
Abstract
Duchenne muscular dystrophy (DMD) is a devastating muscle-wasting disease caused by mutations in the dystrophin gene. Utrophin is a homologue of dystrophin that can compensate for its absence when overexpressed in DMD animal models. Utrophin upregulation is therefore a promising therapeutic approach for DMD. Utrophin is regulated at both transcriptional and posttranscriptional levels. Transcriptional regulation has been studied extensively, and assays have been described for the identification of utrophin promoter-targeting molecules. However, despite the profound impact that posttranscriptional regulation has on utrophin expression, screening assays have not yet been described that could be used to discover pharmaceuticals targeting this key phase of regulation. We describe the development and validation of a muscle cell line–based assay in which a stably expressed luciferase coding sequence is flanked by the utrophin 5′- and 3′-untranslated regions (UTRs). The assay was validated using the posttranscriptional regulation of utrophin by miR-206. The assay has a Z′ of 0.7, indicating robust performance in high-throughput format. This assay can be used to study utrophin regulatory mechanisms or to screen chemical libraries for compounds that upregulate utrophin posttranscriptionally via its UTRs. Compounds identified via this assay, used alone or in a synergistic combination with utrophin promoter-targeting molecules, would be predicted to have therapeutic potential for DMD.
Introduction
Duchenne muscular dystrophy (DMD) is a fatal, X-linked muscle-wasting disorder, caused by mutations in the dystrophin gene.1,2 Dystrophin provides structural integrity to skeletal and cardiac muscle by linking the subsarcolemmal actin cytoskeleton to the extracellular matrix, via the dystrophin-associated protein complex. In the absence of dystrophin, muscles are unable to transmit force efficiently and become susceptible to damage during contraction and relaxation, leading to cycles of degeneration and regeneration. Eventually, regeneration fails, and muscle fibers are replaced by fatty and fibrous tissue. 2 For patients, DMD causes progressive muscle weakness, dependence on a wheelchair, cardiomyopathy, respiratory insufficiency, and a shortened life span.3,4 There is currently no effective treatment available.
Utrophin is the autosomal homologue of dystrophin. It is expressed in place of dystrophin throughout the sarcolemma in fetal muscle but in adult myofibers is restricted to the neuromuscular and myotendinous junctions. 5 There are two isoforms of utrophin, A and B, that are transcribed from different promoters. 6 Utrophin A is the predominant isoform in the myofiber. 7 Utrophin is highly similar to dystrophin8–10 and can compensate for its absence when overexpressed transgenically in the myofibers of mouse models of muscular dystrophy. 11 Utrophin upregulation is therefore a promising therapeutic approach for DMD. Although numerous preclinical studies have shown functional improvement in dystrophin-deficient mice and dogs following utrophin upregulation,12–18 most of these strategies are still experimental and face technical hurdles. Therefore, a better understanding of the regulation of utrophin expression is crucial for the development of utrophin upregulation strategies that can be used successfully in the clinic.
The regulation of utrophin at the promoter level has been well characterized,19–24 and indeed many of the aforementioned animal studies targeted the utrophin promoter for upregulation. However, it is becoming increasingly clear that posttranscriptional regulation is a major factor in the control of utrophin expression levels and that the 5′- and 3′-untranslated regions (UTRs) of the utrophin mRNA play a central role in this regulation. Specifically, an internal ribosome entry site has been identified in the 5′-UTR, whereas the 3′-UTR contains an mRNA-stabilizing element and is also the target of miRNAs.25–28 Indeed, we have recently shown that the utrophin mRNA exists in a state of translational repression. 28 Therefore, the posttranscriptional regulation of utrophin represents an area of significant potential for the identification of novel therapeutic targets. However, although assays have been developed to screen for utrophin promoter activation,29,30 no screening assay has yet been described that could be used to identify molecules targeting posttranscriptional utrophin regulation.
