
Abstract
Superoxide plays a key role in many pathological processes; however, detection of superoxide by one of the most common methods using dihydroethidium (DHE) may be unspecific because of overlapping fluorescence of the superoxide-specific product, 2-OH-ethidium (2OH-E), and the unspecific oxidation product, ethidium. Here, we show a new optimized fluorescence spectroscopy protocol that allows rapid and specific detection of superoxide in cell-free systems and intact cells using DHE. We defined new optimized fluorescent settings to measure the superoxide-specific product and minimize the interference of unspecific DHE oxidation products. Using this protocol, we studied real-time superoxide production by xanthine oxidase– and menadione-treated cultured cells. Specificity of the plate reader–based superoxide measurements was confirmed by the inhibition of fluorescence with superoxide dismutase and high-performance liquid chromatography (HPLC) analysis. We show that limitations of the HPLC-based analysis can be overcome by the optimized fluorescence spectroscopy.
Superoxide has been implicated in the initiation and development of many pathological conditions including drug toxicity, inflammation, cardiovascular diseases, and diabetes.
1
Despite its importance, there is a limited number of methods for measurement of superoxide in live cells and tissue. Many probes, including dichlorodihydrofluorescein, lucigenin, or dihydrorhodamine, have low specificity and are prone to artifacts.
2
The seminal work of Kalyanaraman’s group
3
has shown that dihydroethidium (DHE) produces a number of unspecific products and a single superoxide-specific product, 2-OH-ethidium (2OH-E;
It has been recently shown that the superoxide-specific product of mitochondria-targeted triphenylphosphonium–DHE conjugate (MitoSOX) has a specific excitation peak at 396 nm, which can facilitate selective superoxide detection. 6 The excitation spectrum of 2OH-E, however, is very similar to the Eth spectrum, and excitation at the 2OH-E peak intensity (480 nm) does not provide specific 2OH-E measurements. 3 Excitation at 480 nm is used in many studies in which total fluorescence is recorded. 7 These settings, however, may generate artifacts due to unspecific Eth fluorescence. 4
In this work, we investigated the fluorescent properties of Eth and the superoxide-specific product of DHE, 2OH-E, and developed an optimized fluorescence spectroscopy protocol that allows rapid and specific detection of superoxide in cell-free systems and intact cells using DHE.
Materials and Methods
HPLC Detection of DHE
Generation of 2OH-E was confirmed by HPLC analysis as described previously. 8 Briefly, DHE and its oxidation products were separated by using a Thermo C18 reverse-phase HPLC Betacil column, 250 mm × 4.6 mm, 5 µm, and a mobile phase containing 0.1% trifluoroacetic acid and an acetonitrile gradient (from 37% to 47% over 23 min) at a flow rate of 0.5 mL/min. Eth and 2OH-E were detected with a fluorescence detector using an emission wavelength of 580 nm and an excitation of 480 nm. A photodiode array detector was additionally used for DHE detection. To identify DHE (Molecular Probes) and its oxidation products, we used Eth (Molecular Probes) and 2OH-E generated as described below. Standards were used in the range of 10 nM to 10 µM with an injection volume of 30 µL. The stock of 2OH-E was generated by complete DHE oxidation for 6 h in a superoxide-generating cell-free enzymatic system containing 10 mU/mL xanthine oxidase (Roche Molecular Biochemicals, Indianapolis, IN) and 0.5 mM xanthine (Sigma Chemical Co., St. Louis, MO) as previously described. 5 The reaction mixture did not contain more than 0.1% of DMSO. Xanthine was prepared in advance as a 50 mM stock solution in 0.9% saline. Progression of DHE oxidation to 2OH-E was tracked with HPLC analysis until complete DHE conversion.
The HPLC system was purchased from Shimadzu and consisted of the central communication and control module CBM-20A, DGU-20A5 vacuum degasser, LC-20AD quaternary solvent delivery unit, LC-20AD pump rinsing kit, SIL-20AC UFLC autosampler, SPD-20A UV-VIS detector, and RF-20Axs fluorescence detector. Data were analyzed using Shimadzu LabSolutions LC/GC Workstation v5.32 SP1 Software.
