
Abstract
Human interleukin 17 (IL-17) is a proinflammatory cytokine derived mainly from activated T cells. Extensive evidence points to a significant role of IL-17 in many autoimmune and infectious diseases, as well as tumorigenesis and transplant rejection, and suggests that targeting IL-17 could be a promising therapeutic strategy. Robust cell-based assays would thus be essential for lead identification and the optimization of therapeutic candidates. Herein, we report a well-characterized two-step assay, consisting of (a) in vitro activation and stimulation of CD4+ T lymphocytes by a defined complex of antibodies and cytokines, leading to T helper 17 (Th17) cell differentiation and IL-17 production, and (b) IL-17 quantification in cell supernatants using a homogeneous time-resolved fluorescence (HTRF) assay. The system was optimized for and shown to be reliable in high-throughput compatible 96- and 384-well plate formats. The assay is robust (Z′ > 0.5) and simple to perform, yields a stable response, and allows for sufficient discrimination of positive (IL-17–producing cells) and negative controls (uninduced cells). The assay was validated by performing dose-response testing of rapamycin and cyclosporine A, which had previously been reported to inhibit IL-17, and determining, for the first time, their in vitro potencies (IC50s of 80 ± 23 pM and 223 ± 52 nM, respectively). Also, IKK 16, a selective small-molecule inhibitor of IκB kinase, was found to inhibit IL-17 production, with an IC50 of 315 ± 79 nM.
Introduction
Interleukin (IL)–17A (also known as IL-17) is the prototype member of a novel family of cytokines, the IL-17 family,1,2 and a signature cytokine defining a new class of CD4+ effector T cells, termed Th17 cells. 3 Historically, two major CD4+ T cell subsets have been defined according to their cytokine production pattern, interferon-γ (IFN-γ)–producing Th1 cells and Th2 cells releasing IL-4, which have long been known to regulate cellular and humoral immunity. 4 Th17 cells have been identified only recently as a Th lineage that is not only distinct from Th1/Th2 cells in its gene expression and regulation but also in terms of its biological function. Th17 cells play the crucial role in mounting the immunity for both intracellular and extracellular pathogens 5 but also, by virtue of their production of two inflammatory mediators, IL-17A and its homologous cytokine IL-17F, 6 are generally thought to be proinflammatory. Whereas IL-17 is predominantly expressed by activated T cells,2,7 this cytokine exhibits pleiotropic biological activities on various types of cells, such as fibroblasts, keratinocytes, endothelial cells, and epithelial cells.7,8 IL-17 has been found to induce the production of many cytokines and chemokines, for example, tumor necrosis factor–α (TNF-α), IL-1β, IL-6, IL-8, IL-12, prostaglandin E2, and granulocyte colony-stimulating factor.1,7–9 Through the induction of a number of cytokines, IL-17 is able to mediate a wide range of responses, mostly proinflammatory and hematopoietic. Consistent with IL-17 wide-range effects, its cognate cell surface receptor, the IL-17 receptor (IL-17R, or IL-17RA),1,2 has been found to be ubiquitously expressed in many tissues and cell types.2,7
Numerous findings suggest that IL-17 plays a pivotal role in the initiation or maintenance of the inflammatory responses associated with disease pathology, and thus Th17 cells and their powerful inflammatory cytokine IL-17 have become a major target in inflammatory and autoimmune diseases. In recent years, the IL-17 family has been linked to many immune/autoimmune-related diseases, including rheumatoid arthritis and osteoarthritis, Crohn’s disease, psoriasis, asthma, inflammatory bowel disease, and multiple sclerosis.2,8–15 Initial reports also propose a role for Th17 cells and IL-17 in tumorigenesis and transplant rejection. 9
Given the strong association between excessive Th17/IL-17 activity and human disease, it has been suggested that targeting IL-17 may be a promising therapeutic strategy. Demonstrated in vivo activities of the IL-17 cytokine family illustrate the enormous clinical potential of and need for antagonists of proinflammatory molecules such as IL-17. Until recently, only antibodies neutralizing the biological activity of IL-17 16 and soluble versions of the IL-17R protein 17 have been investigated in early phase trials as antagonists of IL-17–mediated inflammatory diseases. No other specific inhibitors of IL-17 have been discovered yet, but it has been recently demonstrated that a few known immunosuppressive agents exhibit activity against Th17 and/or IL-17. Thus, rapamycin (sirolimus), an immunosuppressive drug used to counter autoimmunity and to prevent acute graft rejection in human, 18 and another immunosuppressive antibiotic, cyclosporine A, have been shown to suppress in vitro Th17 cell differentiation. 19 Rapamycin, which has been known to exert its action primarily via suppression of IL-2 and other growth factors, 20 has also been shown to inhibit IL-17 production. 21 Glucocorticoids are also commonly used in the treatment of autoimmune diseases, and they exert their role through multiple mechanisms, including inhibition of a variety of cytokines such as IL-2, IL-6, TNF-α, and IFN-γ. 22 The glucocorticoid dexamethasone recently has been shown to inhibit IL-17 production as well 21 but is a much weaker inhibitor than rapamycin.
Convenient and well-performing screening assays for a wide range of targets are in great demand for biomedical research. The lack of specific IL-17/Th17 inhibitors or antagonists is, to a large extent, due to a lack of robust cell-based assay tools for high-throughput screening (HTS). Assays without the need for washing steps while still unaffected when analyzing complex biological samples are especially difficult to develop. Here, we describe the development, optimization, and validation of a new cell-based HTS assay for the identification of inhibitors of IL-17 and/or Th17. CD4+ T cells are stimulated to induce the differentiation of Th17 cells and production of IL-17, and the cytokine is quantified in the cell culture supernatant using a homogeneous assay. The assay has been miniaturized into a 384-well format and validated using known Th17/IL-17 antibiotic inhibitors. Our results demonstrate that the assay is robust and can be applied to HTS for inhibitors of IL-17.
