
Abstract
Despite significant use in basic research, embryonic stem cells have just begun to be used in the drug discovery process. Barriers to the adoption of embryonic stem cells in drug discovery include the difficulty in growing cells and inconsistent differentiation to the desired cellular phenotype. Embryonic stem cell cultures require consistent and frequent handling to maintain the cells in a pluripotent state. In addition, the preferred hanging drop method of embryoid body (EB) differentiation is not amenable to high-throughput methods, and suspension cultures of EBs show a high degree of variability. Murine embryonic stem cells passaged on an automated platform maintained ≥90% viability and pluripotency. We also developed a method of EB formation using 384-well microplates that form a single EB per well, with excellent uniformity across EBs. This format facilitated high-throughput differentiation and enabled screens to optimize directed differentiation into a desired cell type. Using this approach, we identified conditions that enhanced cardiomyocyte differentiation sevenfold. This optimized differentiation method showed excellent consistency for such a complex biological process. This automated approach to embryonic stem cell handling and differentiation can provide the high and consistent yields of differentiated cell types required for basic research, compound screens, and toxicity studies.
Introduction
In recent years, there has been a concerted effort to move compound screening into more biologically relevant cellular assays. This has been seen in the increase of screens performed in primary cells as the pharmaceutical industry moves away from immortalized or genetically modified cell lines. Embryonic stem cells offer the promise of a ready supply of just such relevant cell types through the processes of self-renewal and differentiation. Unfortunately, the most common methods for embryonic stem cell differentiation are labor intensive, inefficient, and costly, and these challenges must be overcome before differentiated cells will become commonplace in drug screening. Furthermore, embryonic stem cell growth can be challenging, as maintenance of pluripotency is important to the overall vitality of any embryonic stem cell screening regimen. To increase the consistency of both our starting murine embryonic stem cell cultures and the resulting differentiated cells, we investigated an automated approach.
Although monolayer-based differentiation of embryonic stem cells offers some advantages, the formation of spherical cell aggregates, known as embryoid bodies (EBs), is still the favored method of embryonic stem cell differentiation. EBs are thought to provide cellular environments and cell-cell interactions that more closely mimic normal development.1,2 However, the two most common formats for EB formation, the hanging drop method and static suspension culture, both have significant drawbacks. Hanging drop formation involves the labor-intensive process of pipetting small volumes of cells onto the underside of a Petri dish lid. 3 Following inversion of this lid, the cells aggregate at the bottom of the drop and ideally form a single EB per drop. Although this method is reasonably efficient in the formation of EBs, the low-throughput nature of this process and the frequent loss of EBs during manipulation, coupled with an inability to change media during this method, leave much room for improvement. By comparison, static suspension culture, which simply involves plating a cell suspension into a low-attachment dish, is much less effort. However, the inefficient and uncontrolled aggregation of large numbers of embryonic stem cells results in high levels of cell death and inconsistent EB size and shape. 4 EB size and shape have been shown to influence the resulting differentiated cells, 5 and therefore high EB variability results in cell lineage variability. An ideal differentiation method would provide greater consistency and control in EB formation than static suspension culture. A number of additional methods have been attempted (thermocycle or low-attachment microplates, 6 lithographic methods, 7 conical tubes 8 ), but all of these suffer from similar drawbacks of high effort, low throughput, or inconsistent EB size/shape. Initiating EB formation in 96-well low-attachment plates, often following centrifugation, has achieved results similar to hanging drops.9,10 However, these cells still require significant manipulations, including eventual transfer to tissue culture plates for EB attachment. Our goal was to improve upon this process through the use of automation, to achieve higher throughput and greater consistency in EB formation.
Another impediment to the use of differentiated cells in drug discovery is the relatively low yield of a given cell type. Typical EB formation involves the removal of leukemia inhibitory factor (LIF), 11 and this is sufficient to form a mixed population of different cell types.12,13 However, for a compound screen, researchers would likely desire a single purified lineage, and thus finding methods to increase the formation of this lineage would be beneficial. A lineage that arguably would provide the most benefit for drug screening is the cardiomyocyte lineage. As a large portion of off-target adverse events are due to cardiotoxicity, differentiated cardiomyocytes may provide the most cost-effective way to perform physiologically relevant toxicology studies. 14 Recognizing the need for reproducible and robust cardiomyocyte differentiation, our second goal was to increase the percentage of cardiomyocytes formed during EB-based differentiation. A significant number of signaling pathways have been shown to be involved in cardiomyocyte differentiation. Cardiomyocyte formation can be influenced by treatment with recombinant proteins such as bone morphogenetic proteins (BMPs), 15 activin, 16 Wnt, 17 fibroblast growth factor (FGF), 18 and vascular endothelial growth factor (VEGF) 19 or other molecules such as ascorbic acid 20 and protein kinase inhibitors, 21 as well as by differentiating cells under hypoxic conditions. 22 Whether the effect on cardiomyocyte formation is positive or negative can often depend on the timing of these treatments. 17 Typically, the effects of these factors are assessed in isolation, but we chose a design of experiment (DOE) approach that would detect multifactor effects that could generate a pronounced enhancement of cardiomyocyte differentiation.