Fluorescent Spectroscopy
To obtain fluorescence spectra and to generate DHE fluorescent products, 20 µM DHE from DMSO stock solution was freshly prepared in 50 mM phosphate-buffered saline (PBS; 0.9% NaCl, 50 mM Na2HPO4, pH 7.4). The final DMSO content was 0.1%. The stock of 2OH-E as previously described 5 was generated by complete DHE oxidation for 6 h in a superoxide-generating cell-free enzymatic system containing 10 mU/mL xanthine oxidase (Roche Molecular Biochemicals) and 0.5 mM xanthine. Xanthine (50 mM; Sigma Chemical Co.) was prepared in 0.9% NaCl as a stock solution, and the appropriate volume was added to the reaction mixture to reach the required concentration. Progression of DHE oxidation was analyzed by HPLC.
Fluorescence intensities were acquired using a BioTek H1 96-well plate reader. For experiments with cultured cells, black glass-bottom plates were used (BD Biosciences, Franklin Lakes, NJ); for all other experiments, polypropylene black plates were used (Nunc, Thermo, Denmark). The instrument was kept at 37 °C during the measurements.
Cell Culture
Human aortic endothelial cells were purchased from Lonza (Basel, Switzerland) and cultured in the EGM-2 medium supplemented with 2% fetal bovine serum. Cells 80% to 90% confluent were used for experiments. Before treatment, EGM-2 medium was replaced with 50 mM Krebs-HEPES buffer (145 mM NaCl, 4.86 mM KCl, 5.7 mM NaH2PO4, 0.54 mM CaCl2, 1.22 mM MgSO4, 5.5 mM glucose; pH 7.4)
Results and Discussion
Oxidation of DHE in biological systems forms a superoxide-specific product, 2OH-E, and an unspecific product, Eth. Other oxidation products do not generate considerable fluorescence. Detection of 2OH-E is, however, complicated because of the overlapping fluorescence of 2OH-E and Eth. 4 We therefore searched for a new optimized fluorescence spectroscopy protocol that allows rapid and specific detection of 2OH-E and eliminates the contribution of the Eth fluorescent signal. To improve the specificity of 2OH-E detection, we analyzed emission spectra of commercially available Eth (Molecular Probes) and 2OH-E generated as described in the Materials and Methods sections. We used different excitation wavelengths to determine the best conditions to specifically detect the signal of 2OH-E. As shown in Figure 1A , excitation at 480 nm results in a slightly shifted emission spectra of 2OH-E compared with Eth. Changing the excitation from 480 nm to 405 nm significantly reduced the emission intensity of Eth compared with 2OH-E. Importantly, changing the excitation from 480 nm to 405 nm increases the specificity of the 2OH-E detection; however, the emission signal of 2OH-E is also reduced. As shown in Figure 1B , the emission signal at 580 nm was 2.5-fold lower at 405 nm excitation compared with 480 nm excitation. Because both excitation at 405 nm and 480 nm gave distinct fluorescent spectra of 2OH-E and Eth ( Fig. 1A ), we calculated the fluorescence ratio between 2OH-E and Eth. As shown in Figure 1B , the fluorescence ratio at 480 nm excitation was maximal at 530 nm emission (52:1 2OH-E:Eth ratio), whereas at 405 nm excitation, the fluorescence ratio was maximal at 570 nm (21:1 2OH-E:Eth ratio). In most studies, when DHE is used, the probe is excited at 480 nm and the emission signal at 580 nm is recorded. 7 With these settings, the ratio of 2OH-E:Eth fluorescence signals is low ( Fig. 1 ); therefore, these conditions do not provide reliable and specific superoxide detection because of the high contribution of Eth to overall fluorescence. Analysis of the fluorescence intensity of 2OH-E and the fluorescence ratio between 2OH-E and Eth ( Fig. 1B ) shows potential to improve 2OH-E detection by changing these settings. The highest selectivity could be achieved by changing the emission detection to 540 nm and excitation to 480 nm; however, this modification results in substantial reduction in fluorescence intensity. For this reason, these selective settings may have limited application under conditions of low-level superoxide or limitations of equipment sensitivity. One possible solution is to find an optimal excitation and emission wavelength that would provide high fluorescence intensity with relatively high specificity. As shown in Figure 1B , the specificity of the assay dramatically improves compared with standard setting when the excitation is changed to 405 nm and the emission is detected at 570 nm. In our cell-free and intact cell experiments using a Synergy H1 fluorescent plate reader (Biotek, Winooski, VT), we found that excitation at 405 nm and emission at 570 nm provide optimal settings. The optimal settings should be confirmed empirically depending on the sensitivity of equipment, the amount of superoxide production in biological samples, and the ratio of 2OH-E and Eth. Using these settings, we investigated superoxide production in a cell-free xanthine oxidase superoxide-generating system and menadione-treated cultured endothelial cells.
