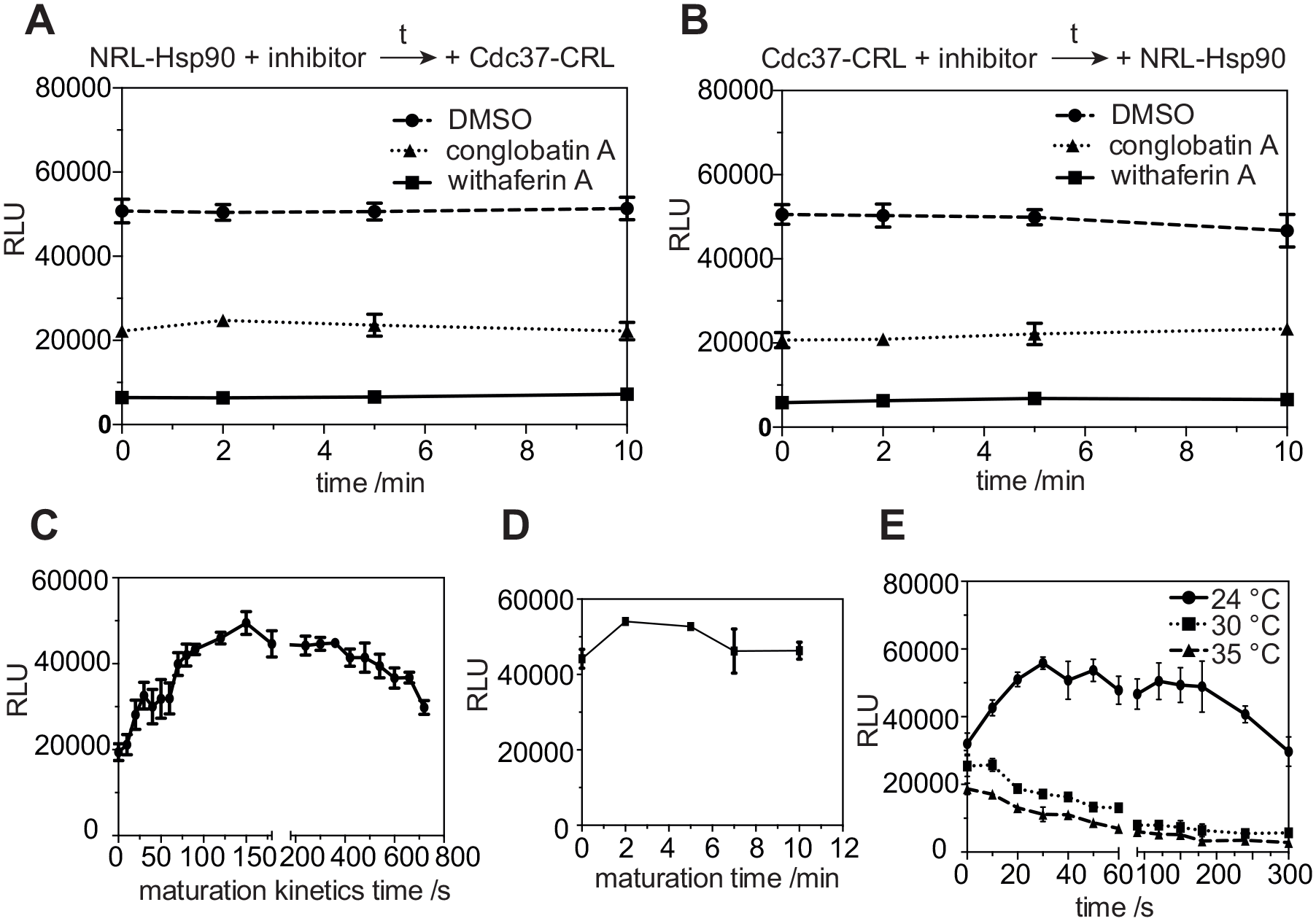
Hsp90/Cdc37 complex maturation, signal stability, and temperature dependence. (
Hsp90/Cdc37 Complex Matures in 2 Min
In order to determine the maturation time of the split luciferase when NRL-Hsp90 and Cdc37-CRL are combined, we recorded the complex maturation kinetics. To this end, Cdc37-CRL-lysate and substrate were injected into the wells containing NRL-Hsp90-lysate in quick succession. After 10 s of mixing time, it took approximately 150 s for the luminescence signal to reach its maximum (
We then validated this complex maturation time within our standard protocol setting (
Substrate Incubation Time and Temperature Dependency
Finally, under standard protocol conditions we tested how the luciferase signal evolves with time at different temperatures. After substrate addition, the NRL-Hsp90 and Cdc37-CRL reaction was mixed for 10 s, before the signal was recorded. At 24 °C the maximum signal was reached after 30 s and remained stable up to approximately 180 s (
Validation of Assay Using Known Hsp90/Cdc37 Inhibitors
To validate the assay for the screening of Hsp90/Cdc37 interaction inhibitors, we tested several compounds with previously reported mechanisms of Hsp90 inhibition (
All compounds were initially tested at the screening concentration of 20 μM to qualitatively determine their activity on Hsp90/Cdc37 interaction. At the tested concentration, N-terminal ATP competitors (geldanamycin, 17-AAG, and luminespib) had no or little