
Abstract
The human commensal pathogen Streptococcus pneumoniae expresses a number of virulence factors that promote serious pneumococcal diseases, resulting in significant morbidity and mortality worldwide. These virulence factors may give S. pneumoniae the capacity to escape immune defenses, resist antimicrobial agents, or a combination of both. Virulence factors also present possible points of therapeutic intervention. The activities of the surface endonuclease, EndA, allow S. pneumoniae to establish invasive pneumococcal infection. EndA’s role in DNA uptake during transformation contributes to gene transfer and genetic diversification. Moreover, EndA’s nuclease activity degrades the DNA backbone of neutrophil extracellular traps (NETs), allowing pneumococcus to escape host immune responses. Given its potential impact on pneumococcal pathogenicity, EndA is an attractive target for novel antimicrobial therapy. Herein, we describe the development of a high-throughput screening assay for the discovery of nuclease inhibitors. Nuclease-mediated digestion of double-stranded DNA was assessed using fluorescence changes of the DNA dye ligand, PicoGreen. Under optimized conditions, the assay provided robust and reproducible activity data (Z′= 0.87) and was used to screen 4727 small molecules against an imidazole-rescued variant of EndA. In total, six small molecules were confirmed as novel EndA inhibitors, some of which may have utility as research tools for understanding pneumococcal pathogenesis and for drug discovery.
Introduction
Streptococcus pneumoniae (pneumococcus) is an asymptomatic colonizer of the human upper respiratory tract. However, the commensal bacteria are also the causative agent of respiratory and life-threatening invasive diseases. Dissemination of pneumococci from the nasopharynx into the lungs or bloodstream leads to otitis media, pneumonia, bacteremia, and meningitis. 1 Pneumococcal diseases are traditionally treated with antibiotics and prevented with polysaccharide-protein conjugate vaccines. Unfortunately, because of the dramatic increase in antibiotic resistance and limitations of the currently available vaccines, the threat from pneumococcal disease remains high. Thus, therapeutics directed against novel S. pneumoniae targets are needed to combat pneumococcal infections.
S. pneumoniae pathogenesis is a complex and dynamic process. The human host continuously deploys an array of innate and acquired immune defenses to prevent pneumococci from traversing epithelial barriers. 2 However, the pathogen expresses numerous virulence factors, which can act alone or in concert to promote invasive pneumococcal disease. The major virulence attribute of pneumococcus is the presence of a thick, capsular polysaccharide layer, which inhibits phagocytosis and complement recognition.3,4 In addition to the capsule, there are many other pneumococcal virulence factors that are involved in the disease process. Elucidating the roles played by virulence factors allows an understanding of the pathogenesis of infection and can provide insights into novel therapeutic options.
The presence of a surface nuclease involved in pneumococcal DNA transport was reported more than 40 years ago. 5 S. pneumoniae is naturally transformable, and the nuclease activity of EndA is necessary for nontransforming strand degradation and DNA uptake. 6 EndA is required for efficient bacterial transformation in pneumococcus; strains with deactivating endA mutations exhibit an efficiency of transformation reduced more than 100-fold compared with wild type.7–9 Through its role in DNA uptake, EndA contributes to genetic plasticity, a defining characteristic of the pathogen. 10 This flexibility of pneumococcus likely facilitates responses to evolutionary pressures that provide a significant advantage during infection, such as evasion of host immune defenses or development of antibiotic resistance. 11
The ability of neutrophils to clear invading microorganisms by phagocytosis is well established. Recently, neutrophils have been shown to produce neutrophil extracellular traps (NETs) that entrap and kill pathogens in the extracellular environment. 12 NETs consist of neutrophil DNA as a backbone with embedded antimicrobial peptides, histones, and proteases. The weblike structure of NETs physically trap bacteria, but unlike Shigella and other pathogens, 13 the pneumococcus is relatively resistant to NET-mediated killing. 14 Although not killed by NET constituents, pneumococcus entrapment by NETs impedes bacterial dissemination. However, the surface endonuclease, EndA, facilitates pneumococcal escape by degrading the DNA scaffolding of the NETs. 15 Mutant S. pneumoniae strains lacking EndA activity do not destroy NETs and show decreased virulence in mouse models of infection. 15
EndA’s importance for S. pneumoniae pathogenesis arises from its cardinal roles in transformation and facilitating NET escape. Deletion of endA diminishes the efficiency of transformation, which could hinder the genetic variation that contributes to pneumococcal virulence. Moreover, pneumococci lacking endA are not able to free themselves from NETs and show reduced invasive infection in mice. Given this evidence, we hypothesize that small-molecule inhibition of EndA could attenuate pneumococcal pathogenesis and offer a novel target for the control of pneumococcal infection. The discovery of potent and selective EndA inhibitors that modulate the target in bacteriological studies and mouse models of pneumococcal infection would be an important step in elucidating EndA’s role in the pathogenesis of S. pneumoniae.
In this study, we sought to develop a simple, inexpensive, and robust high-throughput screening (HTS) assay that directly targeted EndA and its catalytic hydrolysis of double-stranded DNA (dsDNA). The use of the DNA dye ligand, PicoGreen, to monitor nuclease activity in real time had been reported previously. 16 We adapted this approach for HTS with EndA. Following optimization and validation, the PicoGreen nuclease assay was used to screen 4727 compounds for EndA inhibitors. This initial screen identified 32 active compounds, and subsequent assays and counterscreens led to an EndA inhibitor with potential for further study and optimization. This work is a promising start for structure-based drug design using hits reported herein, in concert with additional screening efforts to discover novel EndA inhibitors.
Materials and Methods
Materials
The Quant-it PicoGreen dsDNA Reagent (Cat. No. P7581) was purchased from Molecular Probes by Life Technologies (Grand Island, NY). Lambda DNA-Hind III digest (Cat. No. N3012L) was purchased from New England Biolabs (Ipswich, MA). The cloning, production, and purification of EndA(H160G) were reported previously. 17 Unless otherwise stated, all other reagents used for buffers and assays were purchased from Thermo Fisher Scientific (Waltham, MA).