In this study, we have developed a cell-based luciferase reporter assay for the identification of small molecules, drugs, or other factors that upregulate utrophin via its 5′- and 3′-UTRs. Our assay uses the cytomegalovirus (CMV) promoter to produce a reporter mRNA that consists of the coding sequence of luciferase flanked by the utrophin UTRs. Any tested substance that alters mRNA stability, rate of translation, or targeting by miRNAs would result in a change in luciferase activity. We have validated the assay based on the known regulation of utrophin by miR-206.26,28 We then tested the assay using a 2-O-methyl phosphorothioate (2OMePS) oligomer that upregulates utrophin by blocking certain miRNAs from binding, and determined that the assay is suitable for high-throughput screening based on its Z′ of 0.7. This assay is a valuable tool that could be used for high-throughput screening of chemical libraries for utrophin-activating compounds, as well as testing of other potential therapeutic agents. Such molecules would be useful both from a therapeutic point of view and to further understand the mechanisms of posttranscriptional utrophin expression regulation.
Materials and Methods
Cloning of Construct
The 5′- and 3′-UTRs of utrophin were amplified by PCR and cloned into pGL4:50 (Promega, Madison, WI), to the 5′ and 3′ of the luciferase coding sequence, using the restriction sites HindIII (5′-UTR) and FseI (3′−UTR), to create pGL4:50/5′3′. DNA sequencing was used to confirm successful cloning and absence of PCR errors.
Cell Culture
C2C12 cells (purchased from ATCC) were cultured in high-glucose DMEM with 10% fetal bovine serum, 2 mM L-glutamine, 100 U/mL penicillin, and 0.1 mg/mL streptomycin. Cell culture reagents were purchased from Life Technologies (Grand Island, NY). For the C2C12-5′3′ cell line, 250 mg/mL hygromycin B (Roche Applied Science, Indianapolis, IN) was added to the media. C2C12 cells were plated at 60 000 cells per well in 6-well plates for RNA isolation or 15 000 cells per well in 24-well plates for luciferase assay. C2C12-5′3′ cells were plated at 3 000 cells per well in 96-well plates for DMSO testing or 15 000 cells per well in 24-well plates for all other experiments.
Transient Transfection of Constructs
Equimolar amounts of the pGL4:50 and pGL4:50/5′3′ plasmids (500 and 730 ng per well of a 24-well plate, respectively) were transfected into C2C12 cells using Lipofectamine 2000 (Life Technologies), together with the Renilla luciferase–expressing plasmid pRL-TK as an endogenous control.
Stable Transfection and Selection of Clonal Lines
C2C12 cells were transfected with pGL4:50/5′3′ using Lipofectamine 2000 (Life Technologies) and cultured for 2 weeks in the presence of hygromycin to select for stably transfected cells. Stable colonies were removed from culture dishes using cloning rings and replated in 24-well plates at approximately 1 cell per well. Colonies were allowed to develop and then were trypsinized and replated again at 1 cell per well. This was repeated three times to produce clonal cell lines.
Transfection of Pre-miRNAs and miRNA Inhibitors
Pre-miR-206, anti-miR-206, and equivalent scrambled controls were purchased from Life Technologies and transfected into C2C12-5′3′ cells at 10 nM (pre-miRs) or 300 nM (anti-miRs) using Lipofectamine 2000 (Life Technologies), following the manufacturer’s protocol. Luciferase assays were done after 24 h as described below. In-house manufactured let-7-blocking 2OMePS oligomers or inactive controls were transfected at 300 nM, using Lipofectamine 2000 at a 1:1 ratio. In all cases, analyses were done after 24 h.
DMSO Treatment
For evaluation of sensitivity to DMSO, C2C12-5′3′ cells were seeded in 96-well plates and treated with DMSO (Sigma-Aldrich, St. Louis, MO) at final concentrations of 0 % to 0.5%. Luciferase activity was assayed after 48 h, as described below.
Luciferase Assays
Luciferase assays were done using either the BrightGlo Assay (Promega) for DMSO testing in 96-well plate format or the Dual Luciferase Assay (Promega) for 24-well format experiments, following the manufacturer’s instructions. For experiments using C2C12-5′3′ cells with no Renilla luciferase endogenous control, only firefly luciferase luminescence was measured using the LAR II reagent. For the BrightGlo Assay, luminescence was measured using a Luminoskan Ascent luminometer (Thermo Fisher Scientific, Waltham, MA). For the Dual Luciferase Assay, luminescence was read using a TD 20/20 luminometer (Turner Designs, Sunnyvale, CA).
RNA Isolation and Reverse Transcription
RNA was isolated from cultured C2C12 cells using an RNeasy kit (Qiagen, Valencia, CA), following the manufacturer’s protocol. RNA was reverse-transcribed using a Superscript II First-Strand Synthesis kit (Life Technologies), using random hexamers, according to the manufacturer’s instructions.
