
Abstract
Histone methyltransferases (HMT) catalyze the methylation of histone tail lysines, resulting in changes in gene transcription. Misregulation of these enzymes has been associated with various forms of cancer, making this target class a potential new area for the development of novel chemotherapeutics. EZH2 is the catalytic component of the polycomb group repressive complex (PRC2), which selectively methylates histone H3 lysine 27 (H3K27). EZH2 is overexpressed in prostate, breast, bladder, brain, and other tumor types and is recognized as a molecular marker for cancer progression and aggressiveness. Several new reagents and assays were developed to aid in the identification of EZH2 inhibitors, and these were used to execute two high-throughput screening campaigns. Activity assays using either an H3K27 peptide or nucleosomes as substrates for methylation are described. The strategy to screen EZH2 with either a surrogate peptide or a natural substrate led to the identification of the same tractable series. Compounds from this series are reversible, are [3H]-S-adenosyl-L-methionine competitive, and display biochemical inhibition of H3K27 methylation.
Introduction
Epigenetics refers to the study of heritable changes in genome function that occur without a change in DNA sequence and the mechanisms through which the genome is accessed in different cell types during development and differentiation. 1 Enzyme-mediated epigenetic control of gene transcription is a critical aspect of embryonic development and cellular differentiation where chromatin structure plays a significant regulatory role in gene expression.
Epigenetic modifications occur through DNA methylation and reversible posttranslational modifications of histone proteins, primarily within the positively charged amino-terminal tails that are exposed on the surface of nucleosomes.2,3 Among the many chemical modifications that make up the histone code, lysine methylation can be associated with active or repressed transcriptional states depending on location.4,5
The polycomb group proteins are evolutionarily conserved transcriptional repressors important for the maintenance of cellular identity. The catalytic subunit of the polycomb repressive complex 2 is the histone methyltransferase, EZH2 (enhancer of zeste homolog 2). The enzymatic activity of EZH2 is dependent on other PRC2 protein members, EED and SUZ12, to catalyze the selective mono-, di-, and trimethylation of lysine 27 on histone H3.6,7 Trimethylated H3K27 (H3K27me3) along with other components are thought to cooperate to mediate gene silencing. Recent reports have demonstrated the synergistic influence of PHF1 to stimulate activity of EZH2 within the PRC2 complex, thereby increasing levels of the repressive mark H3K27me3 in vitro and in vivo.7,8 In this context, it is widely held that EZH2 is an active participant in epigenetic signaling.
The misregulation of histone methyltransferase activities has been associated with multiple cancer types. EZH2, which is overexpressed in many different types of cancer, has been proposed as a molecular marker for prostate cancer progression and aggressiveness.9,10 It has been implicated in the progression of bladder tumors 11 and shown to regulate proliferation of pancreatic tumor cells. 12 In addition, EZH2 plays a significant role in the development of adult and pediatric brain tumors 13 and has been linked to breast cancer aggressiveness.3,14,15 More recently, heterozygous mutations in EZH2 at Y641 and A677 have been identified in lymphoma that alter substrate specificity, thereby increasing H3K27me3 levels.6,16–18 Taken together, these data have revealed EZH2 as an attractive target for drug discovery efforts.
Methyltransferase assays that use [3H]-S-adenosyl-L-methionine (herein referred to as SAM) are extremely sensitive, display lower rates of compound interference, and can generally be adapted to other enzymes of this target class.19,20 Scintillation proximity assay (SPA) and filter binding methodologies enabled the characterization of EZH2 kinetics with substrates and inhibitors. SPA methodology was expanded to EZH2 inhibitor discovery efforts using two different methylation-capable substrates with either a peptide, based on the histone H3 lysine 27 sequence, or purified HeLa nucleosomes. The peptide and nucleosome SPA formats were optimized to run in miniaturized high-throughput environments to screen approximately 2 million compounds in the GlaxoSmithKline (GSK) collection, through which inhibitors of EZH2 activity were identified. One such inhibitor, GSK-A, was identified that demonstrates on-target and cell-based activity through reduction of H3K27 methylation. GSK-A was further characterized to be competitive with the SAM substrate through kinetic methods and by using a complementary [3H]-SAM binding assay that was developed to assess the ability of inhibitors to displace substrate from EZH2 independent of the presence of peptide or nucleosome.
Experimental Procedures
Materials
Unless stated otherwise, all reagents were obtained from Sigma (St. Louis, MO) and were at a minimum of reagent grade. Custom syntheses of C-terminus–appended histone H3 peptides (residues 21–44; ATKAAR
SPA reactions for assay development, optimization, and enzyme characterization were evaluated in 384-well low-volume assay plates (784075; Greiner Bio-One, Monroe, NC); high-throughput screening (HTS) reactions were evaluated in 1536-well low-volume assay plates (782075; Greiner Bio-One). The SPA luminescence signal was detected in a PerkinElmer Viewlux plate imager, using a 613/55-nm emission filter. Additional detailed information regarding the generation of reagents, assay methods development, and filter binding reactions is provided in supplemental information.
EZH2 Peptide SPA
If required, compounds were predispensed into assay plates prior to the addition of 5 nM EZH2. Unless stated elsewhere, reactions were initiated with an equal-volume addition per assay well of the substrate solution containing 0.3 µM H3K27 peptide that was preconjugated with 1.5 mg/mL streptavidin-coated PS imaging SPA beads (RPNQ0261) and 0.25 µM [3H]-SAM. Timed end-point reactions were terminated with an equal-volume addition per well of unlabeled SAM (0.1 mM, final concentration); reaction plates were sealed and dark adapted for 20 min, and a 3-min end-point luminescence image was acquired in a Viewlux. Where indicated, in the absence of added quench solution, a reaction plate was read continuously with images acquired every 3.5 min for up to 2 h. 21
EZH2 Nucleosome SPA
Similar methodology to the peptide SPA protocol was followed with the indicated modifications. Equal-volume additions of 10 nM EZH2 and the substrate solution (containing 5 µg/mL HeLa nucleosomes and 0.25 µM [3H]-SAM) were dispensed into assay plates. Timed end-point reactions were terminated with an equal-volume addition of 0.5 mg/mL PS-PEI Imaging Beads (RPNQ0098) containing 0.1 mM unlabeled SAM. Reaction plates were sealed and dark adapted for 20 min, and a 5-min end-point luminescence image was acquired in a Viewlux. The PEI coating minimized the nonspecific background and has proven to be a standard SPA imaging bead for many GSK nucleosome-based methyltransferase assays through enhanced substrate capture between the negatively charged DNA and positively charged polyethylenimine coating. 22
Adaptations and Execution of the Peptide and Nucleosome HTS
Assay quality and validation studies were conducted to miniaturize the EZH2 assays and to ensure 1536-well compatibility and tolerance to production-scale liquid handling. Signal-to-background ratio, Z′, 23 and DMSO tolerance to 5% were assessed, and upon miniaturization, quality was either retained or enhanced in 1536-well plates. An assay validation set of approximately 10 000 compounds was tested in triplicate using HTS assay conditions to assess assay performance and to obtain a data set indicating predicted hit rate and robust 3-standard deviation (SD) cutoff. A more complete description of the use of robust statistical analysis for the hit selection process has been detailed previously.24,25 Briefly, a robust 3SD cutoff is defined as [(the robust mean % response of the sample population) + (3× the robust standard deviation of the sample population)].