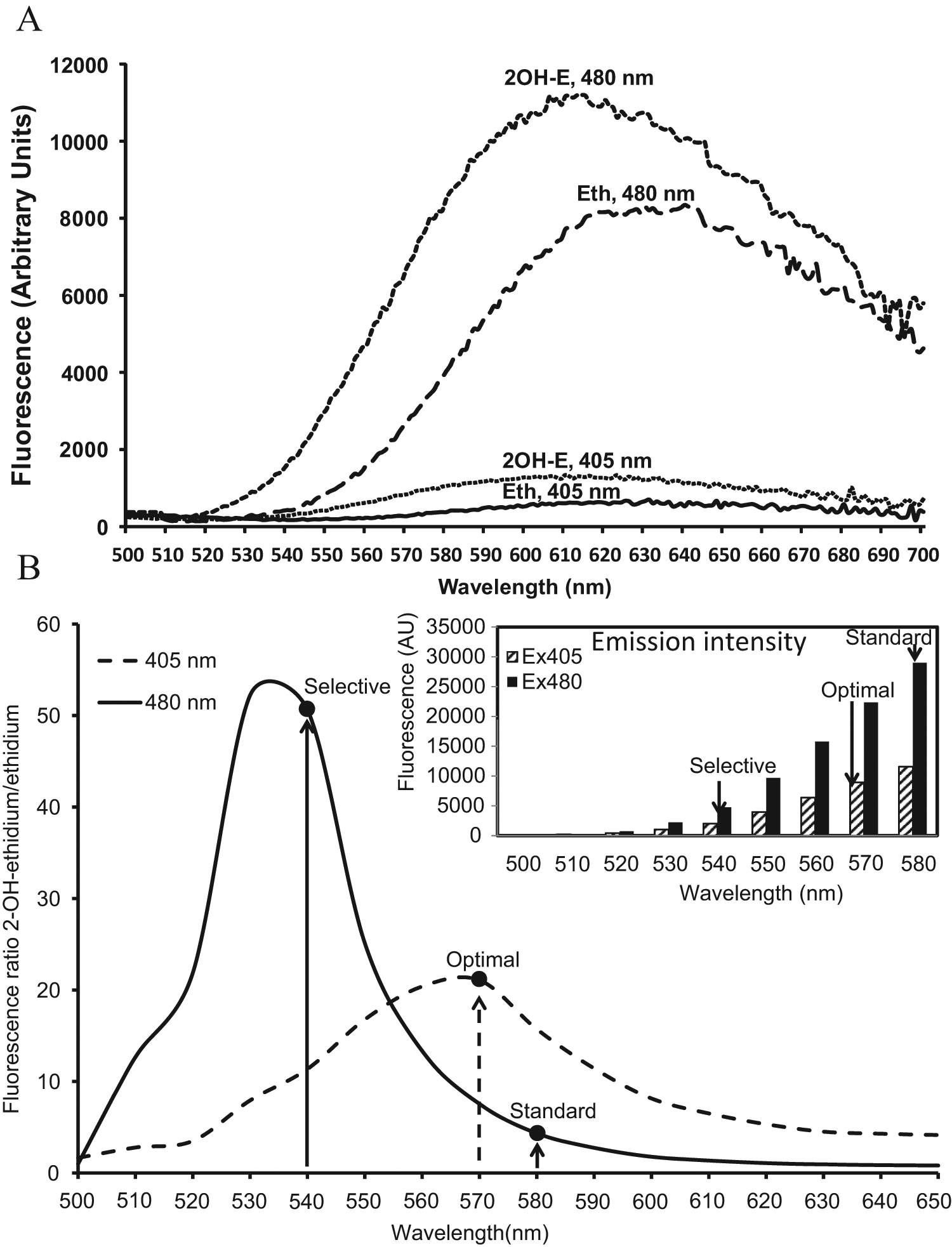
Fluorescence spectra of ethidium and 2-OH-ethidium. (
First, we measured superoxide generation in a cell-free xanthine oxidase (0.1 mU/mL) plus xanthine (0.2 mM) enzymatic system using optimized fluorescence settings. As shown in Figure 2 , this method allowed kinetic measurements of superoxide by following the fluorescence signal at 570 nm using excitation at 405 nm. Superoxide dismutase (SOD) is an enzyme that dismutates superoxide to hydrogen peroxide; therefore, it is frequently used in experimental models to scavenge superoxide and demonstrate specificity of an assay. It is important to note that SOD is a much better negative control then SOD mimetic compounds because of potential unspecific reactivity of fluorescent probes, reaction mixture components, and metabolites. 9 In measurements presented in Figure 2 , the fluorescent signal was inhibited in the presence of 50 U/mL SOD, confirming the superoxide-specific origin of the signal. Data from this experiment appear to exhibit a delay in the fluorescence that may be a result of the limited sensitivity of the method. We used a very low activity of xanthine oxidase (0.1 mU/mL) to mimic physiological production of superoxide, whereas in previous publications, 10 mU/mL xanthine oxidase was used. In the first 5 min, the ratio between signals from xanthine oxidase + xanthine and xanthine oxidase + xanthine + SOD samples is low and thus limits the capability of detecting an increasing fluorescence signal. This ratio increases with time and thus overcomes the limitation. To further confirm the specificity of detected fluorescent products, we analyzed these samples using HPLC with a fluorescence detection system. As shown in the inset of Figure 2 , HPLC analysis confirmed formation of the superoxide-specific product 2OH-E in the presence of xanthine oxidase/xanthine and DHE. Detection of superoxide in samples containing low activity of xanthine oxidase suggests that a new protocol is both selective and sensitive for measuring superoxide production in real biological samples.