effect on Hsp90/Cdc37 complex formation, as expected. By contrast, the allosteric, C-terminal nucleotide pocket binder EGCG inhibited the complex. Novobiocin was very inefficient to inhibit complex formation at the tested concentration. Overall, allosteric, C-terminal Hsp90/Cdc37 interaction inhibitors (celastrol and withaferin A) seemed to have the highest inhibitory effect on complex formation; however, as the counterscreen below revealed, the celastrol results were confounded. The direct PPI inhibitor conglobatin A potently inhibited complex formation, while platycodin D had no effect at the tested concentration (
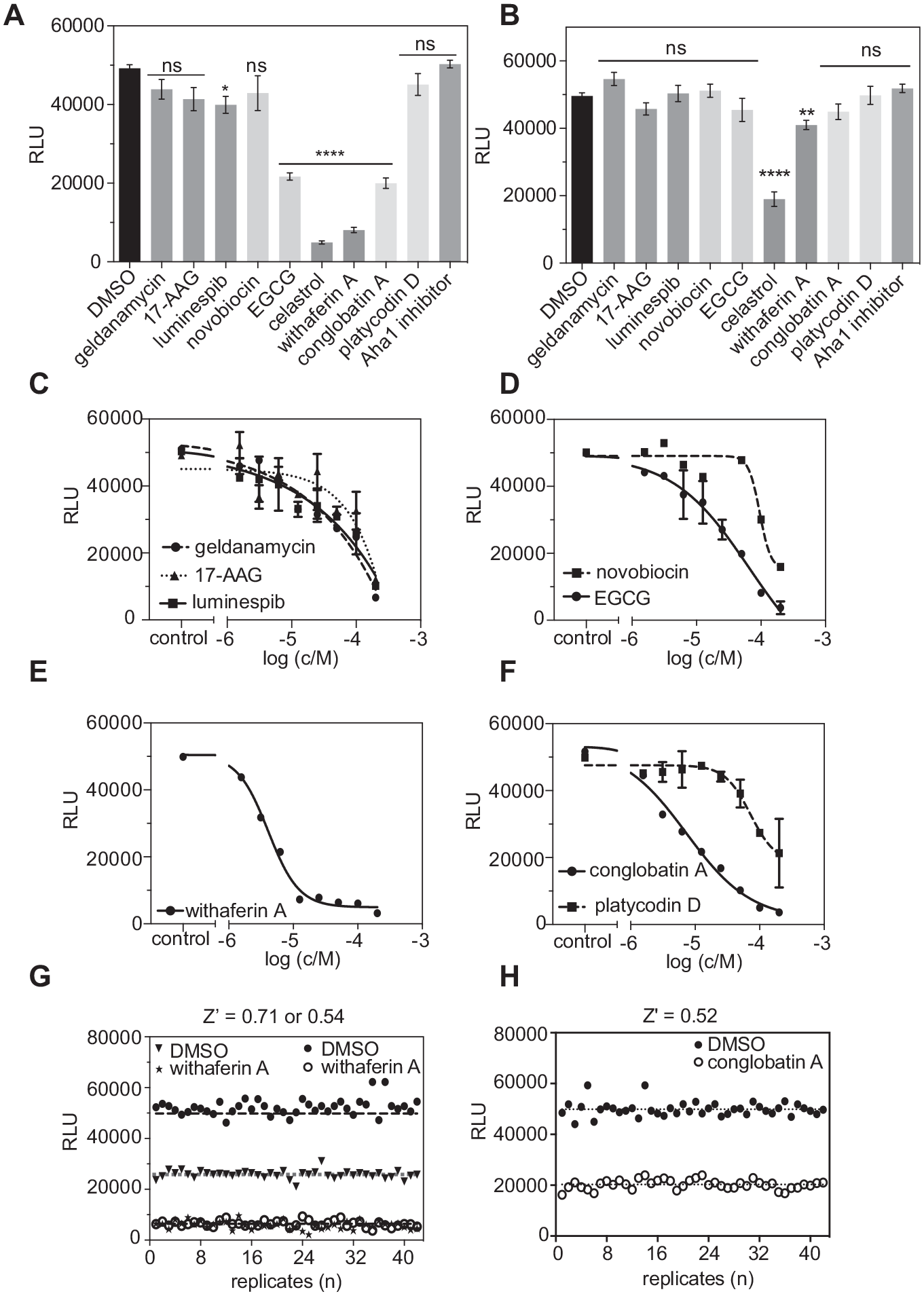
Validation of Hsp90/Cdc37 split Renilla luciferase assay for compound characterization and screening. (
In order to ensure that loss of luciferase signal is due to the disrupted complementation and not to effects on the luciferase itself, we performed a counterscreen using full-length Renilla luciferase. Celastrol strongly decreased the luminescence signal, while all other compounds had no effect; some significant loss was, however, observed with withaferin A (
The screening concentration results were corroborated by determining the IC50 values of these Hsp90 inhibitors for the Hsp90/Cdc37 interaction in a twofold dilution series from 200 to 1.56 μM (
Finally, we evaluated the high/medium-throughput compatibility of the assay by determining Z′. Z′ typically ranges between 0 and 1, with Z′ > 0.5 being required for HTS assays.