HTS assay-ready plates were prepared by dispensing 50 nL DMSO or 1 mM compound solution (in 100% DMSO) using acoustic dispensing technology (Labcyte Echo 555; Labcyte, Inc., Sunnyvale, CA) into 1536-well plates. Working stock reagents were dispensed for negative control, enzyme, and substrate solutions with a Multidrop Combi (Thermo Scientific, Waltham, MA) to provide a 100-fold dilution of dispensed compound into a 5-µL assay reaction mixture. Quench solution was similarly dispensed to all wells at 60 min. Plates were sealed and centrifuged for 1 min at 500 rpm, and following a 1-h dark adaptation, plates were imaged and data acquired as described above. Plate statistics, such as signal-to-background ratio, Z′, and robust 3SD activity cutoff, were calculated using AbaseXE (IDBS, Guilford, UK). Similarly, HTS hit confirmation and dose-response plates were prepared and evaluated using assay conditions identical to the primary screen. The hit selection process during hit confirmation, however, differs from the analysis of the primary screen (a random sample population; see above). 23 The robust mean and robust standard deviation used for the robust 3SD calculation are based on the control population and not the sample population as the latter is highly enriched for active compounds. 23 The hit selection cutoff during hit confirmation experiments is therefore defined as [(the robust mean % response of the control population) + (3× the robust standard deviation of the control population)]. Compounds were considered confirmed and progressed to dose-response testing if their activity was above the robust cutoff in at least two of three experiments (a robust hit in the primary HTS and a robust hit in at least one replicate of the duplicate confirmation evaluations). IC50 values were obtained using AbaseXE’s full-curve analysis bundle. Hit population analysis and visualization, activity-weighted diversity selection (AWDS), 26 and inhibition frequency index (IFI, defined as the relative frequency with which a compound has scored more than 50% inhibition in multiple HTS assays, excluding kinases) 27 were conducted using Spotfire DecisionSite (Tibco Software, Palo Alto, CA). AWDS (nucleosome HTS only), IFI, and chemical property analysis were used to rationally reduce the number of compounds tested in follow-up studies while maintaining the chemical diversity identified in the primary screen.
[3H]-SAM SPA Binding Assay
[3H]-SAM binding to EZH2 was determined independently of added peptide or nucleosomes. Where indicated, inhibitors were added to reaction plates prior to the addition of assay components. For the determination of the [3H]-SAM dissociation constant (K d ), SAM (0–10 µM) was added to 384-well low-volume assay plates containing DMSO (1%), followed by an equal-volume addition of EZH2 (50 nM) prepared in buffer containing 1 mg/mL PS-PEI–treated WGA (Type B) imaging beads (RPNQ0289; PerkinElmer). Luminescence measurements were obtained after 2 h and fit to a single-site ligand-binding equation (GraFit, version 5.0.12; Erithacus Software, Horley, UK). Inhibitor potency, assessed as a decrease in the end-point luminescence in the presence of 0.25 µM [3H]-SAM and 25 nM EZH2, was fit to a four-parameter IC50 model (XLfit, version 5.1.0.0; ID Business Solutions Ltd., Alameda, CA). Multiple SPA imaging beads were evaluated for high efficiency of protein binding with minimal background; although no glycosylation of EZH2 was expected, the PEI-WGA beads provided the highest level of capture possibly through ionic interactions.
Determination of EZH2 Substrate and Inhibition Kinetic Parameters
Reaction velocities were obtained from the slopes of the linear portion of reaction rate profiles (analyzed by linear regression) determined from a series of time-quenched end-point reactions in the filter binding assay format. Binding and kinetic rate data were fitted to the appropriate rate equations by using the nonlinear regression function of GraFit. Initial velocity data obtained from substrate co-titrations were fitted to a sequential kinetic mechanism (equation (1)). Initial velocity data obtained from substrate-inhibitor co-titrations were fitted to models for either competitive or noncompetitive inhibition (equations (2) and (3), respectively). For equations (1) to (3), V is the maximal velocity; k cat represents V normalized to enzyme concentration; A and B represent substrate concentrations; I is inhibitor concentration; Kia is the dissociation constant for A; Ka and Kb are Michaelis constants for substrates A and B, respectively; and Kis and Kii are the slope and intercept inhibition constants, respectively.
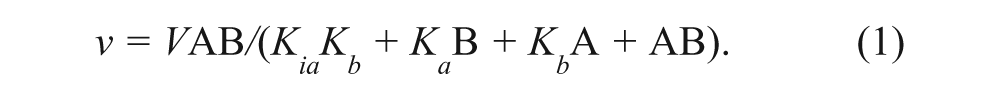
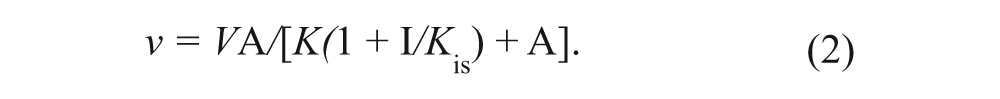
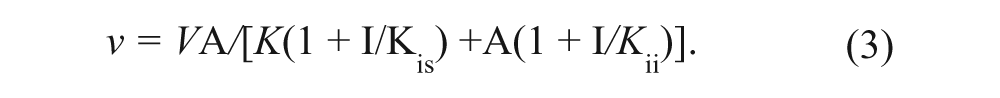
Results
Reagent Generation
A homogeneous enzyme supply was provided to support assay development and to enable each HTS. Individual baculovirus constructs were generated for each protein component of the five-member EZH2 complex (EZH2, SUZ12, EED, AEBP2, and RbAp48). Coordinated expression in Sf9 cells resulted in purification of up to 1 to 2 mg of the EZH2 complex/liter cell culture. Each high-throughput screen required 50 mg of the catalytically active five-member EZH2 complex that was isolated with purity estimated at >80%. Although the molar ratio of the five proteins of EZH2 cannot be judged by band density on the stained denaturing gel ( Fig. 1A ), the EZH2 complex migrated as a single entity on a native non-denaturing polyacrylamide gel with a molecular weight (MW) of approximately 400K corresponding to the proper assembly of a five-member protein complex ( Fig. 1B ).