To increase the consistency of our starting murine embryonic stem cell cultures, we adopted an automated approach to cell maintenance and passaging. For automated EB formation, a noncontact dispenser was used to plate cells into 384-well polypropylene plates, and the cells formed a single EB per well with excellent consistency and roundness. Transfer of EBs to gelatin-coated plates using a 96-channel head allowed attachment while maintaining the single EB/well format that facilitated high-throughput optimization screens. Ultimately, the optimized microplate differentiation method produced a consistent cardiomyocyte yield in excess of 40% of the total differentiated cells.
Materials and Methods
Murine embryonic stem cell (mESC) reagents
The 129 line of mESCs (Invitrogen, Carlsbad, CA) were cultured at 37 °C, 5% CO2 in a humidified incubator. Culture medium consisted of KnockOut Dulbecco’s modified Eagle’s medium (KO-DMEM) + 15% (v/v) KnockOut Serum Replacement (KSR), glutamine, and nonessential amino acids (NEAA) (all Invitrogen) and 1000 U/mL ESGRO (Millipore, Billerica, MA). Basal differentiation medium replaced KSR with 15% (v/v) fetal bovine serum (FBS) and contained no ESGRO. Cells were cultured on six-well tissue culture plates that had been coated with 1% (w/v) gelatin (Millipore) for 15 min at 37 °C. Cells were passaged with TrypLE (Invitrogen) every 2 to 3 days.
Automation of embryonic stem cell culture
Cells were incubated in a Cytomat 2C incubator (Thermo Scientific, Rockford, IL) that was integrated to a Biomek FXP liquid handler (Beckman Coulter, Indianapolis, IN).* The liquid handler was located in a HEPA-filtered enclosure to maintain sterility during cell manipulations. Addition/removal of reagents (gelatin, TrypLE, media) and cell manipulation were accomplished using the Span-8 Pipettors on the Biomek FXP. Following trypsinization on the Biomek, cells were counted on an integrated Vi-CELL XR Cell Viability Analyzer (Beckman Coulter), and the viable cell count was used to drive the liquid-handling steps for addition of 800 000 viable cells per well. Additional aliquots of cells were added to microcentrifuge tubes, and these samples were removed from the automated system and manually stained for SSEA-1 as a marker of pluripotency. 23
Automation of embryonic stem cell differentiation
For differentiation experiments, the BioRAPTR FRD workstation (Beckman Coulter) was used to dispense 500 cells/well into 384-round bottomed polypropylene plates (Nunc, Thermo Scientific, Rockford, IL) in a culture volume of 40 µL/well. After 2 days, an additional 80 µL/well of media was added, and on day 5, the EBs were imaged using an ImageXpress Micro (Molecular Devices, Sunnyvale, CA) to determine their circularity (shape factor). Shape factor is the ratio of the shortest radius to the longest radius, so a perfect circle has a shape factor of 1. The 5-day EBs were transferred to 96-well gelatin-coated tissue culture plates containing 100 µL fresh media (~200 µL total volume), using the Multichannel Pipettor of the Biomek FXP workstation. The EBs adhered and spread across the well surface. Adherent cultures were maintained for 3 days and were assayed for cardiomyocytes after 8 total days of differentiation.