Materials and Methods
Materials
CD4+ T cells (donors 104, 105, 106, and 165) were purchased from Astarte Biologics (Redmond, WA), and Poietics CD4+ T cells (donor 20386) were provided by Lonza (Allendale, NJ). DMSO, rapamycin, and cyclosporine A were obtained from Sigma (St. Louis, MO). N-(4-Pyrrolidin-1-yl-piperidin-1-yl)-[4-(4-benzo[b]thiophen-2-yl-pyrimidin-2-ylamino)phenyl]carboxamide hydrochloride (IKK 16) was from Tocris Bioscience (Ellisville, MO). RPMI-1640 cell culture medium was obtained from American Type Culture Collection (Manassas, VA). Media supplements and antibiotics, as well as Dynabeads Human T-Activator CD3/CD28, were purchased from Invitrogen (Carlsbad, CA). Cytokines were obtained from R&D Systems (Minneapolis, MN). All cell culture and assay plates were from Corning (Lowell, MA). The Human Interleukin 17 HTRF kit was obtained from Cisbio (Gifsur-Yvette, France).
Cell Culture Conditions and Treatment
A frozen vial of CD4+ T cells was thawed in a 37 °C water bath, and the cells were washed twice with 10 mL of the complete medium (RPMI-1640 supplemented with 10% fetal bovine serum [FBS], 100 U/mL penicillin, and 100 µg/mL streptomycin), followed by centrifugation (800 rpm, 10 min). Cells were resuspended in 4 mL of the complete medium and incubated in a 6-well plate at 37 °C, 5% CO2. Following an overnight recovery, the cells were washed with the complete medium, diluted to the desired cell density, and cultured in 6-, 96- or 384-well nontreated surface sterile plates in a total volume of 2 mL, 200 µL, or 50 µL per well, respectively. Each plate included wells with induced and uninduced cells and also cell-free control wells that had the same volume of the culture medium but no cells. Induced cells were activated in the complete medium with Dynabeads Human T-Activator CD3/CD28 at a bead-to-cell ratio of 1:2.5 and stimulated with the following cytokines: transforming growth factor–β (TGF-β, 5 ng/mL), IL-6 (20 ng/mL), IL-23 (20 ng/mL), and IL-1β (10 ng/mL). Uninduced cells only received cytokine IL-2 (50 ng/mL). Plates were incubated at 37 °C, 5% CO2 for up to 7 days during initial assay development and for 3 days thereafter. Cell supernatants/media were collected at the indicated time points, spun down, and used immediately for measuring IL-17 or stored at −20 °C.
For compound inhibition testing, cells were stimulated in 384-well plates in the presence of compounds at the desired concentrations. First, 0.5 µL of a test compound dissolved in 100% DMSO or DMSO alone was transferred to the plate by a Hummingbird pin tool (Digilab, Holliston, MA), followed by plating of 49.5 µL of cells or culture medium, thus resulting in a 100-fold compound and DMSO dilution. Three types of controls (16 wells each), in which compounds were replaced with 1.0% DMSO, were included on each plate: cell-free control wells, induced (positive or 0% inhibition control), and uninduced cells (negative or 100% inhibition control).
IL-17 HTRF Assay
IL-17 levels in cell supernatants and cell-free control samples were measured using the Homogeneous Time-Resolved Fluorescence (HTRF) assay kit from Cisbio, following the manufacturer’s instructions. The assay was run at room temperature in low-volume, solid white, nonbinding surface 384-well plates (cat. 3673; Corning, NY). The plates were read in the HTRF mode (excitation 380 nm and emission 665 nm and 620 nm) using the Analyst GT reader (Molecular Devices, Sunnyvale, CA) after overnight incubation of samples with the antihuman IL-17A (hIL-17A) antibody conjugates.
Data Analysis
The activities were calculated as % of Delta F and % inhibition, based on the HTRF ratio (A665/A620 × 104), using the following formulas:
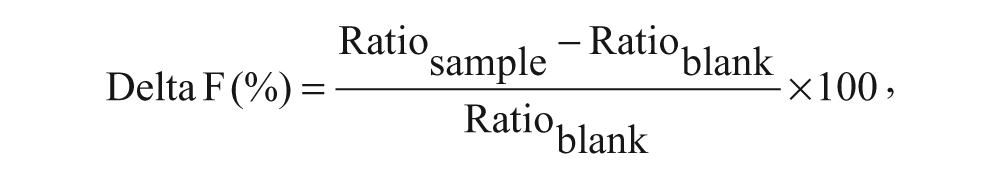
where Ratioblank is the mean HTRF ratio for the cell-free control wells (or zero IL-17A wells for calculating a standard curve).
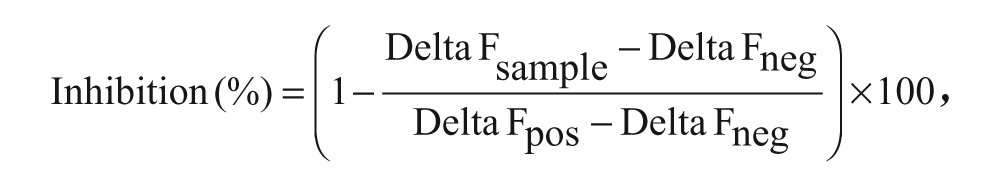
where Delta Fneg and Delta Fpos are the mean Delta F values calculated for the control wells with uninduced and induced cells, respectively.
For estimation of the assay performance, Z′ values 23 were calculated for each plate comparing positive and negative controls. A Z′ value ≥0.5 was chosen as the acceptance cutoff. The signal-to-background (S/B) ratio was calculated for each plate as a quotient of the mean maximal divided by the mean minimal signal (HTRF ratio) from induced and uninduced control wells, respectively. Coefficients of variation (CVs) for controls were also calculated based on the HTRF ratio, and intra- and interassay CVs were calculated as averages of positive and negative control CVs. The XLfit 5.2.0.0 software (IDBS, Guildford, UK) was used for curve fitting. IC50 values were calculated using a four-parameter logistic fit model, model 205, and IL-17 standard curves were fitted using a linear polynomial model, model 100.