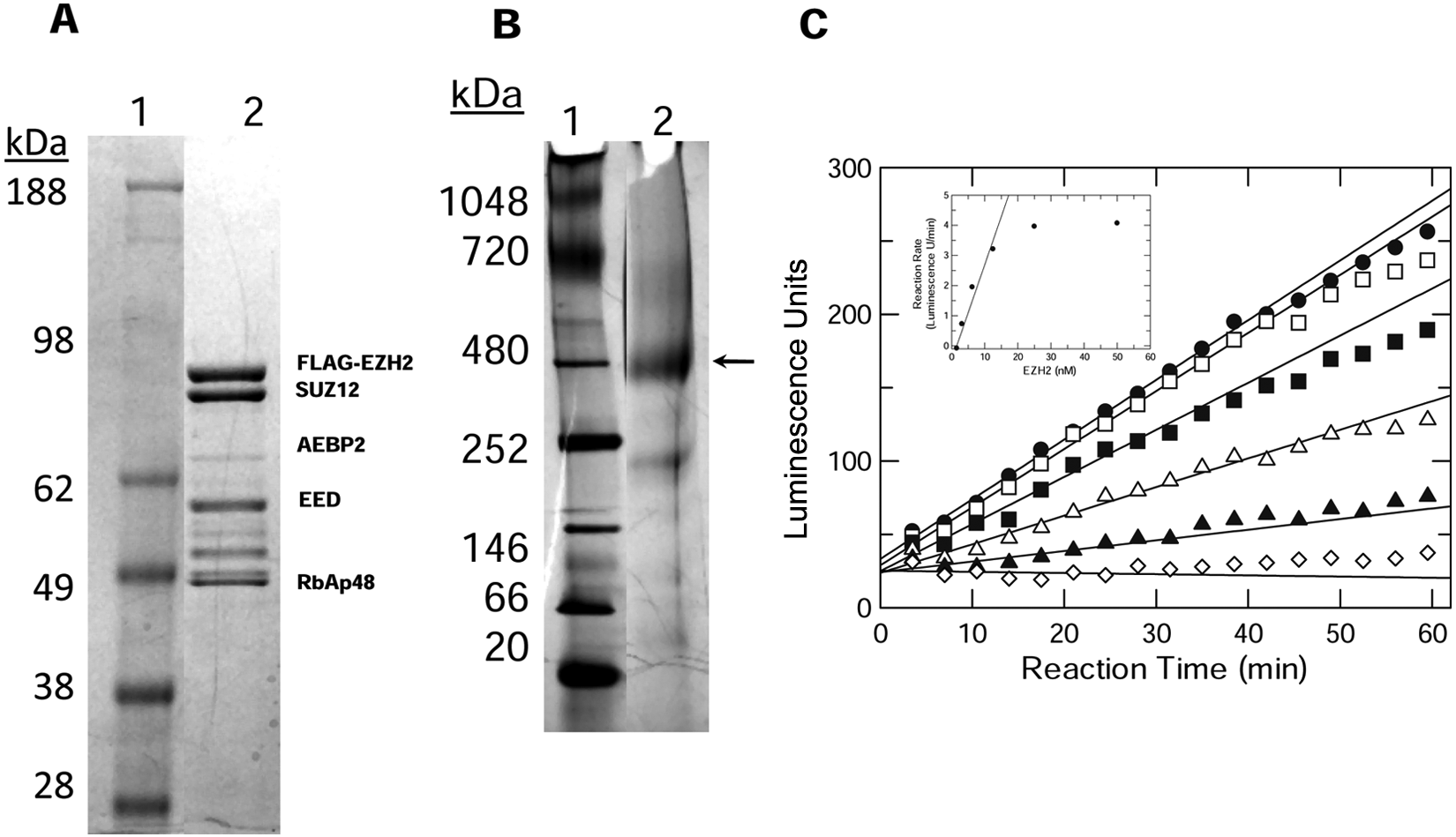
Characterization of purified EZH2 five-member complex. (
Previously, activity evaluations of the EZH2 five-member complex indicated no difference between mono- and dinucleosomes but did indicate a preference for either mono- or dinucleosomes rather than core or trimmed nucleosomes. 22 Optimized methods were adapted to enable a 50-L HeLa cell fermentation capable of producing up to 5 to 10 mg mono- or dinucleosomes/L cell culture. 22 Approximately 90 mg was required to support the assay development, HTS, and mechanism of action studies.
Determination of EZH2 Activity
The continuous SPA methodology was modified to measure EZH2 methyltransferase activity in a low-volume kinetic 384-well format using the biotinylated peptide preconjugated to the streptavidin-coated SPA beads. 21 Product formation was linear for 1 h; EZH2 activity was linearly dependent on enzyme concentration up to 12 nM ( Fig. 1C ). Similar results were also observed in the nucleosome SPA where product formation was determined by a series of timed quenched reactions. Therefore, the assay concentration of EZH2 was maintained at ≤10 nM unless specified otherwise.
Determination of EZH2 Kinetic Parameters
Filter binding methods were used to determine reaction rates for substrate co-titrations. Although comparable substrate kinetic parameters were obtained for the EZH2 three-member (
Kinetic Parameters for EZH2 Five-Member Complex
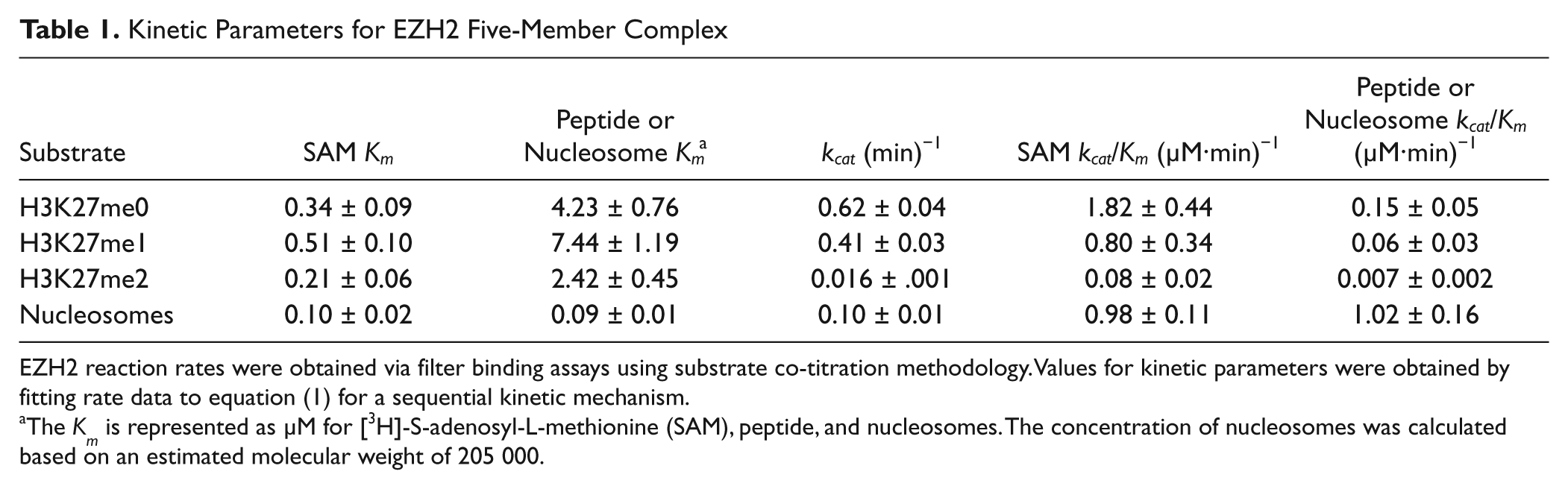
EZH2 reaction rates were obtained via filter binding assays using substrate co-titration methodology. Values for kinetic parameters were obtained by fitting rate data to equation (1) for a sequential kinetic mechanism.
The K m is represented as µM for [3H]-S-adenosyl-L-methionine (SAM), peptide, and nucleosomes. The concentration of nucleosomes was calculated based on an estimated molecular weight of 205 000.
Determination of Inhibition Kinetic Parameters
Evaluating a mono- or dimethylated peptide product as an EZH2 inhibitor is complicated by the fact that it is also a substrate for additional methylations, as evidenced by the kinetic parameters in
Table 1
. Similar to a previously published report, inclusion of the trimethylated (H3K27me3) peptide product resulted in a 2.5× and 3.4× maximal stimulation of EZH2 nucleosome or peptide methyltransferase activity, respectively (
To demonstrate that inhibitors of EZH2 could be identified within the context of the assays described, IC50 evaluations for the product inhibitor S-adenosylhomocysteine (SAH), sinefungin, and the H3K27A peptide were determined with either peptide or nucleosomes ( Table 2 ). To further characterize their respective mechanism of inhibition, product or inhibitor co-titrations versus SAM and peptide were conducted. SAH was best fit to a competitive inhibition model (equation (2)) versus varying SAM. When peptide was the varied substrate versus SAH, rate data were best fit to a noncompetitive inhibition mechanism (equation (3)), where K is = K ii ( Table 2 ). The H3K27A peptide demonstrated noncompetitive inhibition versus SAM and competitive inhibition versus the H3K27 peptide substrate. The generic methyltransferase inhibitor, sinefungin, demonstrated competitive inhibition versus SAM in the activity and binding assays ( Table 2 ). In contrast to the competitive product inhibition results observed for SAH versus SAM in the activity assays, SAH poorly displaced [3H]-SAM in the EZH2 binding assay. As anticipated, the mutant peptide was not competitive versus SAM in the binding assay.