38
The allosteric, C-terminal Hsp90/Cdc37 interaction inhibitor withaferin A and the direct Hsp90/Cdc37 interface inhibitor conglobatin A were used as the positive controls for the validation. Z′ was determined to be 0.71 and 0.52 for withaferin A (
Discussion
The cell lysate-based, split luciferase assay reported here allows for the identification of two broad types of Hsp90/Cdc37 interaction inhibitors, allosteric C-terminal binders and direct PPI inhibitors. The assay has a very good Z′ score and is therefore medium- to high-throughput amenable. By using mammalian cell lysates, we can study proteins that are difficult to express and/or purify from bacteria. When comparing our test set of inhibitors, it appears that C-terminal nucleotide pocket inhibitors possess high inhibitory potential. However, nucleotide binding to the N-terminus of Hsp90 negatively affects C-terminal domain binding, 43 which may reduce the efficacy of inhibitors acting on this domain. Besides, cross-reactions between the nucleotide-binding domains may occur, as C-terminal binders have been shown to disrupt both C- and N-terminal nucleotide binding. 44
In line with previous reports, inhibition of Hsp90/Cdc37 by novobiocin was very inefficient.45,46 However, the suggested direct PPI inhibitor platycodin D did not inhibit Hsp90/Cdc37 complex formation in our assay. This seems to contrast with previous findings, which demonstrated in immunoprecipitation experiments that platycodin D reduces levels of Hsp90-bound Cdc37, while it does not affect Hsp90 or Cdc37 protein levels. 40 Considering the absence of a response in our assay, the described activity of platycodin D may be due to indirect effects of the compound.
In general, cell extract/lysate-based immunoprecipitation assays can suffer from high false-positive results when searching for direct protein–protein interaction inhibitors, as indirect mechanisms of action can remain undetected in the complex protein mixtures. Other biophysical methods can interrogate direct protein–protein interactions in the cellular environment. Förster resonance energy transfer (FRET) methods employing steady-state or time-resolved fluorescence intensity (TR-FRET) measurements, as well as the bioluminescence energy transfer (BRET) method, are commonly used to study protein–protein interactions. Most of the high-throughput-compatible assays involving these methods are carried out by ratiometric measurements of the donor and acceptor emission intensities. Thus, these assays require a plate reader that is capable of measuring both donor and acceptor signals either simultaneously or sequentially.47,48 Therefore, it is not possible to perform such assays with low-end plate readers that lack appropriate filters or a monochromator.
In recent years, other Hsp90 screening assays have been published. Thomas et al. introduced a yeast-based time-dependent turbidity-measuring liquid culture assay to detect Hsp90 antagonists. This high-throughput assay can identify compounds that modulate Hsp90 through different mechanisms. 49 However, the assay requires establishment of diverse, thoroughly characterized yeast strains and the differences between human and yeast protein interaction networks may affect the outcome. Here the presented split luciferase assay could be employed as a secondary screening assay to this chemogenomic screening platform in defining the accurate mechanism of action. In another assay, high-performance liquid chromatography (HPLC)-based affinity chromatography was combined with mass spectrometry, allowing the detection of compounds that bind Hsp90 in a multicomponent mixture without a preceding purification process. While the assay holds promise, it requires access to liquid chromatography/mass spectrometry (LC/MS) equipment. 50 The amplified luminescence proximity homogenous (Alpha) technology was used to identify Hsp90/Aha1 inhibitors. 51 Alpha screening technology is a widely adopted industry standard, due to its versatility and sensitivity. 52 The assay principle is similar to ours; however, it requires purified proteins, which makes its implementation more time-consuming. This recent boost in the development of screening assays to detect Hsp90 inhibitors supports its significance as a highly interesting drug target. Different assays can be used as complementary or alternative techniques depending on the objectives of the studies. We believe that given its advantageous characteristics in terms of sensitivity and usability, the presented split luciferase assay will be useful for the discovery of novel Hsp90/Cdc37 PPI inhibitors with applications in cancer and other diseases.
Supplemental Material
SupplementaryInformation_Siddiqui – Supplemental material for Medium-Throughput Detection of Hsp90/Cdc37 Protein–Protein Interaction Inhibitors Using a Split Renilla Luciferase-Based Assay
Supplemental material, SupplementaryInformation_Siddiqui for Medium-Throughput Detection of Hsp90/Cdc37 Protein–Protein Interaction Inhibitors Using a Split Renilla Luciferase-Based Assay by Farid Ahmad Siddiqui, Hanna Parkkola, Ganesh babu Manoharan and Daniel Abankwa in SLAS Discovery
Footnotes
Acknowledgements
Supplemental material is available online with this article.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: D.A. acknowledges support from the Academy of Finland (no. 304638), the Sigrid Juselius Foundation, and the Cancer Society Finland. F.A.S. acknowledges support from the Finnish National Agency for Education and Åbo Akademi University.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.