Results and Discussion
In Vitro T Cell Stimulation for the Induction of IL-17 Production
IL-17A is produced primarily by activated CD4+ T cells.2,7,9 Differentiation of proinflammatory IL-17–producing cells (Th17) can be induced in vitro from naive CD4+ cells by stimulation through the T-cell receptor (TCR), contingent upon the cytokine environment. However, the optimal conditions for the development of a robust Th17 population secreting large amounts of IL-17 from human T cells remain controversial. In particular, the cytokine requirements for the induction of human Th17 differentiation, as well as the impact of serum cytokines present in culture media, have been the focus of much debate, due primarily to inconsistent findings from studies in humans.24-27 It has also been suggested that generation of primary human Th17 cells secreting IL-17 in vitro may be donor dependent. 25
In this study, populations of negatively selected CD4+ T cells from five different donors were tested, which were purchased from two different suppliers (Lonza and Astarte Biologics). The standard conditions for induction of Th17 differentiation and IL-17 production included TCR activation using Dynabeads coated with monoclonal antibodies against the CD3 and CD28 cell surface molecules of human T cells. Activation was carried out in the presence of four cytokines. The selection of cytokines was based on the comprehensive study by Volpe et al., 24 which demonstrated that TGF-β, IL-23, and proinflammatory cytokines IL-1β and IL-6 were all essential for human Th17 differentiation and high production of IL-17, irrespective of the presence of serum in growth media. The specific concentrations of the cytokines and Dynabeads used in our study are described in Materials and Methods, and the cell culture medium contained 10% FBS. The conditions we selected led to efficient induction and did not seem to require any neutralizing anti–IFN-γ and anti–IL-4 antibodies that were used in some previous studies 26 to prevent the suppression of in vitro Th17 differentiation.
Under these conditions, significant amounts of IL-17 were first detectable by the HTRF assay in cell culture supernatants after 48 h of cell stimulation ( Fig. 1 ), and IL-17 accumulation in the media progressively increased by days 5 to 7, except when daily media changes were tested ( Fig. 1B ). No measurable IL-17 was detected in either uninduced cells ( Fig. 1 ) or cells stimulated with TGF-β, IL-23, IL-1β, and IL-6 in the absence of anti-CD3/CD28 beads (data not shown) or in cell-free media (data not shown). This agrees with the observations by Volpe et al., 24 who found that a standard 5-day T-cell assay is more specific than systems with longer, 10- to 12-day culture duration,26,27 in which substantial IL-17 production was detected in control medium or IL-2 alone. There were some differences in the levels of IL-17 induction between the cell populations from the two donors used in the initial experiments ( Fig. 1A , B ). Thus, the calculated S/B ratios for the 3-day time point in a 6-well plate format were about 24.8 for Astarte Biologics’ donor 105 but only 10.5 for Lonza’s donor 20386. It is not clear, however, how reproducible these differences are since no additional experiments were run with the latter cells. The CD4+ T cells that originated from donor 105 (Astarte Biologics) were used in all subsequent assay optimization experiments.
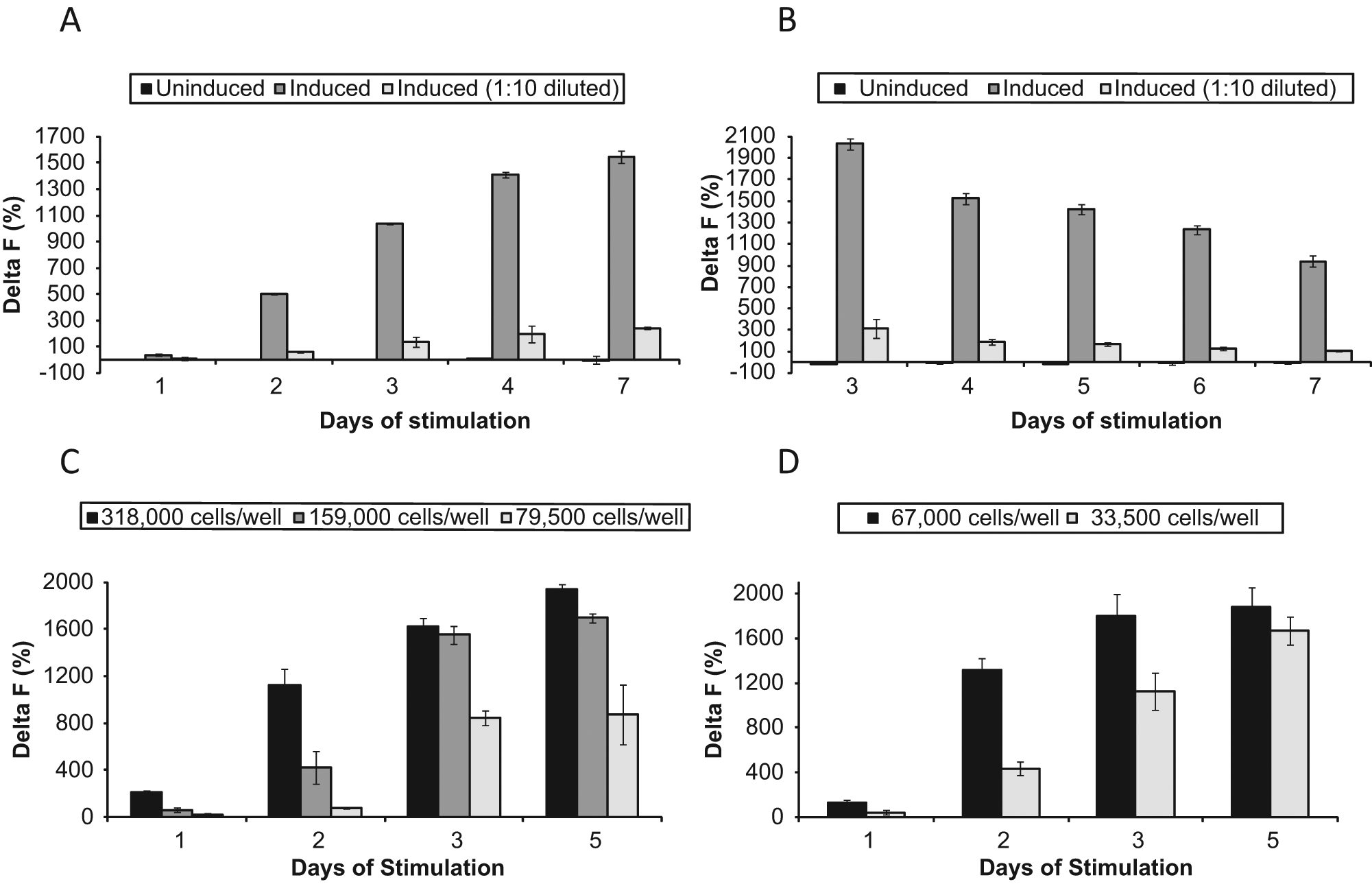
Time courses of interleukin (IL)–17 induction upon incubation of CD4+ T cells in the presence of anti-CD3/CD28-coated beads and cytokines transforming growth factor–β (TGF-β), IL-23, IL-6, and IL-1β. (
Assay Miniaturization and Optimization