Characterization of EZH2 Inhibitors: Potency and Mechanism of Inhibition
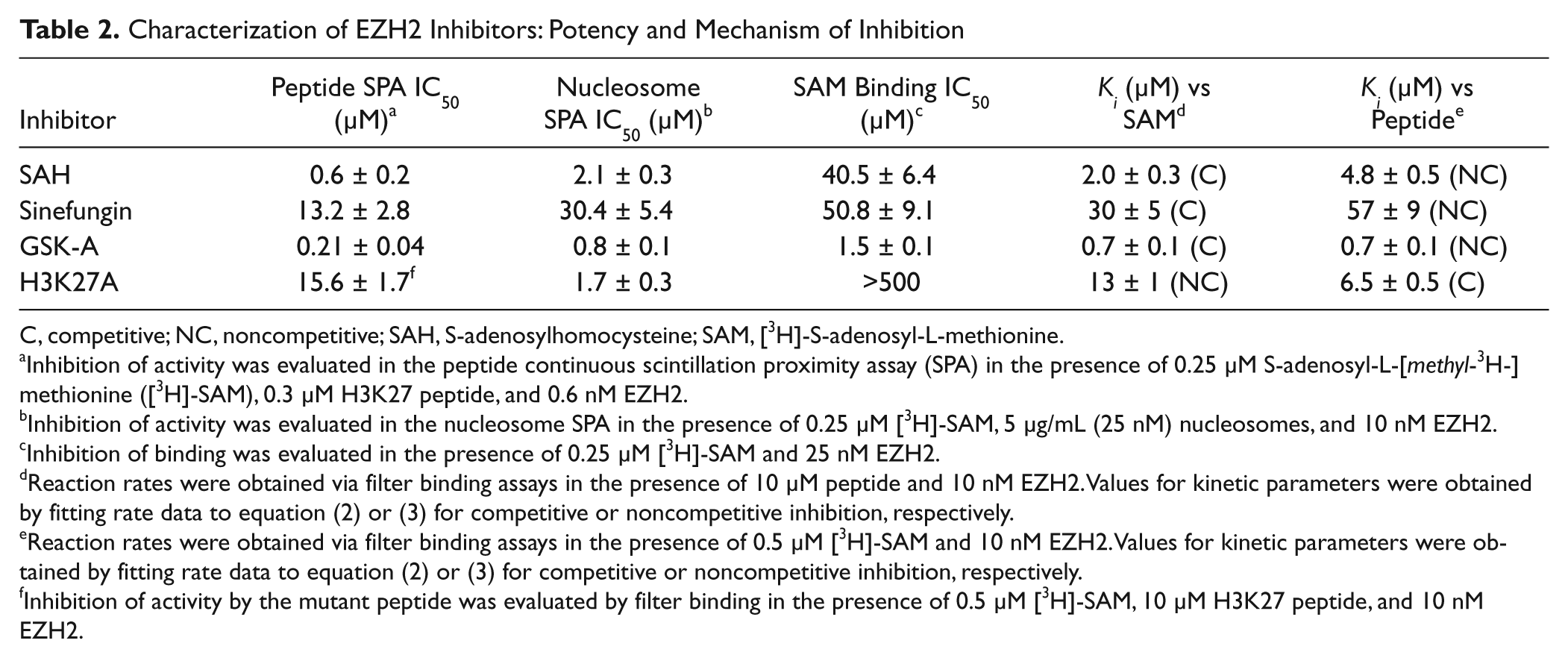
C, competitive; NC, noncompetitive; SAH, S-adenosylhomocysteine; SAM, [3H]-S-adenosyl-L-methionine.
Inhibition of activity was evaluated in the peptide continuous scintillation proximity assay (SPA) in the presence of 0.25 µM S-adenosyl-L-[methyl-3H-]methionine ([3H]-SAM), 0.3 µM H3K27 peptide, and 0.6 nM EZH2.
Inhibition of activity was evaluated in the nucleosome SPA in the presence of 0.25 µM [3H]-SAM, 5 µg/mL (25 nM) nucleosomes, and 10 nM EZH2.
Inhibition of binding was evaluated in the presence of 0.25 µM [3H]-SAM and 25 nM EZH2.
Reaction rates were obtained via filter binding assays in the presence of 10 µM peptide and 10 nM EZH2. Values for kinetic parameters were obtained by fitting rate data to equation (2) or (3) for competitive or noncompetitive inhibition, respectively.
Reaction rates were obtained via filter binding assays in the presence of 0.5 µM [3H]-SAM and 10 nM EZH2. Values for kinetic parameters were obtained by fitting rate data to equation (2) or (3) for competitive or noncompetitive inhibition, respectively.
Inhibition of activity by the mutant peptide was evaluated by filter binding in the presence of 0.5 µM [3H]-SAM, 10 µM H3K27 peptide, and 10 nM EZH2.
Peptide HTS Statistics and Results
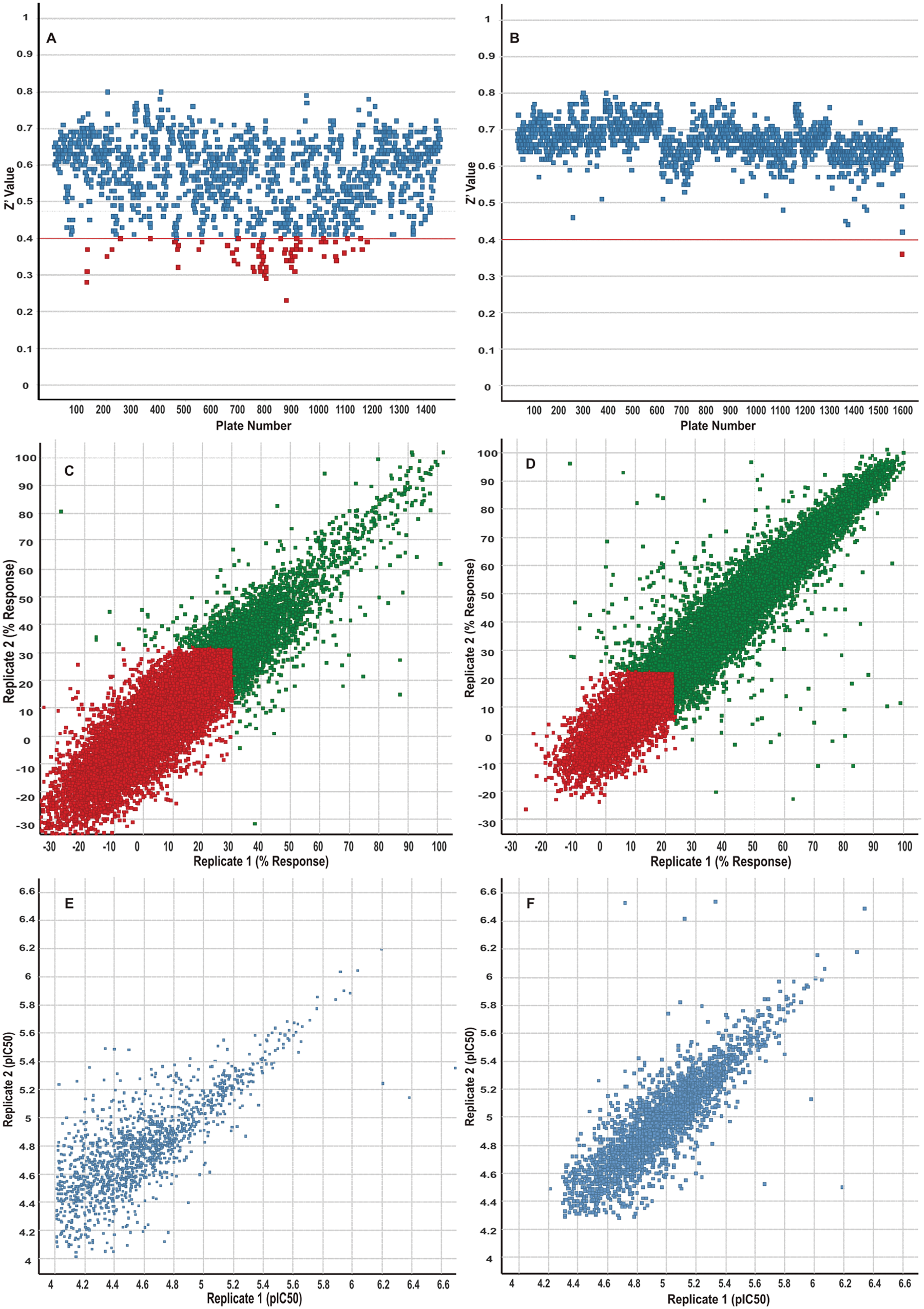
As part of the assay assessment in preparation for HTS, a validation set was screened in triplicate. Data analysis predicted a hit rate of 0.6% and a robust 3SD cutoff of 34%. A total of 1.96 million unique compounds were screened at 10 µM with an average Z′ value of 0.57 across 1432 plates ( Fig. 2A ). Analysis of the single-dose inhibition data gave an average robust 3SD cutoff of 44.2%, resulting in a 0.6% hit rate, and 14 135 primary robust active hits ( Table 3 ). These actives were tested for confirmation in duplicate with a robust 3SD cutoff of 31%, and the replicate correlation is shown in Figure 2C . Only 2860 of these hits confirmed in duplicate testing, and all were selected for dose-response evaluation. To obtain potency data for weaker compounds, dose-response testing was conducted with a twofold serial dilution beginning with a 100-µM top concentration. Duplicate 11-point dose-response data were obtained, and 1818 compounds generated dose-dependent inhibition resulting in IC50 curve fits; the correlation plot is shown in Figure 2E . The HTS statistics, as well as the IC50 potency bins, are summarized in Table 3 . Chemical cluster analysis, IFI, and physical properties of the peptide HTS hits were used to prioritize compounds that were subsequently selected for orthogonal assay testing (i.e., nucleosome activity, filter binding, SAM binding assays), compound repreparation, and structure-activity relationship (SAR) expansion efforts.