LOPAC Compound Collection
The Library of Pharmacologically Active Compounds (LOPAC) was purchased from Sigma-Aldrich as 10 mM stocks in DMSO. The library was previously prepared as 1 µL samples in 384-well V-bottom polypropylene microplates (Greiner, Monroe, NC), sealed by a ALPS 3000 microplate heat sealer (Thermo Fisher Scientific) and stored at −20 °C. On the day of use, the compounds were thawed and diluted to 100 µM (10× final concentration) in R Buffer (reaction buffer: 25 mM imidazole, 20 mM Tris-HCl pH 8.0, 10 mM sodium acetate, 10 mM MES, 25 mM NaCl, 5 mM MgCl2, 1.5 µM BSA, and 0.01% v/v Triton X-100) over two steps using a Thermo Fisher Scientific Multidrop Combi Reagent Dispenser and Multimek NSX-1536 assay workstation system fitted with a 384-well head (Nanoscreen, Charleston, SC). Finally, 1 µL of this stock was spotted into the wells of a 384-well black PerkinElmer Proxiplate (Waltham, MA) for assay use, as described below.
Kinase Focus Set
The Kinase Focus Set was designed and purchased by the Center for Integrative Chemical Biology and Drug Discovery (CICBDD). Design of this library was based on selection from vendor kinase-directed sets. Initially, more than 100 000 compounds were received from various vendors, in the form of SD files. The vendors had originally designed these kinase-focused libraries using one or more of the following approaches: (1) searching virtual and physical general purpose libraries for compounds similar to known kinase inhibitors; (2) selecting or synthesizing compounds having a hinge-binding motif, that is, adenosine bioisosters with a high likelihood to bind the backbone in the kinase hinge-binding motif conserved in every kinase structure; and (3) structure- or ligand-based virtual screening on representative kinase structures. To reduce the size of the library, while preserving its efficiency in screening, we removed structural duplicates, filtered out compounds that did not satisfy the “rule-of-five,” 18 and, finally, performed a cluster-based selection. Clustering was performed by means of the Pipeline Pilot software (Pipeline Pilot, v. 8.5, Accelrys Software Inc., San Diego, CA) in such a way that compounds belonging to the same cluster had pairwise similarity of 50% or more (according to Tanimoto metrics with ECFP4 descriptors). Once clustered, we selected 3 to 10 representative compounds, depending on the cluster size. In the end, the CICBDD acquired 4727 representative compounds that constitute the Kinase Focus Set.
The compound plates of the Kinase Focus Set were prepared by resuspending the powder stock to 1 mM in 100% DMSO in barcoded glass vials with sonication using a Covaris S2 sonicator (Covaris, Woburn, MA). Compounds were plated at 1 mM in 100% DMSO in 384-well V-bottom polypropylene microplates using a Tecan Genesis 200 (Münnedorf, Switzerland). A Multimek spotted 1 µL of the 1 mM compounds into 384-well V-bottom polypropylene microplates, and plates were heat sealed and stored at −20 °C. During screening, these previously prepared plates were thawed and diluted to 100 µM by a single addition of R Buffer, and 1 µL of this stock was spotted into the wells of a 384-well Proxiplate.
PicoGreen Nuclease Assay
The assay was performed by using a Multidrop dispenser to add 1 µL of 10% (v/v) DMSO in the left two columns (columns 1 and 2) for the positive control reactions. In the right two columns (columns 23 and 24), 1 µL of 500 mM EDTA was spotted for the negative control reactions. Assay reactions were carried out directly in the Proxiplate wells containing 1 µL compounds or controls. The reaction volume in each well was 10 µL, and final concentrations of compound and DMSO were 10 µM and 1% (v/v), respectively. A 2x solution of EndA(H160G), containing 30 nM EndA, was prepared in R Buffer. To all wells of the assay plates, 4.5 µL of the 2x EndA(H160G) in R Buffer was added using a Multidrop dispenser. EndA was allowed to preincubate with compounds and controls for 20 min in a 37 °C air incubator. Following preincubation, a 2x solution of lambda DNA-Hind III digest (2 µg/mL) in R Buffer was added at a volume of 4.5 µL to each well. Reaction time began immediately upon addition of the dsDNA substrate and was allowed to proceed for 30 min at room temperature. The plates were quenched by adding 10 µL of 2x PicoGreen Reagent (1.6 µM or 1/200 dilution) in Q Buffer (Quench Buffer: 10 mM Tris-HCl pH 8.0, 20 mM EDTA) to each well. Plates were incubated at ambient temperature in the dark for 5 min prior to reading on an EnVision Multilabel Reader (PerkinElmer) using an excitation wavelength of 480 nm and emission wavelength of 520 nm.
Compounds being evaluated for dose-response runs were attained from frozen 10 mM stocks in 100% DMSO. The Kinase Focus Set hits were plated as threefold dilutions over 10 points using the Tecan in 384-well V-bottom polypropylene microplates. The dose-response curves for LOPAC hits were prepared as threefold dilutions over 10 points using a multichannel pipette. Compound titration series of all hits for IC50 evaluation were spotted at 1 µL by a Multimek instrument into 384-well V-bottom microplates and diluted 10-fold in R Buffer using a Multidrop dispenser. The diluted titrations were then spotted at 1 µL into 384-well black Proxiplates (PerkinElmer) in duplicate, and reagents were added to initiate the assay as described above. The final top concentration of dose-response curves was 30 µM compound (1% v/v DMSO).
Data Analysis
The fluorescence count of each well was measured using an excitation wavelength of 480 nm and an emission wavelength of 520 nm on the EnVision plate reader. Screening data were processed using ScreenAble software (Screenable Solutions). The relative inhibition, expressed as a percentage of the inhibition of assay signal change elicited by 50 mM EDTA, was determined on a plate-to-plate basis by comparing the fluorescence count per compound well (F) with the plate-averaged control wells (n = 32 for each control), using the following relationship:
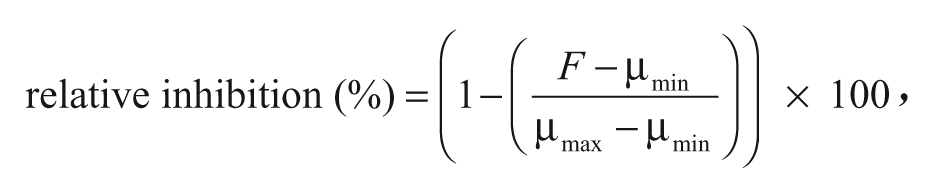
where µmin and µmax are the plate averages of the minimum signal (maximum EndA activity, 1% v/v DMSO added) and maximum signal (fully inhibited EndA, 50 mM EDTA added) controls, respectively.