To be able to support extensive screening for IL-17 inhibitors, the assay needs to be amenable to an HTS format and, ideally, to automation; have as few steps as possible; and be amenable to miniaturization to reduce consumable costs. There are currently no known IL-17 assays satisfying these requirements. To test the possibilities for assay miniaturization and increasing its throughput, cells were stimulated in 6-, 96-, and 384-well plates for up to 7 consecutive days, without changing the media or feeding cells, and IL-17 was quantified in the cell supernatants. As Figure 1 shows, there was no dramatic loss of signal measured by the IL-17 HTRF assay upon assay conversion from 6- to 96- to 384-well cell plates. When data for the same donor, donor 105 ( Fig. 1B – D ), and the same time point (3 days) were compared, the maximum calculated S/B ratios for the 6-, 96-, and 384-well plate were 24.8, 15.9, and 19.2, respectively (i.e., all plate formats generated good assay windows). The experiments clearly demonstrated that the assay is amenable to miniaturization and can be run in an HTS format.
It is also important that, as our results show, no additional steps such as media changes or cell feeding are required during the cell culture phase for at least 3 days, even in the case of 384-well plates, at which time IL-17 production reaches substantial levels in all plate formats and begins to slow down ( Fig. 1 ). In an attempt to maintain the initial rate of IL-17 production beyond 3 days of stimulation, we tested complete daily media changes. The entire previous day’s cell supernatant was carefully removed and replaced with the respective fresh media for induced and uninduced cells. As Figure 1B shows, this resulted in progressively lower levels of IL-17 in the culture supernatants of cells stimulated for 4 to 7 days in a 6-well plate. Possible explanations for that can be that not only media but also some cells were removed every time from the wells upon media changes or that the cells were stressed on a daily basis and did not recover quickly enough. No matter what caused the decline in IL-17 levels, there seems to be no benefit in changing media or performing any other additional manipulations of the cells beyond 3 consecutive days of stimulation. We certainly did not find that for the purpose of this assay, CD4+ T cells, to produce measurable IL-17, needed to undergo multistep and multiday treatment regimes as, for example, the one that included priming for 5 days with anti-CD3/anti-CD28 beads in the presence of various combinations of cytokines, followed by incubation for 7 more days in IL-2 and final stimulation for 5 h with phorbol myristate acetate and ionomycin. 27 With the combination of antibodies and cytokines we selected, the one-step treatment of CD4+ T cells, as described in Materials and Methods, leads to substantial IL-17 production, and the stimulation time can be reduced to only 3 days (72 h).
To further optimize the cell stimulation phase of the assay, several parameters were evaluated, including cell plating density and DMSO tolerance. The effect of cell density on assay performance was tested in 96- and 384-well formats using a range of plating densities. A robust assay window was observed with cell plating densities from 1.6 to 3.2 × 105 cells/well in a 96-well plate ( Fig. 1C ) and 6.7 × 104 cells/well in a 384-well plate ( Fig. 1D ). The 1.6 × 105 cells per well plating density with a final assay volume of 200 µL and 6.7 × 104 cells per well plating density with a final assay volume of 50 µL were selected as the basis for further assay optimization in 96- and 384-well plate formats, respectively, because they provided both good assay windows and minimal variability as exemplified by a CV below 10% at the maximal response.
To satisfy the HTS requirement for compounds to be dissolved in DMSO, a cell-based assay needs to be tolerant to certain concentrations of DMSO. DMSO was titrated from 0% to 8% final concentration in the culture medium, and cell and assay tolerance was tested by measuring IL-17 in the cell supernatants after 3 days of incubation. The testing results ( Fig. 2 ) indicated that no significant decrease in the IL-17 levels occurred as DMSO concentrations were raised from 0% to 2%. However, the IL-17 levels were very low in the presence of 4% DMSO, and no IL-17 was detected in the supernatants of stimulated cells in the presence of 8% DMSO. A final DMSO concentration of 1% was selected and consistently used in all other experiments described here.