Genomic DNA Isolation
Genomic DNA was isolated using an ArchivePure DNA Cell/Tissue kit (5 Prime, Gaithersburg, MD).
PCR
PCRs were done using Taq DNA polymerase, reaction buffer, and dNTPs from Sigma-Aldrich. For PCR from genomic DNA, 100 ng template DNA was used. For PCR from cDNA, 6 ng template DNA was used. In both cases, the luciferase-specific primers 5′-GAGCTATTCTTGCGCAGCTT-3′ and 5′-GTCGAAGATGTTGGGGTGTT-3′ were used.
Results
Generation and Validation of C2C12-5′3′ Cell Line
The construct pGL4:50/5′3′ was generated by cloning the 5′- and 3′-UTRs of utrophin A at the 5′ and 3′ ends of the firefly luciferase coding sequence of pGL4:50. This costruct produces an mRNA consisting of the luciferase coding sequence flanked by the utrophin UTRs, such that luciferase acts as a reporter for regulation of utrophin expression at the posttranscriptional level via the UTRs (
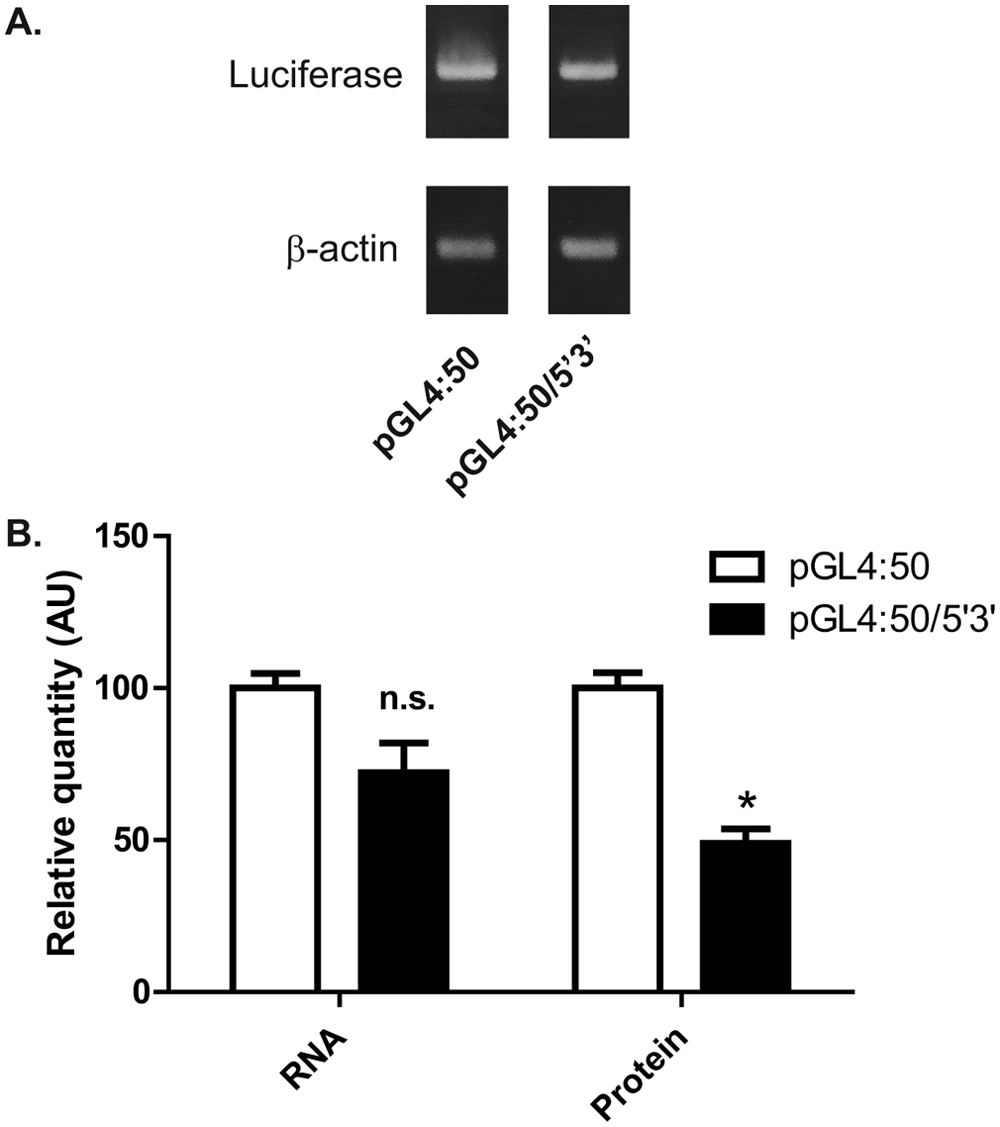
Luciferase translation is repressed by utrophin 5′- and 3′-UTRs. (
To generate the cell line C2C12-5′3′, pGL4:50/5′3′ was transfected into C2C12 mouse myoblasts and stably transfected clones were isolated by antibiotic selection and cellular cloning. Integration of the construct into the cell line genome was verified by PCR from genomic DNA using primers specific to luciferase. In addition, robust luciferase expression was confirmed using a luciferase activity assay (
Development and Validation of Utrophin Posttranscriptional Upregulation Assay