Compound IC50 values were calculated by first converting the F count to percentage inhibition with respect to on-plate controls and then fitting the percentage inhibition values to curve equations using ScreenAble or Prism (GraphPad Software, San Diego, CA). The IC50 values of the Kinase Focus Set hits were calculated using ScreenAble with a three- or four-parameter curve fit. The LOPAC hits were analyzed in Prism using a one-site specific binding with adjustable Hill slope and B min parameters.
Plasmid DNA Conversion Assay
EndA was diluted stepwise to 100 pM in activity buffer (20 mM Tris-HCl pH 8.0, 5 mM MgCl2, 50 mM imidazole) and incubated with 15 ng/mL of supercoiled pBluescript II SK(+) plasmid (Stratagene), in the presence of DMSO or inhibitor. At scheduled time points, the reactions were quenched by addition of loading dye containing EDTA (250 mM). Reaction products were separated by electrophoresis on an agarose-TBE gel (0.8 % w/v). DNA species in the gel were visualized by ethidum bromide staining and scanned with the Gene Genius Bio imaging system (Syngene).
Real-Time PicoGreen Nuclease Assay
All reactions were carried out in 96-well plates (Corning 3991 black flat-bottom) at 25 °C inside the POLARstar Ultima plate reader (BMG labtech). Fluorescent intensity counts were recorded at an excitation wavelength of 480 nm and emission wavelength of 520 nm. Reaction mixtures (100 µL) consisted of 5 µL of 100% DMSO or 20× inhibitor in 100% DMSO and 90 µL of 16.6 nM EndA(H160G) in RT buffer (real-time buffer: 25 mM imidazole, 20 mM Tris-HCl pH 8.0, 10 mM sodium acetate, 10 mM MES, 25 mM NaCl, 5 mM MgCl). Reaction was initialized by addition of 5 µL of 20× lambda DNA-Hind III digest (1 µg/mL final) and 20× PicoGreen (0.3 µM final) in RT buffer. After initialization, shaking for 30 seconds mixed the plate and plate data recorded every 1 min for 70 min. Initial velocities were calculated and used in further analysis. The counterscreen assay was conducted following the same protocol used in the real-time PicoGreen nuclease assay excluding EndA(H160G). In detail, inhibitors were diluted to 20× concentrations (1, 30, 100 µM final) in 100% DMSO, and 5 µL was added to each well followed by 95 µL of lambda DNA-Hind III digest (1.5 µM final) and PicoGreen (0.3 µM final). The fluorescent intensity counts were recorded and averaged over 10 to 15 min for use in further analysis. The rapid-dilution assay was also performed using the real-time PicoGreen nuclease assay with EndA(H160G) preincubated with a high concentration of inhibitor. Specifically, 2 µL of a solution of 1 µM EndA mixed with 90 µM inhibitor for 20 min at room temperature was diluted into 95 µL of RT buffer. Reaction was started with 5 µL addition of 20× lambda DNA-Hind III digest (1 µg/mL final) and 20× PicoGreen (0.3 µM final) in RT buffer and proceeded as described above for the real-time PicoGreen nuclease assay.
Results and Discussion
Assay Development and Validation
Our main objective in developing an HTS assay for EndA nuclease activity was to identify selective chemical probes that would enhance our understanding of EndA’s role in pneumococcal virulence. However, we were confronted with a challenge given the lack of HTS-compatible methods for detecting nuclease activity. Traditional nuclease activity methods such as viscometry, gel electrophoresis, and assays using radioactive or fluorescent-labeled DNA susbstrates 19 are time-consuming, laborious, and can be very expensive. In comparison, a label-free fluorescent nuclease assay would be continuous, convenient, and HTS conducive.16,20 Here, we developed and optimized an EndA nuclease assay using the fluorescent dye ligand PicoGreen. We also assessed the robustness, reproducibility, and HTS compatibility of the PicoGreen assay to ensure its success in screening diverse druglike small molecules for the inhibition of EndA.
The use of fluorescent DNA dye provides both a precise and cost-efficient method of monitoring nuclease activity in vitro. PicoGreen is a sensitive fluorescent dye widely used in analytical protocols in which dsDNA detection is needed. PicoGreen associates with dsDNA through intercalation, minor-groove binding, and electrostatic interactions,
21
resulting in a significant increase in fluorescence compared with free dye in solution. During reaction with EndA, the dsDNA is cleaved into small fragments, which have a weakened interaction with PicoGreen, resulting in lower fluorescence (
Standard fluorescence versus DNA concentration curves using several dsDNA substrates with 0.8 µM PicoGreen were produced. Comparing the standard curves for calf thymus DNA, salmon testes DNA, and lambda DNA digested with Hind III, we selected the latter as our substrate. The standard curve of lambda DNA showed a linear signal range over three orders of magnitude and at 1.0 µg/mL DNA was still in the linear range with sufficient fluorescence to achieve an adequate signal window ( Fig. 1A ).