Presentation of the outcomes for the peptide (
Screening Statistics
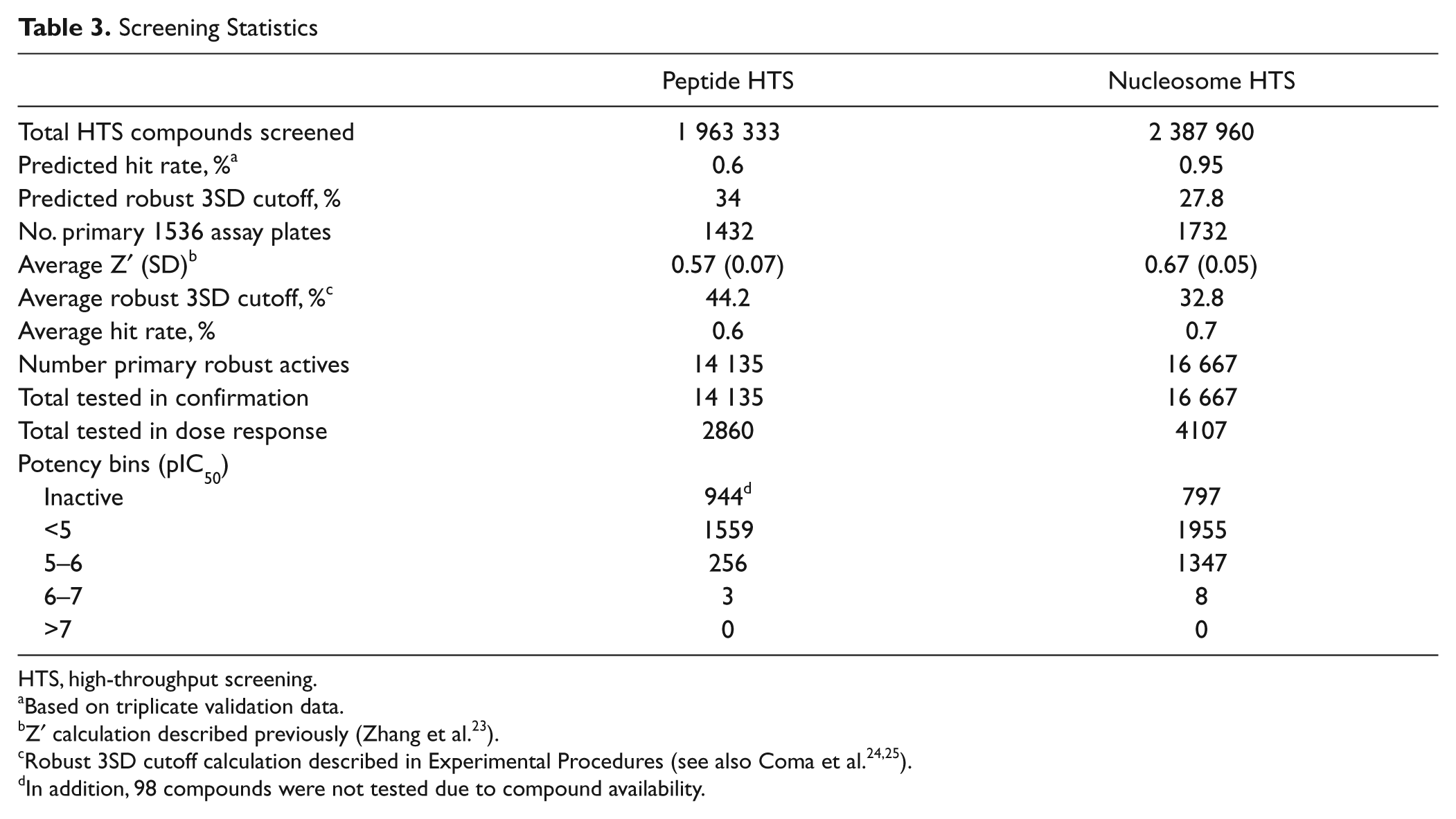
HTS, high-throughput screening.
Based on triplicate validation data.
Z′ calculation described previously (Zhang et al. 23 ).
In addition, 98 compounds were not tested due to compound availability.