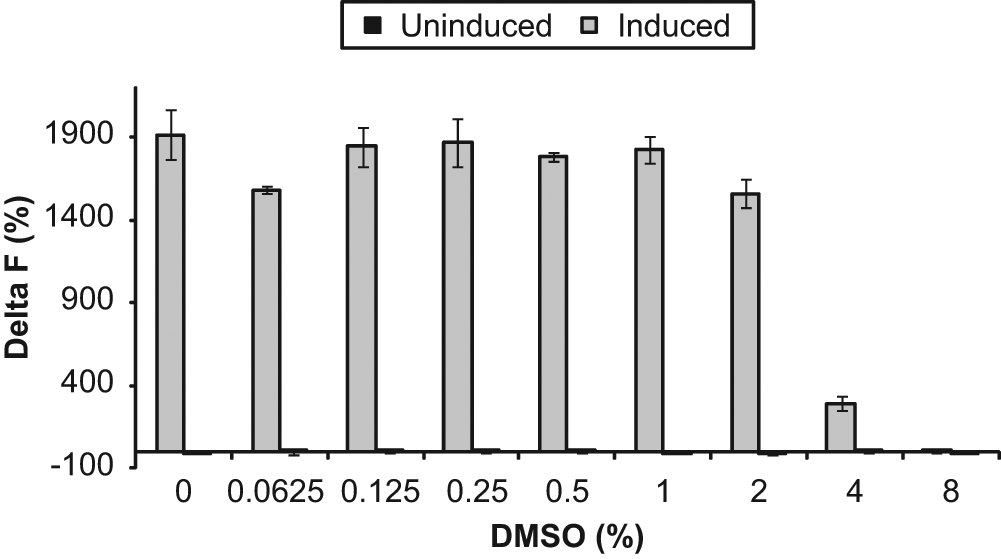
DMSO tolerance of interleukin (IL)–17 production by uninduced and induced CD4+ T cells (donor 105, Astarte Biologics). Cells (67 000 per well) were cultured in a 384-well plate for 3 days in the presence of the DMSO concentrations shown, using duplicate wells per condition. Cells were removed by centrifugation, and IL-17 was measured in cell supernatants. Data represent mean ± SD of duplicate data points.
IL-17 is typically measured in cell culture supernatants by an enzyme-linked immunosorbent assay (ELISA).21,24 To avoid multiple wash steps, as required in ELISAs, we tested the IL-17 HTRF assay kit from Cisbio, which allows experiments to be conducted using simple mix-and-read protocols, and found that the assay was quite sensitive and needed very little optimization. The only optimization step performed was comparing assay performance in solid white (cat. 3673; Corning) and black plates (cat. 3676; Corning). The HTRF signal was measured after parallel incubation of the same serial dilutions of recombinant human IL-17A (0–5000 pg/mL in culture medium) with the anti-hIL17A antibody conjugates in two low-volume 384-well plates, white and black, respectively ( Fig. 3A ). The white plate showed a lower background level as compared with the black plate. The average HTRF ratio in the media-only wells containing 0 pg/mL of hIL-17A was about 1.7-fold lower in the white versus black plate. Accordingly, the relative signal increase at the maximum hIL-17A concentration tested (5 ng/mL) was 6.7-fold above the background in the white plate and 5.2-fold above the background in the black plate. Thus, white plates were selected to measure IL-17 using the HTRF kit.
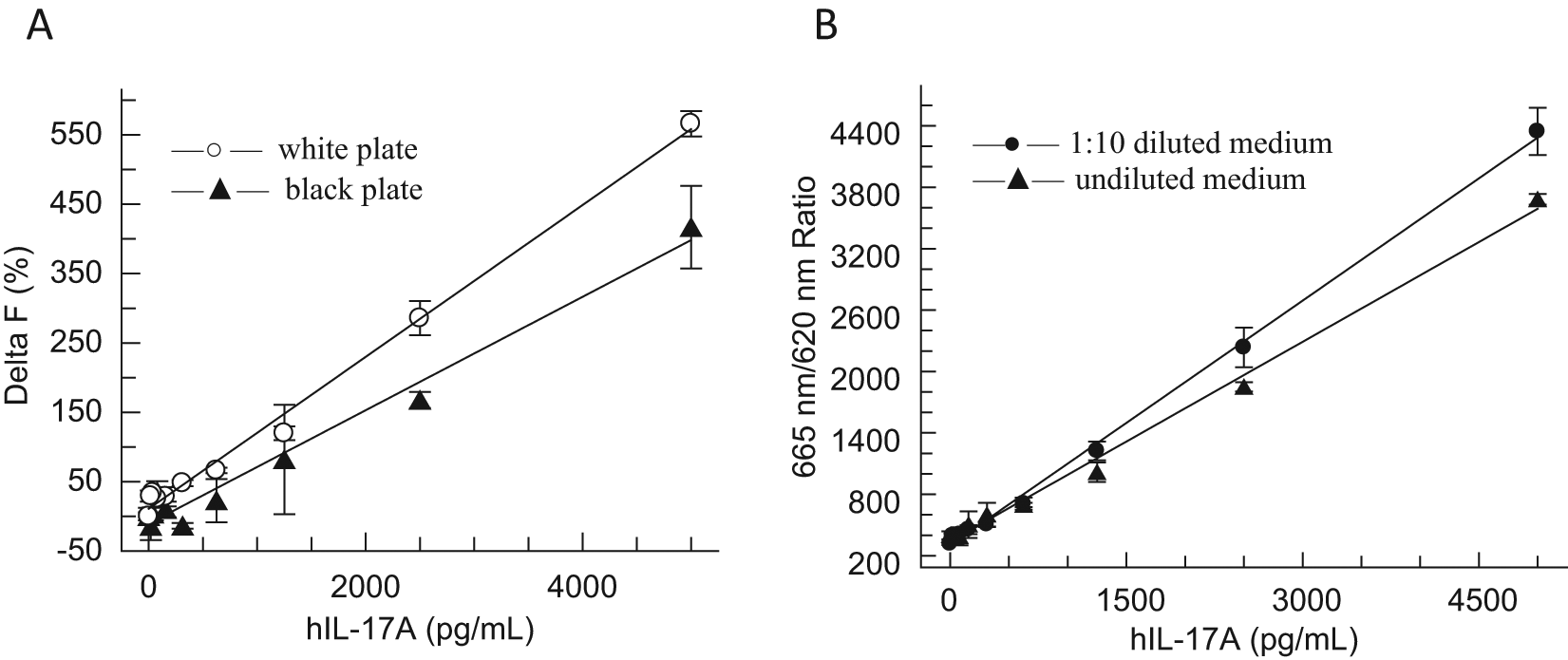
Optimization of the interleukin (IL)–17 Homogeneous Time-Resolved Fluorescence (HTRF) assay using standard curves of recombinant human IL-17A. (
The last parameter evaluated was quenching of the HTRF signal by the culture medium. It was noticed during initial evaluation of the HTRF kit that dilution of the hIL-17A standard in the culture medium resulted in a reduced signal compared with dilution of the cytokine in the diluent recommended in the assay manual (100 mM phosphate buffer [pH 7.0], 0.1% bovine serum albumin, 0.1% Tween 20). It was also found that to completely eliminate the quenching effect, the culture medium needed to be diluted with the buffer approximately 10-fold (data not shown). Experiments were performed to determine the impact of quenching on assay performance and whether diluting cell supernatants was a viable option. Figure 3B shows hIL-17 standard curves generated after diluting the cytokine in the original culture medium and the same medium prediluted 1:10 with the diluent. For the highest IL-17 concentration tested (5 ng/mL), the raw signal (665 nm/620 nm HTRF emission ratio) increased by about 15% when dilutions were prepared in the 1:10 diluted medium versus the standard medium. Obviously, this relatively small signal gain would not be sufficient to compensate for the dramatic reduction of the assay window resulting from a 10-fold dilution of supernatants from induced T cells as compared with undiluted supernatants ( Fig. 1A , B ). Based on the data and because the assay window was robust upon testing of undiluted cell culture supernatants, it has been concluded that undiluted supernatants can be used for IL-17 quantification in the HTRF assay.