We and others previously demonstrated that utrophin is a target of the miRNA miR-206, which binds to the utrophin 3′-UTR and represses translation.26,28 Therefore, we tested the ability of exogenously transfected pre-miR-206 (a double-stranded precursor RNA hairpin that is processed by Dicer in the cell to give rise to mature miR-206) to decrease the luciferase signal of the C2C12-5′3′ cell line. In line with previous findings, transfection of pre-miR-206 reduced the luciferase activity of the cells by 25%, whereas a scrambled pre-miR did not significantly reduce luciferase activity ( Fig. 2 ).
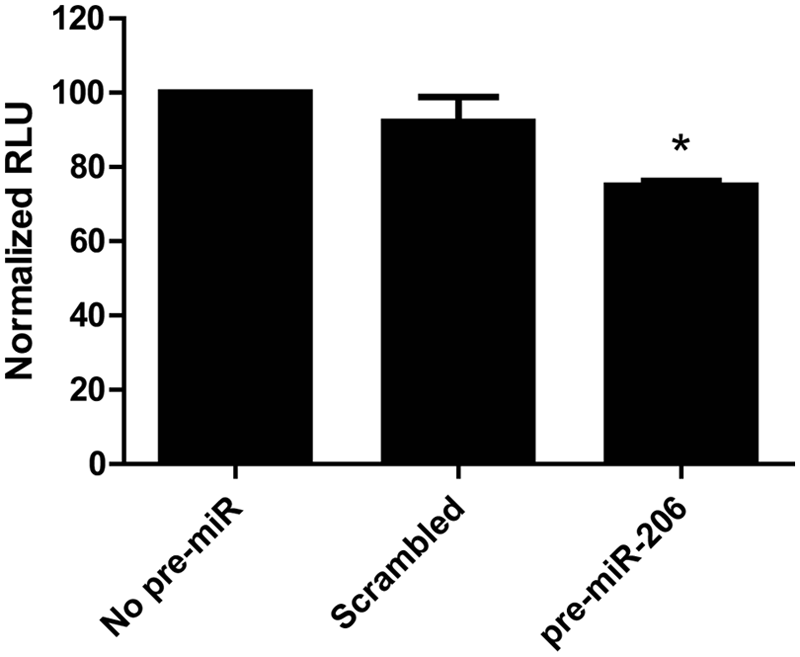
Luciferase activity of C2C12-5′3′ cell line is repressed by miR-206. C2C12-5′3′ cells were transfected with 10 nM pre-miR-206, scrambled pre-miR or no pre-miR, and luciferase activity assayed after 24 h. Transfection with pre-miR-206 produced a 25% decrease in luciferase activity, whereas transfection with a scrambled pre-miR did not produce a statistically significant reduction. Bars represent the mean of 2 independent experiments. Error bars represent the standard deviation. *Significantly different from no pre-miR by Student t test, P < 0.05. RLU, relative luminescence units.
In our previous work, we demonstrated that an antisense inhibitor of miR-206 increased the activity of a transiently transfected reporter construct equivalent to pGL4:50/5′3′. Therefore, we determined whether an antisense inhibitor of miR-206 could increase the luciferase activity of C2C12-5′3′ cells. As expected, transfection of anti-miR-206 produced a 50% increase in luciferase activity compared with a scrambled control ( Fig. 3 ).