Nucleosome HTS Statistics and Results
To further explore the possibility of identifying chemical diversity that was not revealed by the peptide HTS and to identify compounds with potentially unique mechanisms of inhibition (i.e., nucleosome competitive), an additional full diversity screen using nucleosomes as a substrate was conducted. Validation set analysis indicated a predicted hit rate of 0.95% and a robust 3SD cutoff of 27.8% (
Table 3
). A total of 2.39 million unique compounds were screened at 10 µM. The nucleosome screening statistics were improved compared with the peptide HTS, with an average Z′ value of 0.67 across 1732 assay plates (
Fig. 2B
). Analysis of the single-dose testing gave an average robust 3SD cutoff of 32.8%, resulting in a 0.7% hit rate, and 16 667 primary robust active hits were selected for duplicate confirmation screening at 10 µM. The robust 3SD cutoff for the hit confirmation was 23%, and the replicate correlation is shown in
Figure 2D
. Approximately 63% of these hits confirmed in duplicate, and analysis of the hit population was conducted to rationally reduce the number of compounds to be evaluated in dose response. The analysis used for compound selection included AWDS, IFI, chemical reactivity, and chemical property markers. Finally, 4107 compounds were tested in an 11-point dilution series; 3310 compounds gave dose-dependent inhibition resulting in an IC50 (
Characterization of an EZH2 Inhibitor
GSK-A was identified in the peptide and nucleosome screening campaigns and demonstrated similar levels of potency in both activity assays and the SAM binding assay (
Figure 3
and
Table 2
, respectively). Complementary kinetic mechanism of inhibition studies demonstrated that GSK-A is a SAM-competitive inhibitor and a noncompetitive inhibitor versus either peptide (
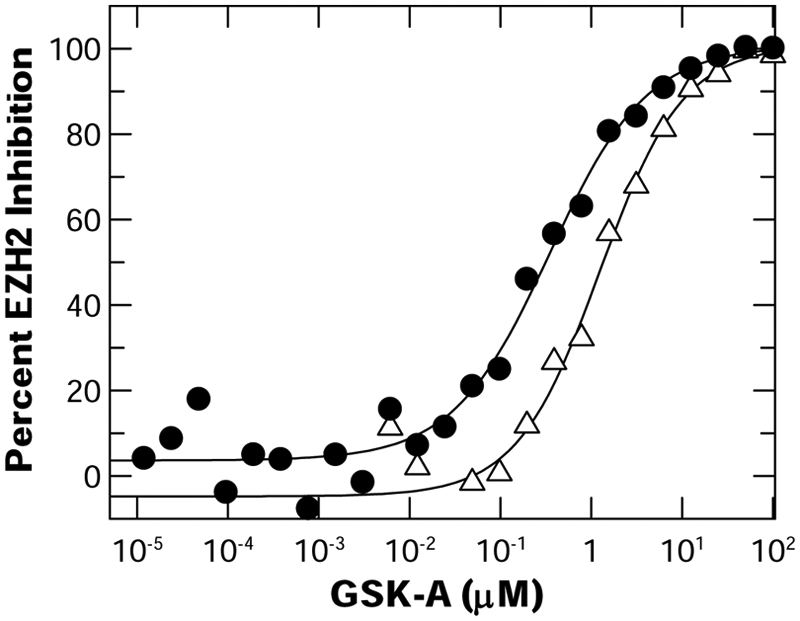
The potency of GSK-A was assessed in the EZH2 peptide or nucleosome scintillation proximity assay (SPA). EZH2 activity was evaluated in the peptide continuous SPA (•) (0.25 µM S-adenosyl-L-[methyl-3H-]methionine [[3H]-SAM] and 0.3 µM peptide); percent activity remaining was assessed from the slope of the reaction rate profile for reactions evaluated in the presence of compound normalized to the DMSO control reaction. Activity monitored in the nucleosome SPA (Δ) (0.25 µM [3H]-SAM and 5 µ/mL nucleosomes) was assessed from reaction end points normalized to the DMSO control reactions. IC50 values determined through XLfit are indicated in
Global suppression of H3K27 methylation is a relatively slow process whereby maximal effects are often observed after several days of drug treatment (M. McCabe, H. Ott, G. Ganji, S. Korenchuk, C. Thompson, G. Van Aller, Y. Liu, E. Diaz, L. LaFrance, M. Mellinger, C. Duquenne, X. Tian, R. Kruger, C. McHugh, M. Brandt, W. Miller, D. Dhanak, S. Verma, P. Tummino, C. Creasy, unpublished data) Exposure of breast cancer (Sk-Br-3) cells to GSK-A for 3 days resulted in a dose-dependent reduction of global H3K27me3 whereby 50% reduction of H3K27me3 was observed at approximately 8 µM ( Fig. 4 ). These data indicate that GSK-A is cell permeable and capable of specifically inhibiting EZH2 in a cellular context. Taken together, the development and validation of reagents as well as kinetic and cellular assay methods have been established to further characterize inhibitors of EZH2 methyltransferase activity identified from these screening campaigns.
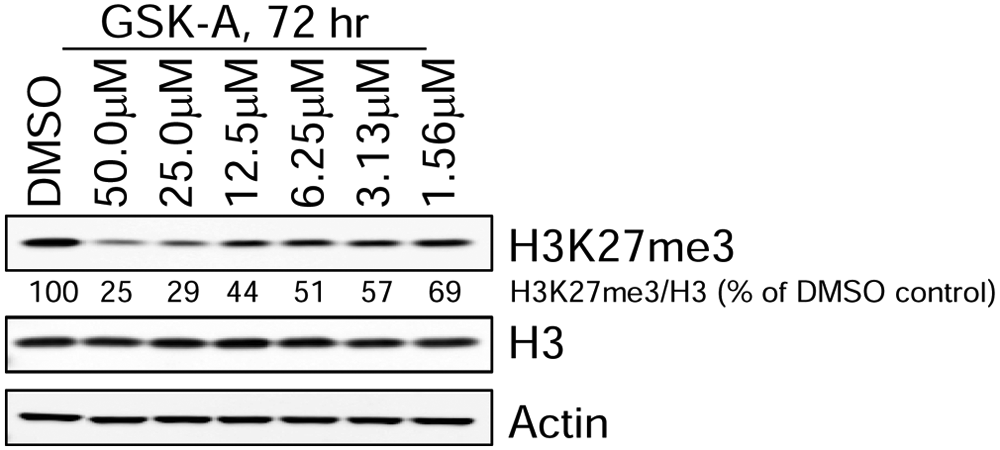
Analysis of Sk-Br-3 cells treated for 3 days with the indicated concentrations of the EZH2 inhibitor, GSK-A. Protein lysates from cells were analyzed by Western blotting with antibodies against the indicated proteins. The relative levels of H3K27me3 (normalized to total histone H3) are indicated for each sample.
Discussion
The coordinated upregulation of EZH2 protein and its associated activity are important biological markers for the aggressive progression of breast and prostate malignancies and as an indicator of poor patient prognosis.9,14 The results of this work highlight the inherent challenges and successes with reagent production, assay development, and HTS in identifying and characterizing newly discovered compounds that inhibit this target.
Absorbance, fluorescence, and luminescence methodologies have been shown to be viable assay formats for assessing methyltransferase activity in a low-throughput capacity. 29 To initiate a new lead identification effort for EZH2 via HTS, an assay capable of directly measuring methyltransferase activity must be adaptable to miniaturization without compromising detection sensitivity and assay quality. Assays that use [3H]-SAM and capture [3H]-methylated product are extremely sensitive and have been used to detail kinetic mechanisms for other methyltransferases with inherently low rates of catalysis. 30 In addition, radioactive assays can be easily adapted to the HTS environment and are less prone to optical interference.19,20 The inclusion of SPA imaging beads, which emit at 615 nm, is more optimal for CCD imagers that are most sensitive to light from the red region of the spectrum, thereby minimizing the concerns of potential quenching issues associated with colored library compounds. 31 The expansion of SPA methodology with [3H]-SAM provided significant advantages toward directly monitoring EZH2 activity using either a synthetic peptide or isolated nucleosomes as the methyl acceptor. Assays were configured for low-, medium-, and high-density capacity; required fewer reagent manipulations; and were less prone to assay complications that may be associated with other antibody, colorimetric, fluorescence, and coupled assay detection methodologies.29,32 Direct detection of nanomolar quantities of the reaction product permitted accurate characterization of EZH2 kinetic parameters with substrates and inhibitors without the aid of additional product detection coupling reagents. The inherently low level of compound interference made SPA an ideal format translatable to HTS. 20 Complementary filter binding methodology with peptide and nucleosome substrates provided direct orthogonal evidence for on-target inhibition. Thus, the development of two independent SPAs, using a synthetic surrogate and a natural substrate, provided a unique opportunity to prosecute GSK’s diverse chemical library against EZH2.