Assay Quality and Reproducibility
To evaluate the reproducibility and quality of the assay in an HTS format, we screened ten 384-well plates under optimal assay conditions in five independent experiments that were performed over the course of several weeks. CD4+ T cells from four different donors—donors 104, 105, 106, and 165 (all males, Caucasian, ages 31–56 years)—were used in these experiments. All plates had positive (cytokine-induced cells), negative (uninduced cells), and blank (cell-free medium) controls in 16 wells (one column) each. The average intraplate CV values were 10.8% for the positive control wells and 6.3% for the negative control wells, and thus intra-assay variability was quite low (CV 8.6%). The overall interassay variability was calculated to be 14.9%, which is acceptable for a cell-based assay, especially since the value is a cumulative result of both technical and biological variability at different levels (plate to plate, day to day, donor to donor, and stimulated vs unstimulated cells). Figure 4A shows the average normalized values calculated for the negative and positive controls on each plate. The Delta F values varied between −6.1% and 6.9% (mean 0.08%) for the negative controls and from 673% to 1243% (mean 1020%) for the positive controls, thus showing good separation between the two types of controls and a large assay window.
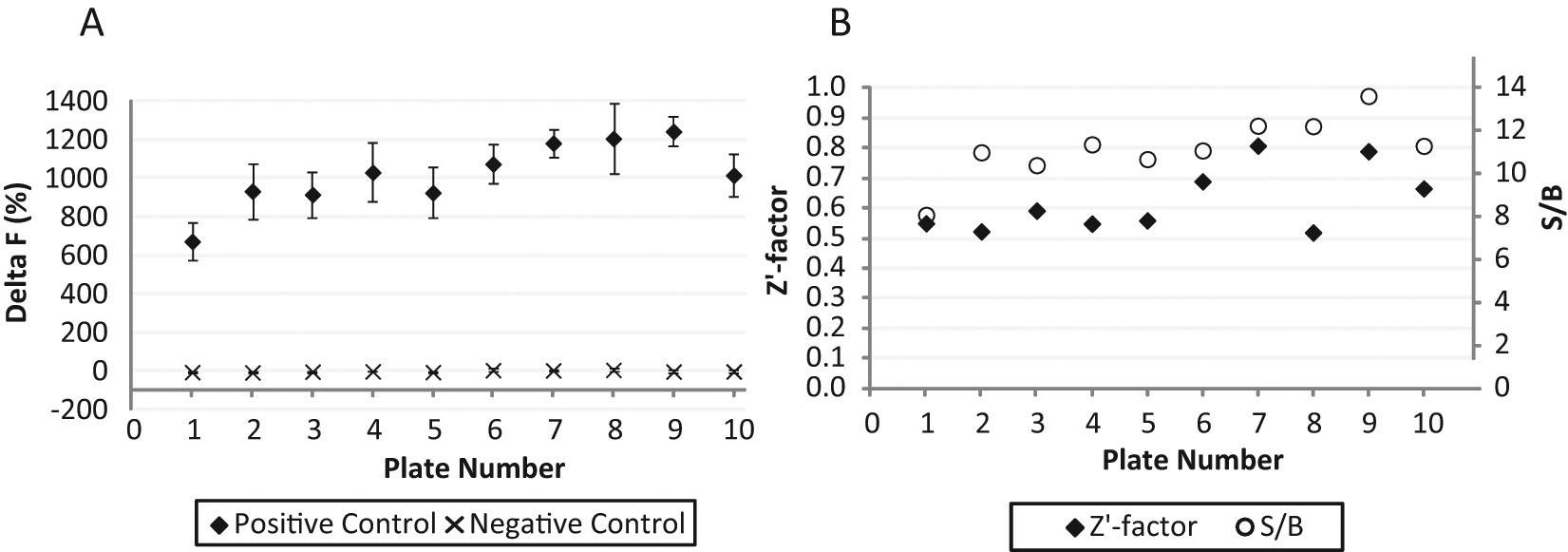
Assay quality parameters. Ten 384-well plates were incubated and tested as described in Materials and Methods, using CD4+ T cells provided by Astarte Biologics, which originated from the following four different donors: plates 1 to 4, donor 104; plates 5 to 8, donor 105; plate 9, donor 106; and plate 10, donor 165. (
The Z′ scores varied for these plates from 0.52 to 0.81, and the average Z′ score was calculated to equal 0.63 ± 0.11, thus representing a robust screening parameter. The S/B ratios varied from 8.1 to 13.6, and the average S/B value was calculated to equal 11.2 ± 1.4 across the plates run under screening conditions ( Fig. 4B ). These observations demonstrate that both steps of the assay, cell stimulation and IL-17 detection, can be run in a 384-well format and that the entire assay is highly reliable, reproducible, and well suited for HTS purposes. This provides confidence that the assay can be used to test for inhibitors of IL-17.