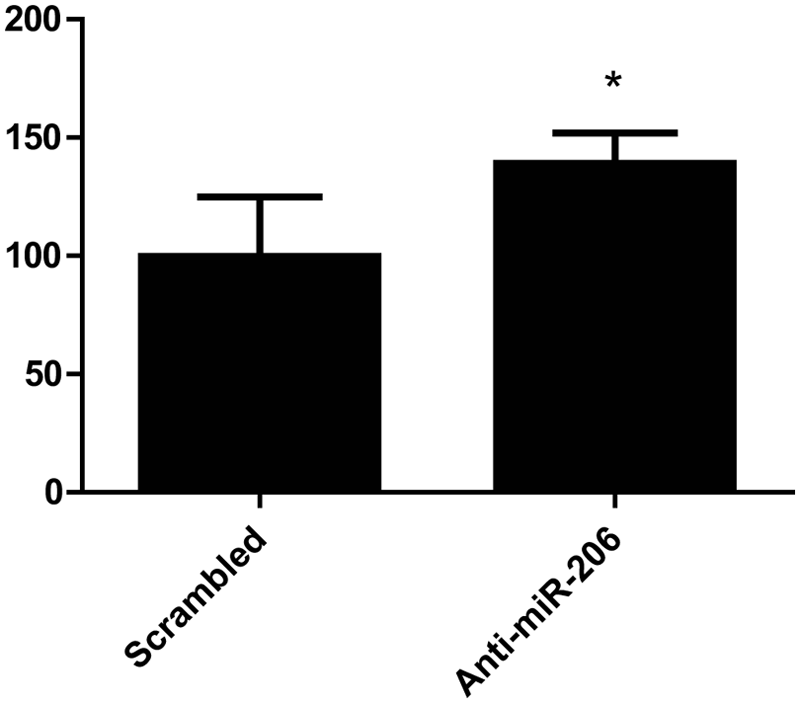
Luciferase activity of the C2C12-5′3′ cell line is upregulated by a miR-206 inhibitor. C2C12-5′3′ cells were transfected with 300 nM of an antisense inhibitor of miR-206 (anti-miR-206), or a scrambled control inhibitor, and luciferase activity was assayed after 24 h. Transfection with anti-miR-206 produced a 50% increase in luciferase activity, compared with the scrambled control. Error bars represent standard deviation. *Significantly different from scrambled inhibitor by Student t test, P < 0.05. RLU, relative luminescence units.
We also tested the response of the C2C12-5′3′ cell line to DMSO, a commonly used solvent in chemical libraries, and found that DMSO concentrations up to 0.5% had little effect on luciferase activity (
Finally, to determine the Z′ for the assay, a measure of statistical robustness and suitability for high-throughput screening, we used a 2-O-methyl phosphorothioate (2OMePS) oligomer that we had been previously shown to upregulate endogenous utrophin protein by preventing the let-7 family of miRNAs from binding to the utrophin 3′UTR, 28 at a range of concentrations from 10 to 600 nM. The 2OMePS let-7-blocker produced a 2.4-fold increase in luciferase activity when transfected into C2C12-5′3′ cells at 300 nM and a 3.9-fold increase at 600 nM compared with an inactive control oligomer ( Fig. 4 ). From the data at 300 nM (–6.5 on the x-axis), we obtained a Z′ of 0.7, suggesting that the assay will perform well in high-throughput format. In addition, the percentage covariances of the inactive control and the positive control at 300 nM were low, 3.0% and 4.5%, respectively, which also indicates that the assay will produce reliable data. Although a 2.4-fold increase could be detected with an acceptable Z′, a 1.7-fold increase (at 100 nM) could not. Therefore, the minimum difference detectable by the assay lies between 1.7- and 2.4-fold. In conclusion, this assay should perform well in high-throughput format and could be used to screen chemical libraries for small molecules that upregulate utrophin at the posttranscriptional level.