Coupling the assay modifications of a miniaturized SPA with imaging detection enabled larger sample populations to be interrogated to further assess enzyme activity under a number of varying reaction conditions. SPA and filter binding methods were used to determine kinetic parameters through global co-titrations of reaction substrates or inhibitors. Comparative kinetic parameters (K
m
and k
cat
) indicated native and monomethylated peptide were preferred EZH2 substrates based on the 10- to 20-fold enhancement in their specificity constant, k
cat
/K
m
, compared with dimethylated peptide (
Table 1
).6,8,16 This finding was further supported through a liquid chromatography/mass spectrometry (LC/MS) analysis of an EZH2-catalyzed methyltransferase reaction with elevated concentrations of enzyme, peptide, and SAM. The primary reaction product identified was H3K27me1, with a reduced but detectable level of H3K27me2 accounting for approximately 17% and 3%, respectively; no production of the H3K27me3 product was detected (
Kinetic parameters were also determined with nucleosomes, a more physiologically relevant substrate for EZH2 ( Table 1 ). The observed k cat with HeLa nucleosomes was lower than with native peptide and was comparable to dimethylated peptide. However, the nucleosome K m was estimated to be 50-fold lower than peptide, and thus the nucleosome specificity constant, k cat /K m , was comparable to native peptide, indicating that it is a suitable substrate for biochemical studies and an HTS. Methylated nucleosomes are very abundant in HeLa cells. It is estimated that 40% of the H3K27 mark is dimethylated, and the H3K27me1 and H3K27me3 marks comprise approximately 11% and 33%, respectively. 33 Thus, HeLa nucleosomes would be expected to be a poorer substrate for EZH2. Although similar values for k cat were observed for nucleosomes and dimethylated peptide, the K m for nucleosomes was approximately 25-fold lower than for peptide, suggesting that other posttranslational modifications to histones, along with exosite recognition distal to the H3K27 mark, may be important binding determinants for enhanced recognition and substrate binding.6,16 The inherently lower substrate concentration and methyltransferase activity observed with nucleosomes compared with peptide required subsequent assays to be modified with higher concentrations of enzyme and a different SPA bead selection to improve assay performance and product analysis.
Although the methyltransferase k cat and substrate K m values listed in Table 1 for SAM, peptides, and nucleosomes are comparable to the values for substrates of other methyltransferases, 30 they differ from values cited in another report for wild-type EZH2. 6 The data in Table 1 and Sneeringer et al., 6 respectively, are in agreement that EZH2 k cat is reduced when comparing an H3K27me0 to an H3K27me2 peptide and that the peptide K m is not influenced by the methylation state of H3K27. However, the 14× and 44× higher values reported in Table 1 for EZH2 k cat for nucleosomes and peptide, respectively, could be attributed to a combination of factors, including the purity and stoichiometry of the components of the EZH2 five-member complex, placement of the Flag affinity tag on the N-terminus of the EZH2 subunit compared with the N-terminus of the EED domain, and differences in the optimized assay buffer, ionic strength, and pH. The values for EZH2 k cat reported in Table 1 for nucleosomes and nonmethylated peptide (0.1 and 0.6 min−1, respectively) are comparable to values reported for other methyltransferases.30,34
To efficiently capture product and maintain a cost-effective use of SPA bead in an HTS capacity, assays were configured with nonsaturating substrate concentrations to enable identification of chemotypes that could be selectively competitive against either of the assay substrates. Complementary SPA and filter binding methodology expanded the capabilities to characterize EZH2 kinetics with substrates, as well as with the two known methyltransferase inhibitors, SAH and sinefungin. Both compounds were confirmed as competitive inhibitors versus SAM and noncompetitive inhibitors versus peptide (
Table 2
). The trimethylated peptide resulted in stimulation of EZH2 activity (
The quality of the EZH2 peptide and nucleosome HTS assays was continuously monitored throughout the miniaturization, automation, and compound testing processes, and both assays displayed acceptable performance standards such as Z′, 3SD cutoff, and hit rate. Figure 2 and Table 3 highlight the screening outcomes and illustrate the reduction in 3SD cutoff and improved Z′, average Z′, and Z′ (SD) for the nucleosome assay. When compared with the peptide HTS, the assay statistics associated with the nucleosome HTS were modestly improved and resulted in a higher assay Z′, lower robust 3SD cutoff, and a comparable average hit rate ( Table 3 ). The improved performance of the nucleosome screen may be counterintuitive based on the simple assumption that a homogeneous and less complex peptide substrate would provide less assay variability. The data presented, however, support the hypothesis that physiologically relevant binding partners (i.e., PRC2 complex components and nucleosome binding) are required to stabilize the integrity and activity of EZH2. 8 These key technical learnings can be applied to subsequent histone methyltransferase (HMT) assay development efforts to improve the quality of future HMT high-throughput screens.
The EZH2 peptide assay full-diversity screen resulted in 1818 compounds with varying potencies and chemical properties. Potency, structural organization, and chemical properties (MW, cLogP, synthetic accessibility) were used to prioritize compound testing in the orthogonal filter binding assay. Inhibitors of high interest were selected for SAR expansion and compound procurement efforts, resulting in the screening of 730 closely related compounds (data not shown).
To further explore the chemical diversity that was not revealed by the peptide screen and to identify compounds with additional mechanisms of inhibition (i.e., nucleosome competitive), an HTS was initiated using an assay based on the natural nucleosome substrate. The nucleosome full-diversity screen resulted in 4107 compounds with varying potencies and chemical properties. The hit population was rigorously analyzed based on chemical organization and clustering, chemical reactivity, IFI, and chemical properties such as cLogP, MW, and ligand efficiency (LE). Although comparative analysis revealed an 11% overlap between the robust actives from each of the screening campaigns, GSK-A was identified by both screens. The correlation replot of the dose-response data from both screens (
Fig. 5
) reveals a slight potency bias of the hits toward the nucleosome assay, whereas similar inhibition potency for GSK-A was observed with a peptide or nucleosome substrate (IC50 values of 0.21 ± 0.04 µM and 0.8 ± 0.1 µM, respectively;
Figure 3
and
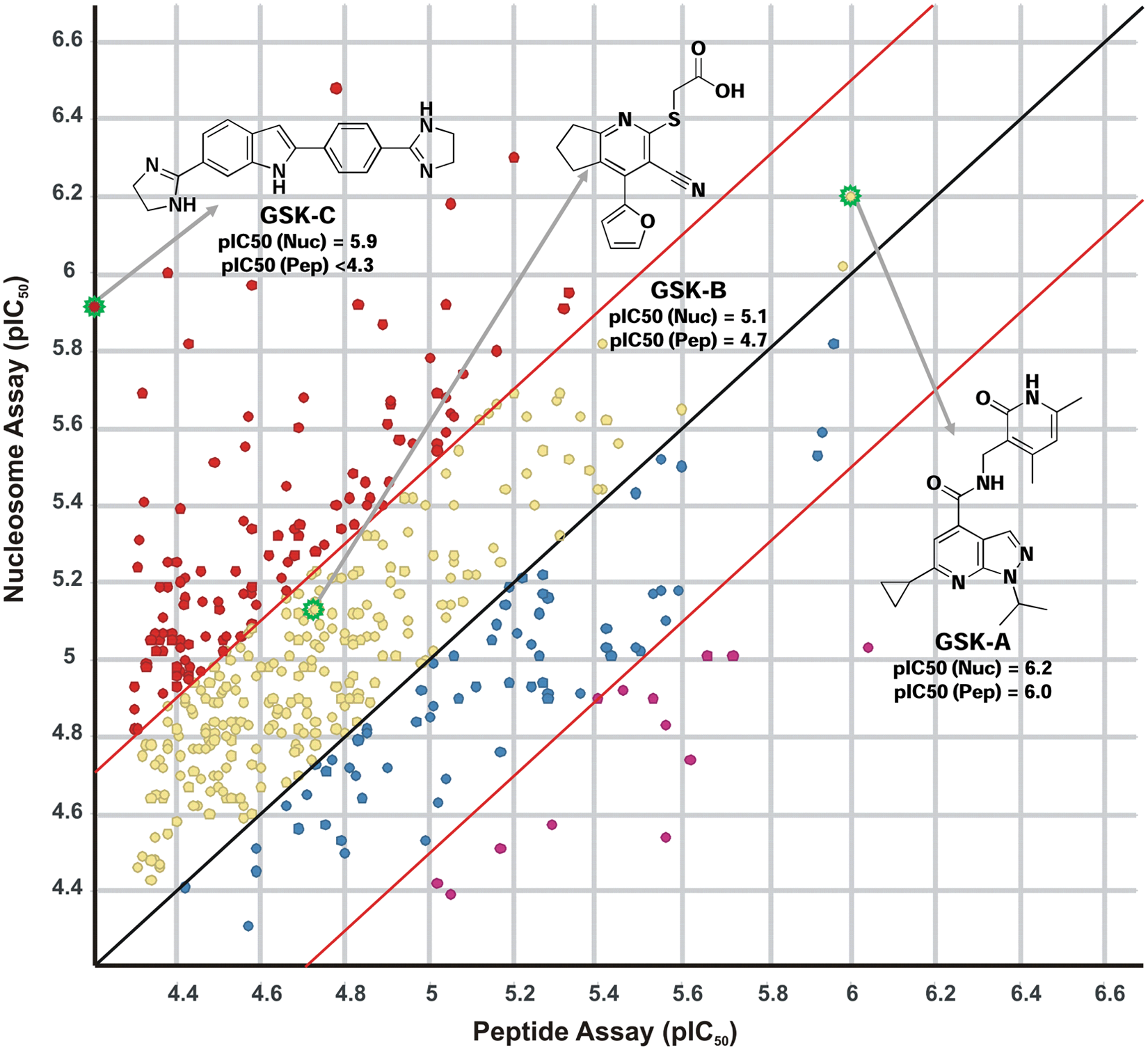
Correlation replot of the average pIC50 values obtained for selected compounds in the peptide and nucleosome EZH2 assays demonstrates a slight potency bias toward the nucleosome assay. Compound pIC50s are colored based on their potency correlation between the two assays: Compounds that demonstrated a >0.5× log more potent shift in the nucleosome assay are indicated in red (•), compounds that were >0.5× log more potent in the peptide assay are labeled in magenta (•), compounds exhibiting a 0–0.5× log potency shift in the nucleosome assay are highlighted in yellow (•), and compounds with a 0–0.5× log shift in potency in the peptide assay are shown in blue (•). The structure of GSK-A, a confirmed lead compound tested in biochemical mechanism of action and cellular assays, is shown. The structures of GSK-B, a confirmed hit of no further interest based on stability issues, and of GSK-C, a confirmed hit of no further interest based on its DNA intercalation activity, are shown.