Inhibitor Testing
It has previously been shown that two immunosuppressive agents, rapamycin and cyclosporine A, inhibited the TGF-β– and IL-6–induced generation of IL-17–producing Th17 cells from CD4 cells and intracellular expression of IL-17. 19 Cells were cultured in a 24-well plate, and the percentage of IL-17–producing cells was estimated using fluorescence-activated cell sorting (FACS) analysis and cell staining with relevant antibodies. Rapamycin and cyclosporine A were only tested at three concentrations each (1, 10, and 100 ng/mL and 1, 10, and 100 nM, respectively). At 10 ng/mL, rapamycin was shown to reduce the proportion of IL-17–producing cells from 5.8% to 2.8% in an unfractioned CD4 cell population and from 7.1% to 4.8% when using FACS-sorted CD4 cells. Cyclosporine A (10 nM) inhibited the generation of IL-17–producing cells to 1.2% and 1.9%, respectively, when using unfractioned and flow-sorted CD4 cells. In another study, 21 it was demonstrated that rapamycin inhibited the production of IL-17, measured in cell supernatants by an ELISA assay, by peripheral blood mononuclear cells (PBMCs) stimulated with anti-CD3 and anti-CD28 antibodies. Even though six concentrations of rapamycin (from 1–1000 ng/mL) were tested, all but the lowest one completely blocked IL-17 production and thus no IC50 was reported.
As a final performance benchmark, the two known IL-17/Th17 inhibitors, rapamycin and cyclosporine A, were evaluated in our assay using the 384-well HTS plate format and optimized conditions ( Table 1 and Fig. 5 ). In these experiments, cells from several donors were incubated in the presence of 8 to 10 concentrations of the compounds or 1% DMSO during the entire period of stimulation (3 days), prior to assaying IL-17 levels. As shown in Figure 5 , rapamycin and cyclosporine A effectively inhibited IL-17 production by stimulated CD4+ T cells, and the cells responded to both antibiotics in a dose-dependent manner with average IC50 values of 80 ± 23 pM and 223 ± 52 nM, respectively ( Table 1 ), further suggesting that this assay can be used to screen for IL-17 inhibitors. In a previous study, 21 it has been shown that rapamycin has no effect on cell viability of PBMCs up to at least 1 µg/mL, which corresponds to ~1.1 µM and thus is much higher than the top concentration we tested (40 nM). Therefore, it is highly unlikely that the inhibitory effects observed are due to cell toxicity of rapamycin. It has been noticed upon fitting the dose-response data that the Hill coefficient for cyclosporine A tends to be higher than the one for rapamycin (2.04 ± 1.00 and 1.20 ± 0.39, respectively). The two antibiotics are known to have different mechanisms of immunosuppressive action;18,20 however, there is currently not enough information allowing any interpretation of the high Hill coefficient for cyclosporine A inhibition of IL-17.
Summary of Concentration-Dependent Inhibition by Rapamycin, Cyclosporine A, and IKK 16 of Interleukin 17 Production by Stimulated CD4+ T Cells from Different Donors
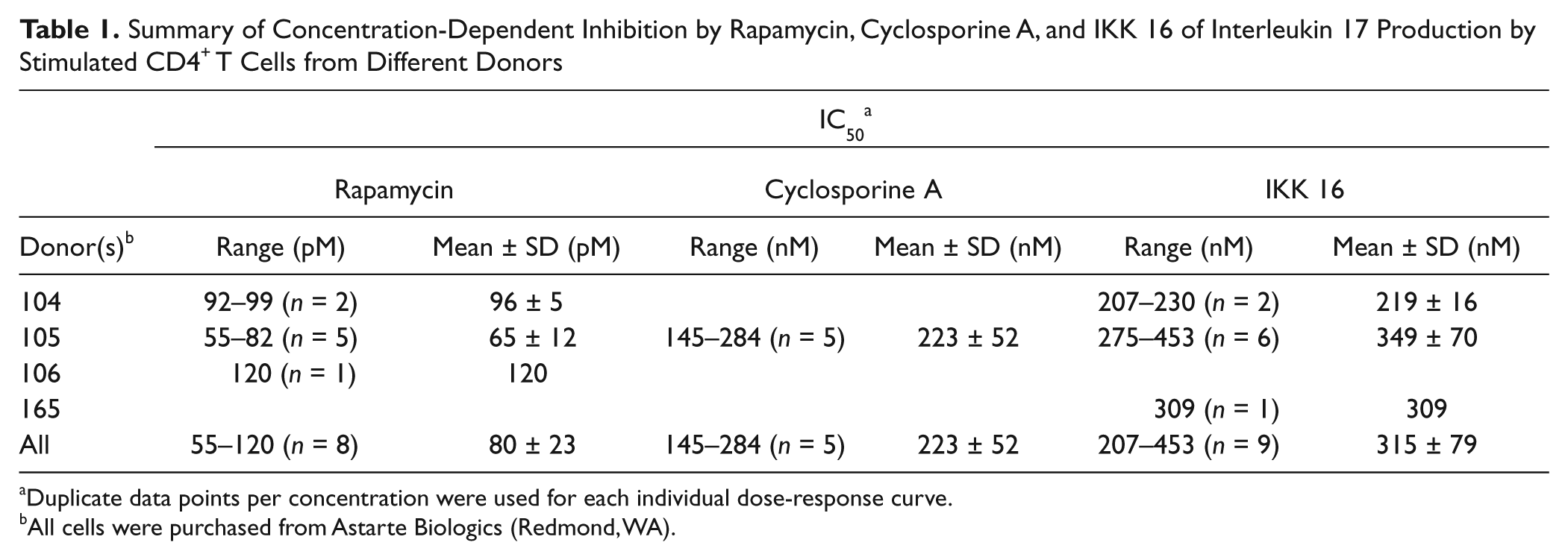
Duplicate data points per concentration were used for each individual dose-response curve.