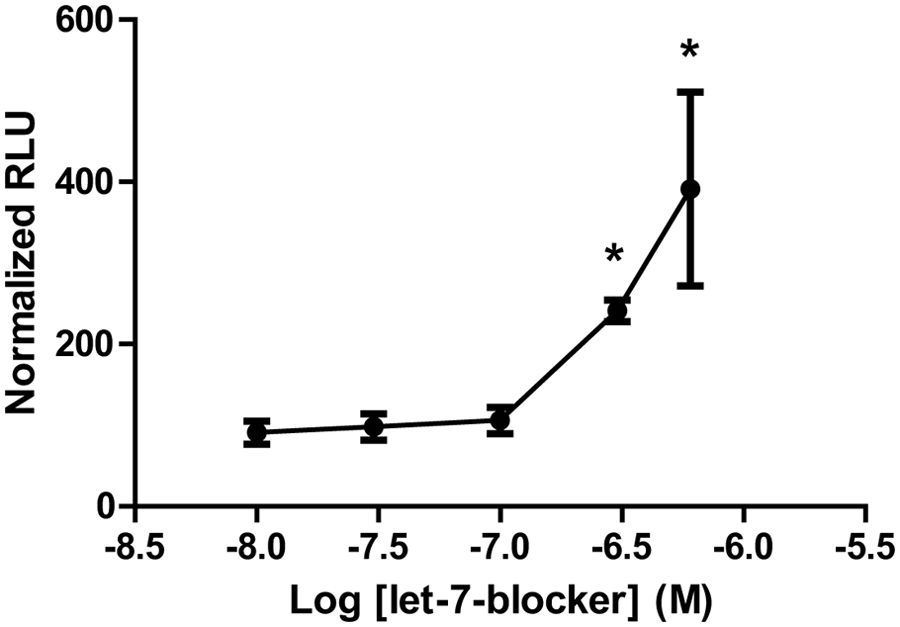
Luciferase activity of C2C12-5′3′ cell line is upregulated by let-7-blocker with Z′ suitable for high-throughput screening. C2C12-5′3′ cells were transfected with let-7-blocker or an inactive control oligomer at a range of concentrations from 10 to 600 nM, and luciferase activity was assayed after 24 h. Transfection with let-7-blocker produced a 2.4-fold increase at 300 nM and a 3.9-fold increase at 600 nM compared with the inactive control. Z′ calculated from data at 300 nM (−6.5 on the x-axis) was 0.7, indicating suitability for high-throughput screening. Error bars represent standard deviation. *Significantly different from inactive control by Student t test, P < 0.05. RLU, relative luminescence units.
Discussion
In this study, we have developed and validated a cell-based assay for posttranscriptional upregulation of utrophin that is suitable for high-throughput screening. In contrast to assays designed to screen for utrophin promoter activation, this assay could be used to screen chemical libraries for small molecules that upregulate utrophin via mechanisms mediated by the 5′- and 3′-UTRs. Based on our previous experience, 29 this assay could be scaled to 384-well format, in which case it is possible to screen about 5000 compounds per day. Compound libraries range in size from about 1000 (e.g., the Prestwick library of Food and Drug Administration [FDA]–approved drugs 29 ) to about 500 000 compounds (e.g., the National Institutes of Health’s MLSMR library 31 ). Using our assay in a 384-well format, it would be feasible to screen libraries in the 1000s to 10 000s range. Larger libraries would require scaling to a higher-throughput format (e.g., a 1536-well format). Hit compounds identified in such screens, once independently validated in vitro, could be tested for therapeutic potential in animal models of DMD, either alone or in a synergistic combination with substances that increase the activity of the utrophin promoter. Mechanistic studies using hit molecules as a starting point could also uncover novel pathways of posttranscriptional utrophin regulation. In addition to screening, the assay is a valuable tool for lower-throughput in vitro studies, as it obviates the need for transient transfection of reporter constructs, providing greater technical flexibility and more consistent results.
In Figure 3 , we saw an increase in luciferase activity of approximately 45% on addition of an antisense miR-206 inhibitor to the C2C12-5′3′ cell line. However, we saw a smaller change (a decrease of approximately 25%) on transfection with pre-miR-206 in Figure 2 . This is probably due to the presence of endogenous miR-206 in C2C12 cells, 28 which is already binding to the miR-206 site on some copies of the luciferase reporter mRNA. Indeed, the miR-206 inhibitor is effective because it alleviates repression by endogenous miR-206. It is also important to note that utrophin is the target of several miRNAs, 28 and hence inhibition of one miRNA will not remove all of the translational repression acting on the utrophin mRNA.