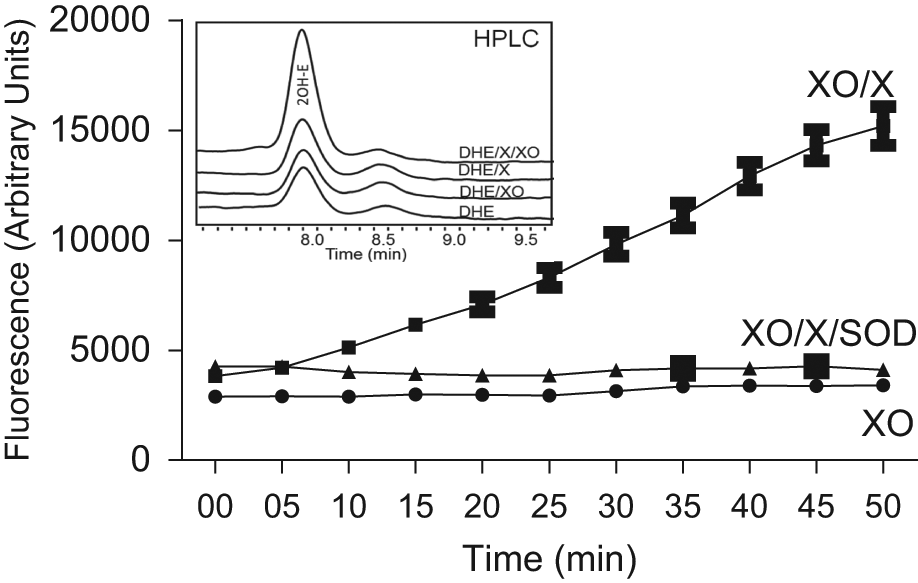
Online superoxide detection by 10 µM dihydroethidium (DHE) in the xanthine oxidase system at 405 nm excitation and 570 nm emission. Freshly prepared solutions of 0.1 mU/mL xanthine oxidase and DHE from DMSO stock were mixed in 50 mM phosphate-buffered saline at pH 7.4. Superoxide production was initiated by 0.2 mM xanthine added from saline stock and inhibited by 50 U/mL superoxide dismutase. The inset shows typical high-performance liquid chromatography (HPLC) analysis of 2-OH-ethidium accumulation in samples containing DHE, xanthine oxidase plus xanthine (DHE/XO/X); DHE, xanthine (DHE/X); DHE and xanthine oxidase (DHE/XO); or DHE. There were no differences in HPLC chromatograms after 10 min.
Next, we tested if this method permits specific 2OH-E detection in intact cells. Toward this aim, we investigated the fluorescence spectra of Eth and 2OH-E in cultured human aortic endothelial cells. Cells grown on fluorescent plates at 90% confluence were loaded with 10 µM 2OH-E (prepared as described in the Materials and Methods section) or Eth (from PBS stocks in Krebs-HEPES buffer) for 60 min and then washed with Krebs-HEPES buffer prior to measurements. The accumulation of Eth or 2OH-E in endothelial cells was confirmed by HPLC using a previously described protocol. 8 As shown in Figure 3 , excitation at 405 nm resulted in diminishing fluorescence emission spectra of the Eth. These data are consistent with cell-free data ( Fig. 1A ), where excitation at 405 nm significantly suppressed the signal of Eth, whereas 2OH-E had a strong fluorescence signal.