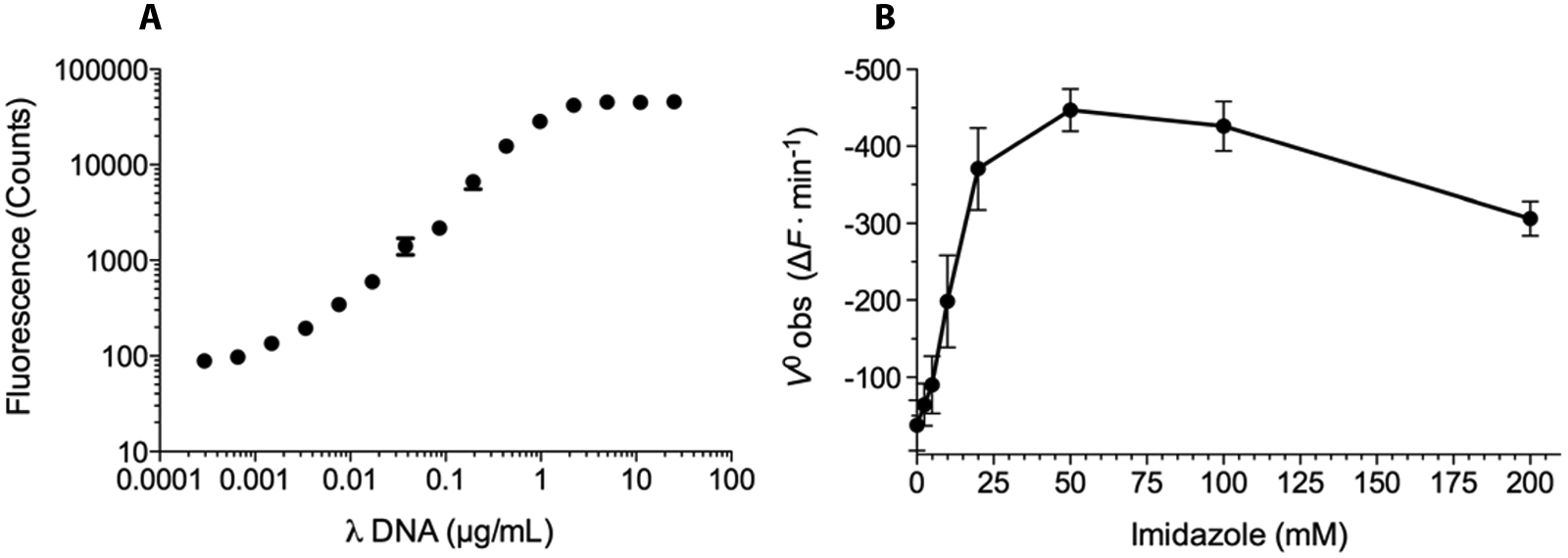
Assay optimization with respect to (
The development of EndA inhibitors requires the production of active and stable forms of EndA for in vitro activity assays. Unfortunately, successful overexpression procedures of recombinant wild-type EndA has not been reported to date, presumably because of uncontrolled nuclease degradation of the host DNA/RNA. Historically, the enzyme has been expressed as an inactive mutant, with the catalytic active site histidine residue replaced with alanine (H160A).17,22 Structure-based modeling suggests that the active site of the EndA variant can accommodate a single imidazole molecule in place of the missing histidine sidechain, leading to rescue of the nuclease activity. Structural information also suggests that the Cβ of H160A might present a steric clash with the imidazole, which could be relieved by a glycine substitution (H160G) at that position. Consistent with this theory, the EndA(H160G) mutant is considerably more active than EndA(H160A). 17 We therefore elected to use EndA(H160G) for assay development, and imidazole can be considered a cofactor in assays measuring the imidazole-dependent nuclease activity of EndA(H160G). 23
To determine the optimum concentration of imidazole, the kinetics of 10 mM EndA(H160G) catalyzed DNA hydrolysis were measured using the PicoGreen assay at imidazole concentrations ranging from 0 to 200 mM ( Fig. 1B ). The observed enzyme velocity reached a maximum at 50 mM imidazole and then decreased at higher concentrations. This inverse effect was reported previously with EndA(H160G) and is likely explained by the elevated ionic strength of the buffer at high imidazole concentrations. 17 An imidazole concentration of 25 mM was chosen as the optimal concentration for the PicoGreen assay.
In the presence of 25 mM imidazole and a saturating concentration of Hind III digested lambda DNA (1 µg/mL), EndA’s nuclease activity increased monotonically with initial EndA concentration (
Another advantage of the PicoGreen assay is its adaptation for real-time monitoring of PicoGreen fluorescence changes, as EndA nuclease activity is unaffected by the presence of PicoGreen dye (data not shown). This allows kinetic and mechanistic parameters of putative inhibitors to be explored using the same assay method.
Given that small-molecule library samples are dissolved in DMSO in preparation for screening, the effect of DMSO on EndA activity was investigated at several different DMSO concentrations, up to 10% (vol/vol;
In other studies, we examined the effect of bovine serum albumin (BSA) and detergent on the signal-to-background window of the PicoGreen assay. In screening assays, BSA provides an alternative interaction surface for promiscuous small-molecule aggregates, and detergent disrupts the formation of colloidal aggregates that can nonspecifically inhibit enzyme activity.
25
The influence of BSA and TritonX-100 were evaluated over three concentration points, and no substantial effects were observed on the signal-to-background window (
To assess the reproducibility of the PicoGreen assay, a number of test 384-well plates were evaluated for interplate and interday variability. Over a span of three days, nine replicate assay plates were run under the optimized conditions described above and used to compile the mean control signals ( Fig. 2 ) for Z′ factor calculation. 26 The maximum signal or inhibited controls (32 per plate, two columns of a 384-well plate) consisted of wells containing EndA, DNA, imidazole, and 50 mM EDTA in buffer. The minimum signal controls (32 per plate, two columns of a 384-well plate) consisted of wells containing EndA, DNA, imidazole, and 1% DMSO in buffer. The PicoGreen reagent was added after 30 min, followed by measurement of fluorescent intensity signal using an EnVision plate reader. The average control signals for interplate assays performed on the same day were analyzed and used to calculate the Z′ values of 0.61, 0.63, and 0.64 for each day. In addition, the interday variability in the maximum signal control was less than 10%. Overall, these results are characteristic of a high-quality assay suitable for HTS, and Z′ values were found to improve with use of liquid-handling equipment.
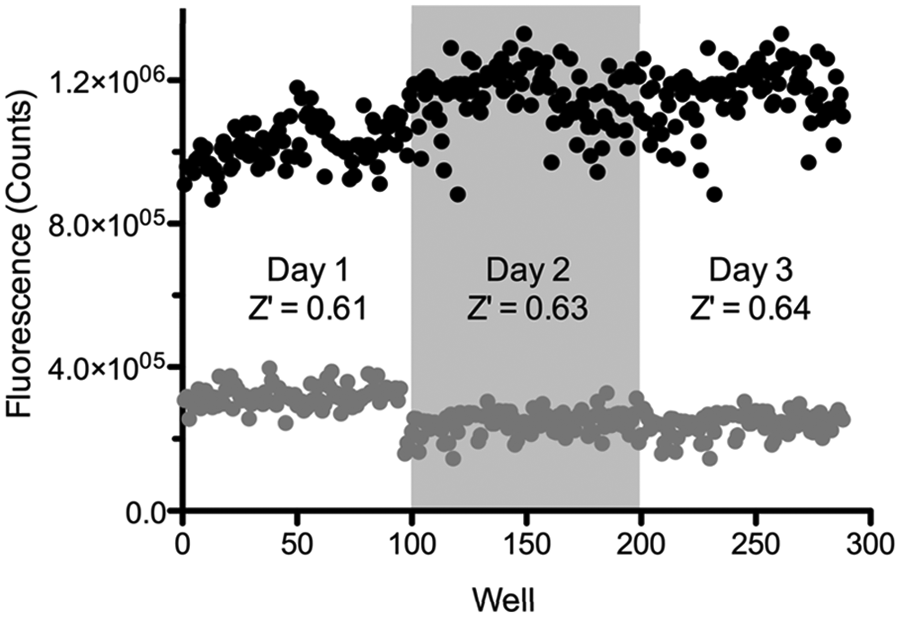
Assay validation over multiple plates and multiple days. The 384-well plates were divided into 32 wells of minimum signal (gray circles) and 32 wells of maximum signal (black signal) control reactions. The variation was measured between three individual runs on the same day and the variation between runs on three separate days. Without using a liquid-handling system, the overall Z′ for 96 wells on days 1, 2, and 3 were 0.61, 0.63, and 0.64, respectively.