GSK-B and GSK-C were also highlighted by the above screening efforts with confirmed and reproducible activity ( Fig. 5 ). GSK-B was identified in the peptide and the nucleosome screening campaigns with pIC50 values of 4.7 and 5.1, respectively. This chemical template was the subject of further studies but was determined to be of no further interest because of issues associated with chemical stability. GSK-C, identified in the nucleosome HTS (pIC50, 5.1), was not revealed in the peptide screen. GSK-C was shown to be a DNA intercalator, which is hypothesized to contribute to its inhibition mechanism possibly through the disruption of the nucleosome integrity and its ability to serve as a viable EZH2 substrate. In addition, the DNA binding activity of GSK-C was considered a general liability and a hurdle for further development.
Although initial priority was placed on characterizing GSK-A inhibition of EZH2 activity using nucleosome and peptide substrates, a more extensive characterization of the hit population revealed by the two HTS campaigns is currently under way. Although outside the scope of this study, a more extensive analysis of the hit population with multiple biochemical assay variations (i.e., varying preincubation and reaction times) as well as nuisance assays has found that many inhibitors, especially time-dependent inhibitors, are false positives. The frequency of susceptibility to false positives does not appear to differ between the two HTS campaigns; further analysis of the chemical similarities and differences revealed by the two screens would be dominated by compounds that have not been repurified or resynthesized for follow-up and would need to be approached with caution.
Generally, the screening platforms have been efficiently used as part of the EZH2 hit identification strategy, but the campaigns have had significant challenges. A significant number of false positives were observed with activities that did not confirm upon resynthesis and repurification from solid. We speculate that many of these false positives were caused by non-UV-active contaminants (i.e., trace metals) that were not present after repreparation and were not detected by common high-performance liquid chromatography (HPLC)–based quality control (QC) methods. In addition, other false positives have been identified with specific liabilities such as the DNA intercalation activity highlighted above.
GSK-A did not exhibit stability or DNA binding liabilities, and based on a favorable SAM-competitive mechanism and synthetic tractability, GSK-A was selected as the starting point for establishing a lead optimization effort and as an essential tool molecule to validate enzymatic and cellular methods. The mechanism of inhibition of GSK-A was studied in greater detail using bivariate steady-state kinetic analysis. Mechanism of inhibition studies demonstrated that GSK-A is a SAM-competitive inhibitor and a noncompetitive inhibitor versus peptide (
Table 2
and
The EZH2 [3H]-SAM binding assay confirmed that SAM-competitive compounds would displace radiolabeled substrate in the absence of catalysis. The H3K27A mutant peptide was not SAM competitive ( Table 2 ) and did not displace [3H]-SAM, whereas sinefungin and GSK-A displaced [3H]-SAM with similar potencies compared with the kinetic activity assays. However, the end product SAH weakly displaced [3H]-SAM despite it being a more potent competitive inhibitor in the activity assays. The discrepancy of SAH inhibition potency between assay formats has been observed across other histone methyltransferases. The more potent inhibition of SAH in activity assays is greatly diminished in the binding assay in the absence of peptide or nucleosomes. One possible explanation is that SAH may require a peptide/nucleosome substrate to form a dead end complex, where tighter affinity is created in a noncatalytic ternary complex. Further evidence for this was obtained through high-resolution crystal structures of SET7/9 and SET8 (an H3K4 and H4K20 methyltransferase, respectively) where both reaction products formed a stable ternary complex.35,36 Horowitz et al. 37 proposed that for the SET domain of SET7/9, coordinated carbon-oxygen hydrogen bonding between the methyl group of SAM, the hydroxyl group of Tyr335, and the main chain carbonyl oxygen of His293 aligns the SAM methyl group in an appropriate geometry with the lysine ϵ-amine group for the SN2 methyl transfer reaction. Cofactor and analog binding affinities, measured by isothermal calorimetry, revealed nanomolar binding affinity of SAM and sinefungin for SET7/9 but approximately 1000× weaker binding of the reaction product, SAH, presumably due to the absence of the methyl group to engage in hydrogen bonding. More recently, stopped-flow fluorescence measurements of PRMT1 have identified a precatalytic conformational transition induced by SAM or SAH binding that changes the affinity for the second substrate. 38
The initial connectivity between enzymatic inhibition and cellular efficacy was established for GSK-A. Breast cancer (Sk-Br-3) cells exposed to GSK-A for 3 days demonstrated a dose-dependent reduction in the levels of trimethylated H3K27 (
In conclusion, the lead identification platforms described were enabled by the development and optimization of relevant reagents and assays and have been used to identify and characterize a novel compound that inhibits the in vitro and in vivo methyltransferase activity of EZH2. The molecule reported represents a starting point for the development and optimization of new tool compounds to further probe the biological function of EZH2 and to understand how the selective inhibition of EZH2 and concomitant reduction of H3K27 trimethylation will affect models of human disease. Finally, GSK-A provides a novel template for drug development and the potential for chemotherapeutic intervention in EZH2-dependent cancers.
Footnotes
Acknowledgements
The authors acknowledge Harjeet VanDer Keyl and Han Trinh for the primary cloning of the PRC2 complex components, Linda Myers and Rob Smith for the generation of viruses for expression, and Dean McNulty’s LC/MS analysis of the peptide methylation products from the first EZH2 reaction. Sample Management Technologies provided >3500 assay ready plates for high-throughput screens and dose-response evaluations. Tony Jurewicz’s assistance with data analysis and screening logistics and Subhas Chakravorty’s Cheminformatics support were greatly appreciated. The authors also thank Jessica Schneck and William H. Miller for their comments and helpful suggestions regarding the preparation of this manuscript.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.