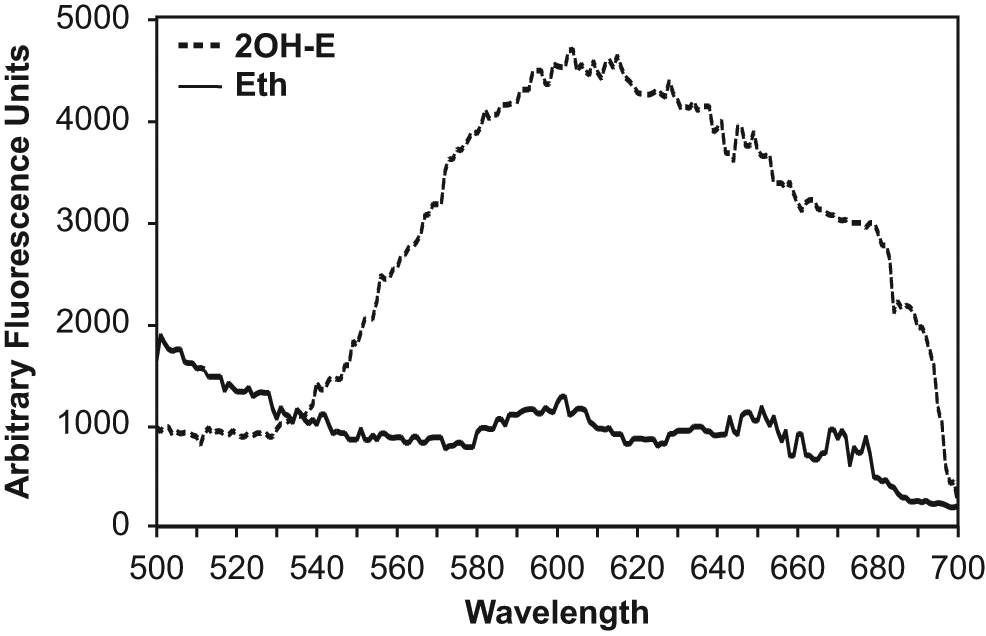
Fluorescence spectra of human endothelial cells loaded with 10 µM ethidium (Eth) or 10 µM 2OH-ethidium (2OH-E) in phosphate-buffered saline, at 405 nm excitation.
Next, we investigated menadione-induced superoxide production in cultured human aortic endothelial cells. Menadione is a well-known redox-cycling quinone used as a model for intracellular superoxide production. 10 For this experiment, cells grown on glass-bottom plates were treated with menadione (15 min, 10 µM), washed, and loaded with DHE (10 µM). Cell-permeable PEG-SOD (50 U/mL) was used to determine the specificity of the fluorescence signal. Accumulation of 2OH-E and Eth in endothelial cells was also confirmed using HPLC. Production of cellular superoxide was monitored online and expressed as a time-dependent increase in the fluorescent signal ( Fig. 4A ) and confirmed with HPLC, as shown in the inset in Figure 4B . Accumulation of superoxide after 30 min of treatment is shown in Figure 4B (plate bottom readings) and was confirmed by HPLC, as shown in the inset in Figure 4B . Both fluorescence spectroscopy and HPLC analysis showed similar increases in superoxide production, indicating the specificity of our improved protocol. As shown in Figure 4A , there was a slight increase of fluorescence in control unstimulated cells, indicating basal superoxide production, whereas menadione caused a robust increase in the fluorescence signal. It is important to note that loading cells with cell-permeable PEG-SOD completely inhibited menadione-induced fluorescent signal, which supports the specificity of superoxide detection.