Pilot Screen of LOPAC Library
Using the optimized and validated PicoGreen assay, we screened LOPAC against EndA(H160G) as a pilot screen study. The LOPAC contains 1280 biologically active compounds that were compressed into four 384-well plates. The LOPAC was assessed in duplicate for EndA inhibition at a final compound concentration of 10 µM. The LOPAC pilot screening results demonstrated a normal distribution centered on a mean of 6% inhibition with a standard deviation (SD) of 8% (
Fig. 3A
). Moreover, the LOPAC pilot screen was characterized by an average Z′ factor of 0.87 (
Fig. 3B
) and demonstrated strong correlation between replicates (
Fig. 3C
). To ensure that we would identify only high-quality hits, we used a selection criterion of 40% inhibition, which represented more than 4 standard deviations away from the minimum signal control. As summarized in
Figure 4
, we identified 16 active compounds from the primary LOPAC screen, and 14 of these compounds were obtained as DMSO stocks for IC50 analysis. In total, 9 of 14 compounds were confirmed as true actives, and 3 compounds, MK-886 (PubChem CID: 4519262), 6-hydroxy-
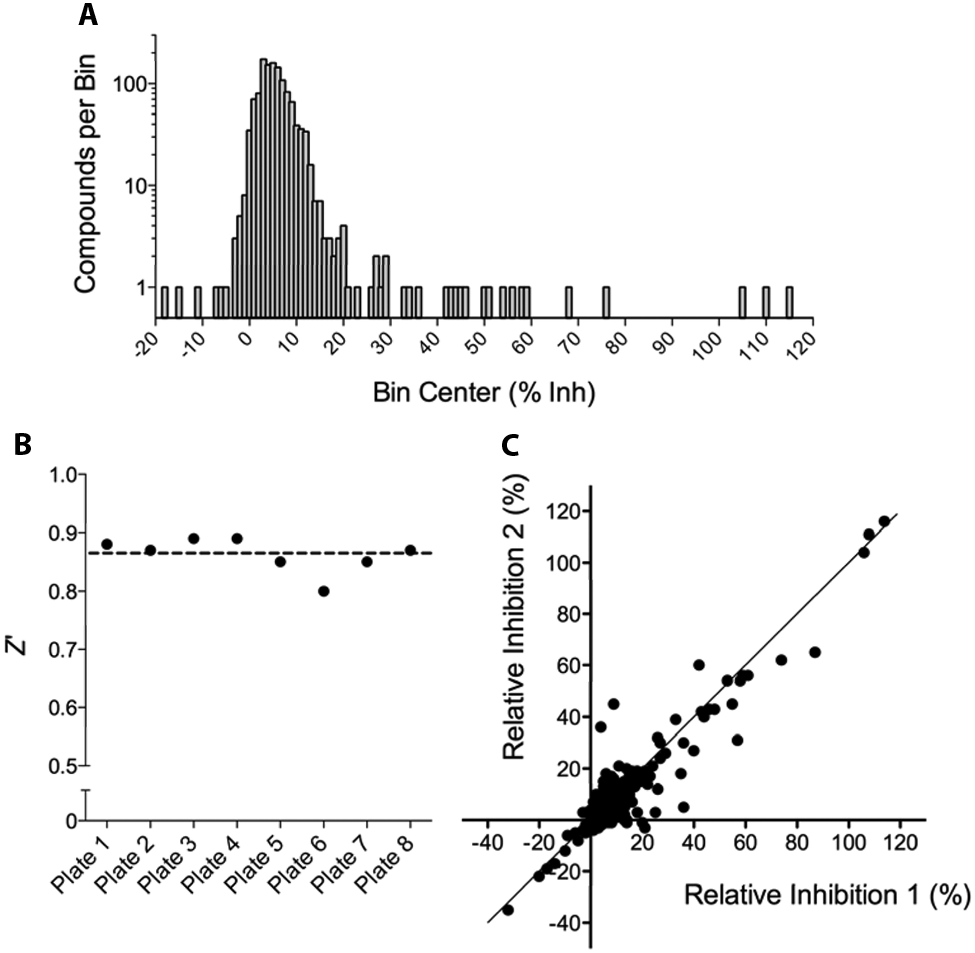
Results from the LOPAC pilot screen. (
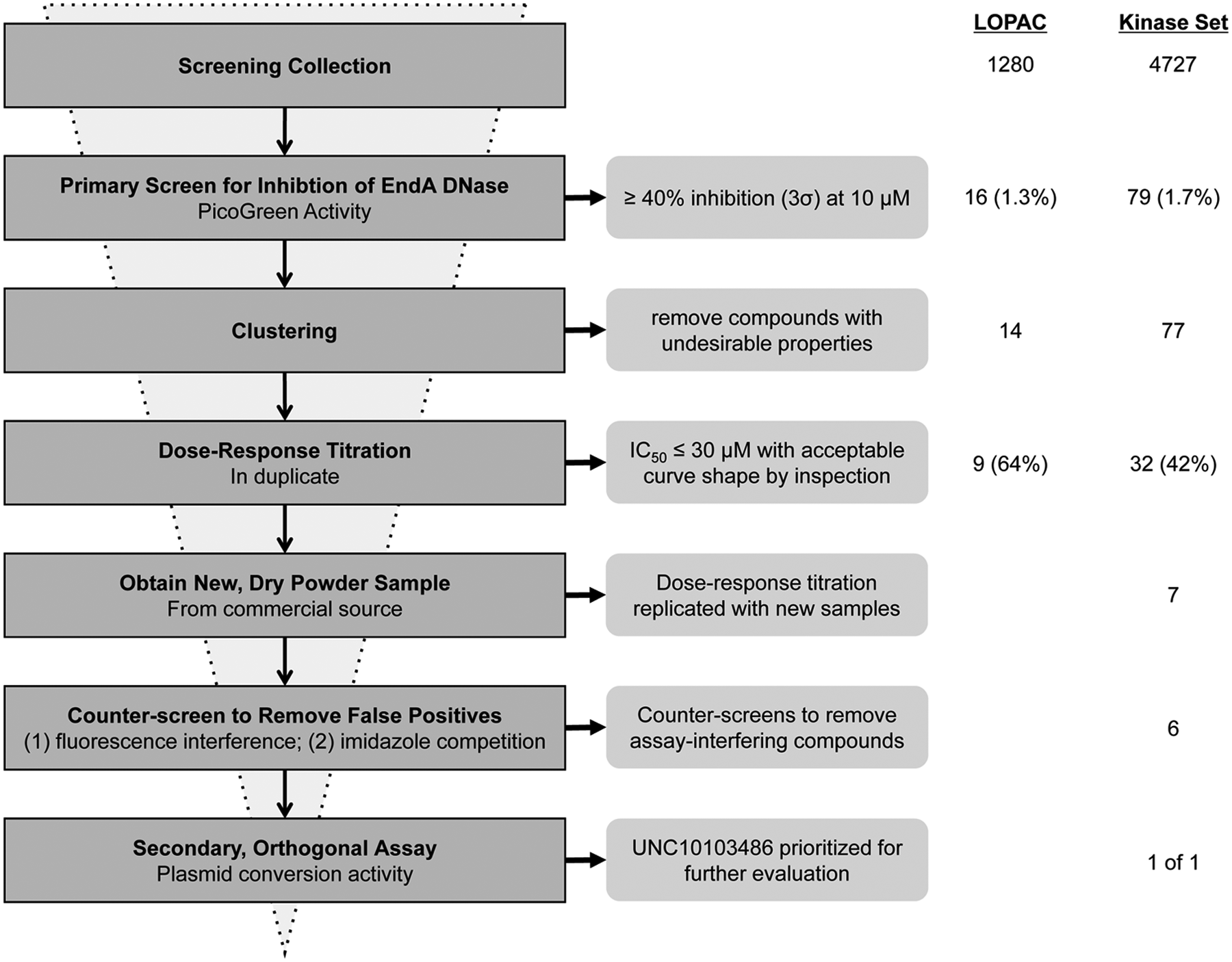
Critical path for EndA inhibitor discovery. In the primary screens, 1280 and 4727 compounds were assessed from the LOPAC and UNC Kinase Focus Set, respectively. In the LOPAC screen, 16 actives were identified (1.6% hit rate), of which 14 were selected to retest in dose-response format. Of these, nine compounds were confirmed as inhibitors (64% confirmation rate). In the Kinase Focus Set screen, 79 actives were identified (1.7% hit rate), of which 77 were selected to retest in dose-response format and 32 compounds were confirmed as inhibitors (42% confirmation rate). Of these, seven were available as dry powder compounds, all seven of which replicated dose-response behavior. Six of the seven compounds passed the counterscreens for interfering compounds and imidazole-competitive compounds. Based on its potent IC50 and other acceptable characteristics, UNC10103486 was prioritized for evaluation in the orthogonal assay for plasmid conversion.