All cells were purchased from Astarte Biologics (Redmond, WA).
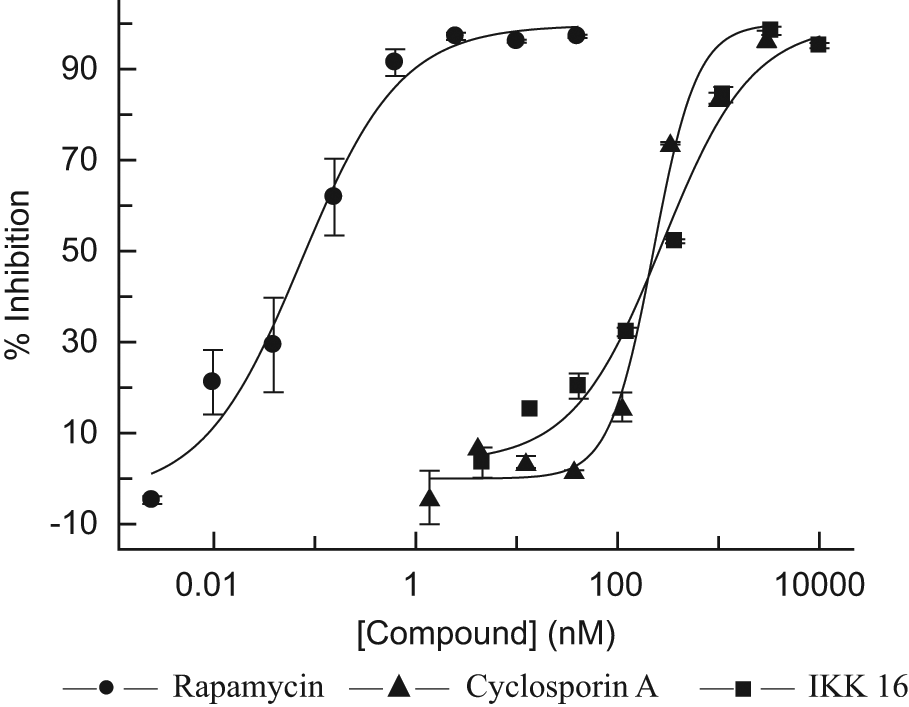
Representative dose-response curves of rapamycin (IC50 = 74 pM), cyclosporine A (IC50 = 226 nM), and IKK 16 (IC50 = 278 nM). Cells (67 000 per well) were stimulated in 384-well plates for 3 days in the presence of the compounds, then removed by centrifugation, and interleukin (IL)–17 was measured in cell supernatants as described in Materials and Methods. Data represent mean ± SD of duplicate data points.
As yet, only limited information is available about the intracellular signaling induced by IL-17. It has been shown, in particular, that engagement of the IL-17 receptor complex triggers activation of the transcription factor nuclear factor κB (NF-κB), 1 which is an essential factor in acute as well as chronic inflammation. Many signaling pathways that activate NF-κB converge at the level of IKK-β, a protein subunit of IκB kinase, which is a component of the cytokine-activated intracellular signaling pathway involved in triggering immune responses and the transduction of proinflammatory signals. Given the tight regulation of NF-κB by IκB molecules and the central role of IKK-β in phosphorylation and degradation of the inhibitor, IKK-β is a very promising target for pharmaceutical substances aiming at interfering with NF-κB activation. 28 We tested in this study IKK 16, a selective and potent inhibitor of IκB kinase, particularly IKK-β (IC50 40 nM), which was also found to inhibit lipopolysaccharide-induced TNF-α release in an animal model. 29 Our results showed that IKK 16 inhibited IL-17 production in a dose-dependent manner ( Table 1 and Fig. 5 ) with an IC50 value of 315 ± 79 nM.
The assay described here is distinct as it allows IL-17 to be measured in a multiwell cell-based assay. We used the IL-17 HTRF assay developed by Cisbio for quantification of IL-17 in cell supernatants because it does not require washing steps but was still found to be unaffected when analyzing complex biological samples. However, any other approaches to IL-17 detection and quantification (e.g., an ELISA) will likely work as well. There are, however, certain limitations that need to be taken into consideration. The assay is not likely to discriminate between selective inhibitors of Th17 cell differentiation, on one hand, and IL-17 production and/or secretion, on the other hand, if such selectivity exists. Inhibitors of other transduction pathways involved in Th17 cell differentiation and IL-17 production may be identified as false positives. Thus, IL-6 is a crucial cytokine for the generation of Th17 cells, 24 and rapamycin has been reported to inhibit IL-6 signal transduction, 30 which may contribute to its inhibitory effect on the generation of Th17 cells. Also, cytotoxic compounds may be identified as false positives, and thus testing compounds of interest in a cell viability or cytotoxicity assay is recommended.
All in all, the assay was found to perform well in cultured cell supernatants with regard to sensitivity, specificity, precision, and dynamic range. Furthermore, the assay possesses several advantages when compared with other assays, including a simple experimental protocol, low sample consumption, HTS format, and the ability to use the commercial IL-17 kit without the need for optimizations of specific reaction conditions. We show that the assay robustly and reproducibly identifies antibiotics rapamycin and cyclosporine A, which have previously been known to inhibit IL-17 production and/or Th17 induction. In addition, we demonstrate for the first time IL-17 inhibitor activity of IKK 16, a selective small-molecule inhibitor of IκB kinase.
In conclusion, the ability to measure IL-17 production in a high-throughput assay represents a significant breakthrough allowing thorough evaluation of IL-17 inhibitors using primary cells producing the cytokine. Moreover, the quantitative nature of the data and the high sample throughput of the assay provide a practical means of measuring inhibitor potencies at multiple concentrations. This should facilitate the calculation of IC50 values for active compounds, as we report here, for the first time, for rapamycin, cyclosporine A, and IKK 16. In addition, the assay possesses the potential to be used to screen large compound libraries and contribute to identification of novel natural product and small-molecule IL-17 and/or Th17 inhibitors.
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.