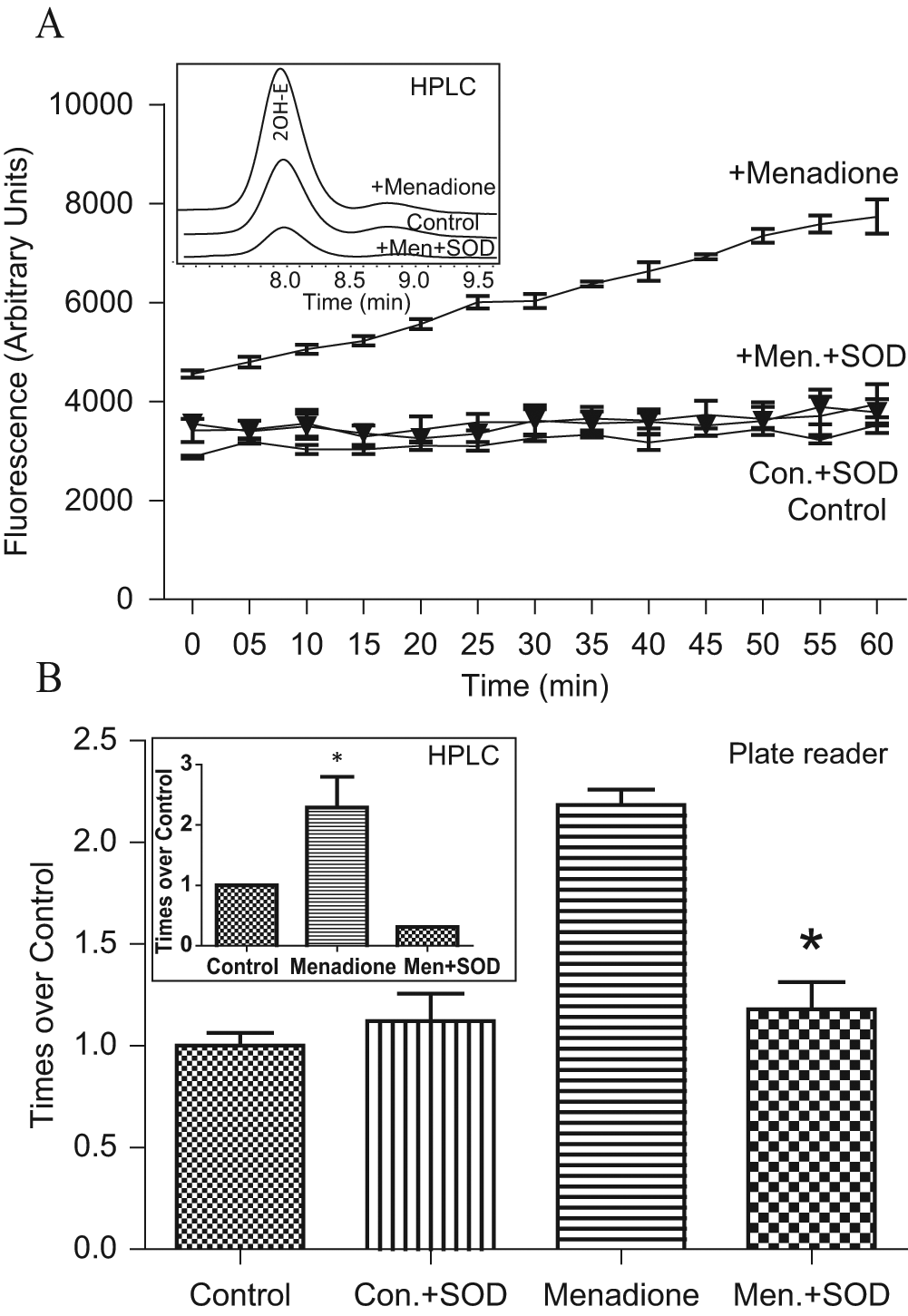
Superoxide detection in cultured endothelial cells. (
Specificity of the superoxide detection using the plate reader was confirmed by HPLC analysis of 2OH-E and Eth in cell-free experiments and in cells treated with menadione. As shown in the insert of Figure 2 , HPLC analysis of the sample with xanthine oxidase plus xanthine and DHE showed specific accumulation of 2OH-E. HPLC analysis of cellular samples showed a significant increase in 2OH-E in menadione-treated cells (insert, Fig. 4B ). These data are in line with our previously reported findings. 10
Although electron spin resonance and HPLC are the most reliable methods for detection of cellular superoxide, 8 these methods do not provide online superoxide detection in attached cultured cells and require time-consuming sample preparation. To overcome these problems, the high-throughput DHE fluorescence assay has been recently suggested; however, according to this method, plate reader data must be confirmed by HPLC analysis. 11 Our work shows that a plate reader–based assay with optimized fluorescence protocol may overcome these limitations and provide a high-throughput method for superoxide detection.
In summary, our data show that optimized fluorescence spectroscopy can be used for rapid and specific measurements of superoxide in cell-free systems and in intact cells to track online kinetic measurements or to determine superoxide production by accumulation of fluorescence signal.
Footnotes
Acknowledgements
We thank Dr. David G. Harrison for his suggestions and fruitful discussion.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was supported by NIH grant PO-1 HL058000.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.