Kinase Focus Set Screening
The Kinase Focus Set was selected as the compound collection for our continuing screening efforts to identify inhibitors of EndA nuclease activity. The Kinase Focus Set was designed at the University of North Carolina at Chapel Hill (UNC) using a combination of kinase pharmacophore-based searching and selection from vendor kinase–directed sets. Although EndA is not a kinase, the small molecules of the Kinase Focus Set were of a manageable number, represented a diversity of scaffolds, and were “rule-of-five” compliant. 18 The Kinase Focus Set has been routinely screened at UNC using nonkinase targets. Moreover, the Kinase Focus Set has yielded selective and potent inhibitors against another nucleic acid-processing enzyme. 29
Compounds from the Kinase Focus Set were screened against EndA in singleton at 10 µM from 1 mM DMSO stocks using the PicoGreen assay. The screening results gave a normal distribution with a mean of 5% inhibition and an SD of 11% ( Fig. 5A ). Overall, the average Z′ = 0.87 for the 16 analyzed plates ( Fig. 5B ). The screen yielded 79 compounds with ≥40% inhibition (3·σ above the mean) and a 1.7% primary screen hit rate ( Fig. 4 ). We eliminated two compounds with unfavorable physiochemical properties and performed dose-response analysis of 77 compounds from their 10 mM DMSO stocks from the CICBDD. Concentration-response evaluations were carried out in duplicate for the 77 initial actives, the results confirmed 32 of the 77 compounds as true actives with IC50 ≤30 µM (i.e., 42% confirmation rate). Raw data for duplicate runs of a validated hit compound demonstrated reproducible dose-dependent inhibition of enzyme activity over the concentrations tested and a mean IC50 = 9 ± 1 µM ( Fig. 6 ).