Typically, in high-throughput screens, the threshold for defining a hit is set at three times the covariance of the controls. For our assay, based on Figure 4 , this would be about 10% upregulation. The threshold is set low enough to capture all true-positives, and it is expected that some false-positives will be present. These can be quickly eliminated by high-throughput dose-response testing, in which true-positives show a dose-dependent increase in assay activity. It is difficult to know how a fold-change in the assay will relate to the fold-change in levels of the endogenous target in vivo. Previous studies have shown that a two- to threefold increase in utrophin protein in dystrophin-deficient mdx mice is sufficient to completely rescue the disease phenotype and that smaller increases also have significant benefits. 11 In our previous work using a utrophin promoter activation assay, our lead compound produced a 1.8-fold increase in luciferase activity in our initial screen, a 2.6-fold increase at its optimum concentration during high-throughput dose-response testing, a 1.8-fold increase in endogenous utrophin mRNA, and a 1.2-fold increase in endogenous utrophin protein in vitro. However, these relationships may not be linear and may vary from compound to compound.
A limitation of the assay, as with other cell-based assays, is that compounds affecting cell growth may produce a change in luciferase readout, as will compounds that activate the CMV promoter. To eliminate compounds that activate the CMV promoter, it would be possible to counterscreen using a cell line stably transfected with the parent construct pGL4:50, which does not contain the utrophin UTRs. It would also be possible to measure effects on cell proliferation using bromodeoxyuridine incorporation followed by immunostaining or enzyme-linked immunosorbant assay. However, this method may not be any less time-consuming than confirmation of the activity of the compound on endogenous utrophin. Ultimately, it is important that any hits from compound screens be independently validated using methods such as Western blotting for changes in endogenous utrophin expression in vitro and that any compounds that are effective in vitro be tested in vivo using animal models of DMD.
To date, much research into the regulation of utrophin and its therapeutic upregulation has focused on activation of the utrophin promoter, and we and others previously developed assays to screen for utrophin promoter activation.29,30 However, recent studies show that regulation of utrophin translation and mRNA stability via UTR-mediated mechanisms is of great importance in the control of utrophin protein levels. Indeed, the repressive posttranscriptional regulation of utrophin may be part of the reason that utrophin upregulating therapies have not yet reached the clinic. The identification of posttranscriptional regulatory mechanisms for utrophin provides promising new targets for DMD therapeutics and suggests that to achieve significant utrophin upregulation, it may be necessary to target both promoter-level and posttranscriptional regulation. The assay presented here will be a valuable tool for the identification of drugs targeting posttranscriptional regulation.
High-throughput screens of compound libraries for activation of utrophin at the promoter level have previously been conducted by ourselves and others.29,30 Because the assay presented here is designed to identify molecules acting at the posttranscriptional level, validated hit compounds identified using the present assay would be complementary to those acting on the utrophin promoter and could potentially be used in combination to produce an additive effect on overall utrophin levels. In addition, utrophin upregulation could be used together with other therapeutic strategies such as gene therapy or exon skipping. Such combinatorial approaches may hold great promise for DMD therapy.
Many therapeutic strategies currently in clinical trials for DMD rely on the use of viral vectors or oligonucleotides, which face hurdles in terms of delivery, stability, safety, and regulatory body approval. We propose to use our utrophin posttranscriptional upregulation assay to screen libraries of small molecules for utrophin upregulating activity. Small, druglike molecules have obvious advantages in terms of delivery and bioavailability. In addition, libraries of FDA-approved drugs, with known safety and pharmacokinetic properties, could also be screened, potentially providing a fast-track route to the clinic.
In conclusion, we have developed and validated a high-throughput cell-based reporter assay for posttranscriptional upregulation of utrophin. We propose that this assay could be used to screen chemical libraries for novel utrophin upregulating compounds. Such compounds, used alone or in a synergistic combination with molecules targeting the utrophin promoter, would hold great promise for the development of an effective utrophin upregulation therapy for DMD.
Footnotes
Declaration of Conflicting Interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: C.M., G.P., and T.S.K. declare a conflict of interest in terms of intellectual property rights related to this study that are owned and managed by the University of Pennsylvania.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by grants from the National Institutes of Health (grant AR-048871 to T.S.K. and 5R01NS044146-06 to S.D.W.), the Parent Project Muscular Dystrophy (to T. S. K.), and from the Muscular Dystrophy Association (grant MDA202682 to T.S.K.).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.