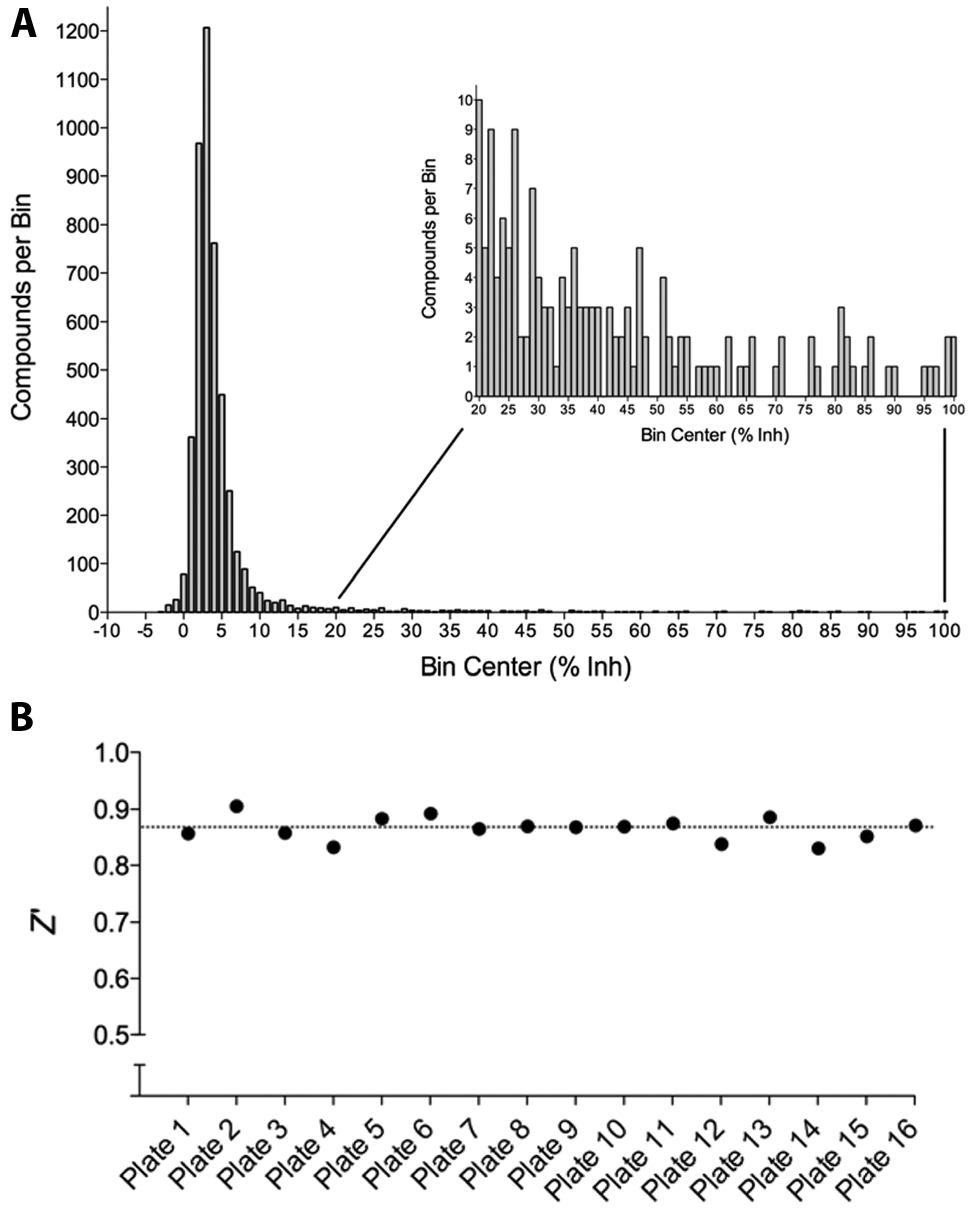
Kinase Focus Set high-throughput screening (HTS) results. (
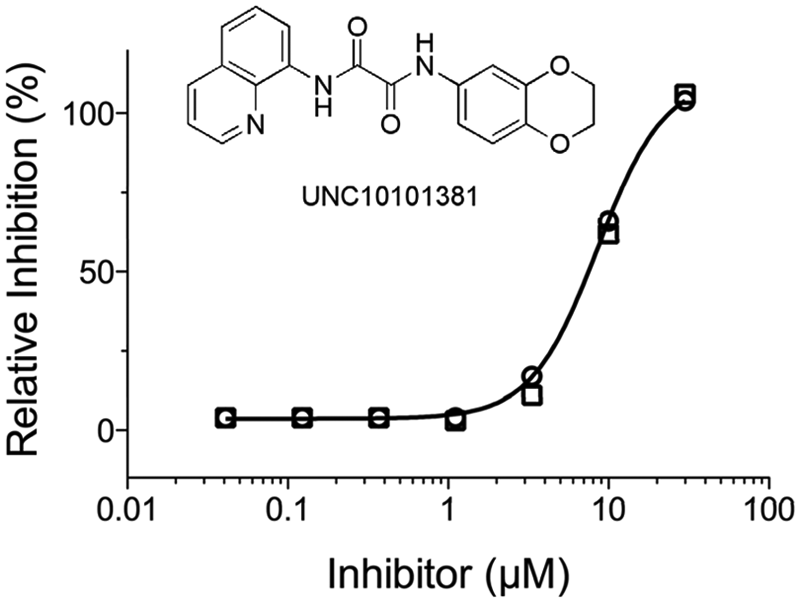
Reproducible concentration-response measurements of a confirmed inhibitor from the Kinase Focus Set screen. The calculated IC50 = 9 ± 1 µM was reproducible for duplicate runs: n = 1 (○) and n = 2 (□).
The primary screen of the Kinase Focus Set using the EndA assay showed a remarkably high overall confirmed hit rate (0.68%), calculated as the percentage of confirmed hits (IC50 ≤30 µM) from the entire Kinase Focus Set library ( Fig. 4 ). With respect to the overall confirmed hit rate, the EndA screen was ranked fourth among 12 screening campaigns against the Kinase Focus Set at UNC. The three screens that showed higher hit rates (between 0.85% and 1.18%) all targeted protein kinases; the eight other screens with lower hit rates were not against kinases. It is unclear why compounds featuring kinase-like chemical motifs (mainly by mimicking the adenosine moiety of ATP) were so efficient on a target lacking a well-defined ligand/cofactor pocket. 17 A plausible hypothesis is that prospective kinase inhibitors may fortuitously possess a chemical feature that also makes them potent against endonucleases. For instance, most known kinase inhibitors are designed to have a common structural fragment called the hinge-binding motif. The hinge-binding motif is a packed set of hydrogen-bond donors and acceptors that bind to a portion of the protein backbone connecting the large and small lobes of the kinase domain. 30 However, it is common for such a motif to also have chelating properties and hence may also bind the Mg2+ ion present in the EndA catalytic site. Regardless, the overall screening statistics indicate a high-quality assay and confirm the utility of the PicoGreen assay to discover novel inhibitors of EndA(H160G) by HTS.
Biochemical Characterization of Confirmed Actives
Of the 32 compounds identified and confirmed as inhibitors, 7 were available from commercial sources. We obtained the 7 compounds, freshly prepared stocks at 10 mM in 100% DMSO, and retested the 7 compounds in dose response and performed further assays to better characterize their biochemical activity and prioritize them for follow-up microbiological experiments. To counterscreen against possible off-target effects, we employed two assays that enabled us to identify false-positives that were identified due to DNA binding by the test compounds or assay interference. We also tested inhibitors to determine whether binding was reversible. Finally, we employed a plasmid conversion nuclease assay17,22 to confirm inhibition of EndA by an orthogonal method.
A compound that binds DNA could produce a false-positive in the PicoGreen assay by sequestering the DNA, thereby preventing EndA nuclease activity. To test for DNA binding, we employed the real-time PicoGreen nuclease assay in the absence of EndA(H160G) at three concentrations (1, 30, 100 µM final) of test compound. True inhibitors of EndA had no effect on fluorescence, whereas compounds that bind DNA or interfere with the DNA-PicoGreen interaction decreased the fluorescence signal. Only one of the seven compounds showed a significant effect in the counterscreen (

Biochemical characterization of a promising EndA inhibitor. (
The PicoGreen nuclease assay measured the imidazole-dependent nuclease activity of EndA(H160G). Therefore, it was possible that some of the identified compounds inhibited the rescued EndA variant by interfering with imidazole binding to the active site. To test compound UNC10103486 for an imidazole-dependent mechanism of action, EndA(H160G) activity was measured by the real-time PicoGreen nuclease assay as a function of imidazole concentration at different inhibitor concentrations. Analysis of the resulting velocity-imidazole concentration curves revealed that UNC10103486 inhibition was not competitive with imidazole ( Fig. 7B ). Moreover, calculation of IC50 from the velocity-imidazole concentration curves determined a constant IC50 = 16 ± 1 µM over increasing imidazole concentrations ( Fig. 7C ) and confirmed UNC1013486 was not competitive with imidazole.
To determine whether UNC10103486 binding was reversible, we adapted the real-time PicoGreen nuclease assay with a rapid dilution assay. Briefly, inhibitor was allowed to bind EndA at a concentration expected to inhibit roughly 95% of EndA activity, then diluted to a concentration expected to inhibit only 5% of EndA activity. A reversible inhibitor would dissociate quickly, allowing immediate recovery of enzyme activity, a slowly reversible inhibitor would allow a gradual increase in activity, and an irreversible inhibitor would completely prevent the recovery of enzyme activity. Dilution of EndA inhibited by UNC10103486 did allow a gradual recovery of EndA, with an initial velocity of 62 ± 22 counts·min−1, compared with 128 ± 6 counts·min−1 with DMSO alone ( Fig. 7D ). This confirmed a reversible binding mechanism for UNC10103486.
We confirmed the inhibition of EndA by UNC10103486 by an alternative nuclease assay for plasmid DNA conversion. In this simple assay, supercoiled plasmid DNA is nicked by EndA and transformed into an open circular structure that migrates significantly more slowly during electrophoresis on an agarose gel. Further plasmid cleavage leads to a linear DNA form that migrates as a band between the supercoiled and open circular forms. Progressively, more cleavage produces oligonucleotides of various sizes, which appear as a smear of faster-migrating products on the agarose gel. We monitored the DNA products in the presence of UNC10103486 and found a dose-dependent inhibition of plasmid DNA cleavage ( Fig. 7E ). EndA inhibition in the plasmid DNA cleavage assay confirmed the activity of UNC10103486 and also that its mechanism was independent of the PicoGreen assay detection method.
In conclusion, we have, to the best of our knowledge, established the first reported HTS assay to identify compounds that inhibit nuclease activity. Using a nuclease assay based on the differential fluorescence output of PicoGreen, we optimized the assay for HTS against an imidazole- rescued variant EndA(H160G). Following stringent assay validation, including a pilot screen of LOPAC, the assay was used to screen 4727 compounds from the Kinase Focus Set. The screen had an average Z′ = 0.87 and resulted in the identification of 32 confirmed actives. From these hits, the compound UNC10103486 was further validated as an inhibitor in an orthogonal EndA nuclease assay and acts via a mechanism independent of PicoGreen and imidazole.
The validation of the PicoGreen nuclease assay and its use in HTS is fundamental to the search for nuclease inhibitors. Specifically, drugs that inhibit nuclease activity may help to improve the outcome of bacterial infection with Streptococcus pyogenes and Staphylococcus aureus, which also express nucleases that facilitate NET evasion.33,34 In addition, a major advantage of the PicoGreen nuclease assay is its versatility as both a screening assay and for monitoring nuclease activity in real time. Such broad application of this technology allows for a consistent transition from primary screening to more in-depth studies of binding properties and mechanism of inhibition.
The S. pneumoniae surface endonuclease EndA has at least two functions that are important in pneumococcal disease. EndA is responsible for DNA degradation and transport during transformation, which contributes to pneumococcal genome variation. Genetic variation, including both de novo adaptation and acquisition of genes conferring antibiotic resistance or virulence factors, is important for the persistence of the pathogen and progression of disease. In addition, the superficial location of EndA permits extracellular degradation of the DNA component of NETs, thereby facilitating pneumococcal dissemination and increasing the risk of invasive infection. These activities of EndA contribute to pathogenesis, making the pneumococcal nuclease an attractive target for controlling S. pneumoniae diseases. Currently, experiments are under way to evaluate UNC10103486 and other promising in vitro inhibitors of EndA using live bacteria assays for transformation and NET degradation. The facile identification of small-molecule inhibitors is an important first step for developing pharmacologic probes of EndA to address its roles in pneumococcal virulence and to establish it as a druggable target for combating pneumococcal infection.
Footnotes
Acknowledgements
The authors thank Emily Hull-Ryde and Chatura Jayakody for their technical assistance.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Institutes of Health, including a grant to S.F.S. from the National Institute of General Medical Sciences (grant number GM058114), a grant to L.C.P. from the Division of Intramural Research of the National Institute of Environmental Health Sciences (grant number 1 ZIA ES102645-03), a grant (ECCPS) to M.M. from the Deutsche Forschungsgemeinschaft, and support from the National Center for Research Resources and the National Center for Advancing Translational Sciences (grant number UL1TR000083).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.