Cardiomyocyte optimization
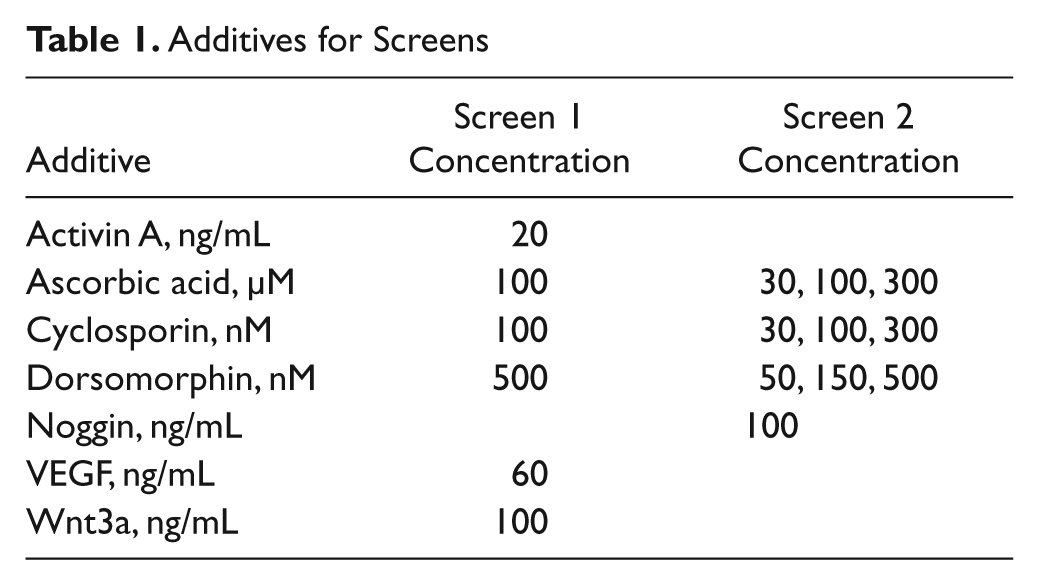
Combinations of compounds and recombinant proteins were tested for cardiomyocyte enhancement using a DOE methodology. Briefly, we tested different concentrations and times of addition of activin a, ascorbic acid, cyclosporin a, 24 dorsomorphin, 25 noggin, 26 VEGF, and Wnt3a, based on published findings. We used statistical design software (Design-Expert 8, Stat-Ease, Minneapolis, MN) to generate test combinations of these factors using a D-Optimal Factorial design. These conditions were converted to BioRAPTR FRD dispense volumes using AAO for BioRAPTR software (Beckman Coulter). Reagents were dispensed into 384-well plates, along with cells and media using the BioRAPTR dispenser as above, and additional reagents/media were added at various time points.
Brightfield/fluorescent microscopy
Brightfield microscopy was used to detect regions displaying the contractile motion that is a hallmark of cardiomyocytes. This method was used as an initial indicator of cardiomyocyte formation in the differentiated cell cultures. Fluorescent microscopy was also used to characterize cardiomyocytes and ensure antibody specificity. Day 11 EBs (6 days postadherence) were trypsinized and readhered in a monolayer for improved visualization. After 48 h, cells were processed using the FIX & PERM Cell Fixation & Permeabilization Kit (Invitrogen) and blocked with 2% (v/v) goat serum. Cells were co-stained for myosin heavy chain (Alexa Fluor 488-MF20, 1:500; eBioscience, San Diego, CA) and α-actinin (1:100 [Sigma, St. Louis, MO] + Alexa Fluor 555 anti-mouse IgG, 1:500 [Invitrogen]), and cell nuclei were labeled with DAPI (Invitrogen). Images were generated on an EVOS fluorescent microscope (AMG, Bothell, WA).
Flow cytometry
To determine pluripotency of automated or manually passaged embryonic stem cell cultures, cells were fixed (Invitrogen) and stained for SSEA-1 (PerCP; R&D Systems, Minneapolis, MN). Stained cells were analyzed on a Gallios flow cytometer (Beckman Coulter). For analysis of differentiated cells during optimization screens, the Biomek FXP Multichannel Pipettor was used to release day 8 EBs from 96-well plates and generate single-cell suspensions through ACCUMAX (Millipore) treatment coupled with repeated pipetting. A Biomek NXP with Span-8 Pipettors (Beckman Coulter) was used to stain cells for myosin heavy chain using the reagents described above. Samples were run on a plate-based FC500 MPL flow cytometer (Beckman Coulter), and the resulting data were analyzed using Kaluza software (Beckman Coulter).
Quantitative PCR
Optimal cardiomyocyte differentiation conditions were validated by measuring the induction of cardiomyocyte-specific gene expression. Differentiated cells from 8 wells per condition were combined on day 8, and total RNA was extracted using the SV Total RNA Isolation System (Promega, Madison, WI). RNA was reverse transcribed using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA), and gene expression was analyzed using TaqMan probes and a 7900 Real-Time PCR System (Applied Biosystems). Relative gene expression was determined using the ΔΔCt method with GAPDH as a reference gene.
Results
Automated maintenance of embryonic stem cell cultures
To alleviate the significant labor required to maintain embryonic stem cells, we developed an automated system to passage cells and exchange spent medium (
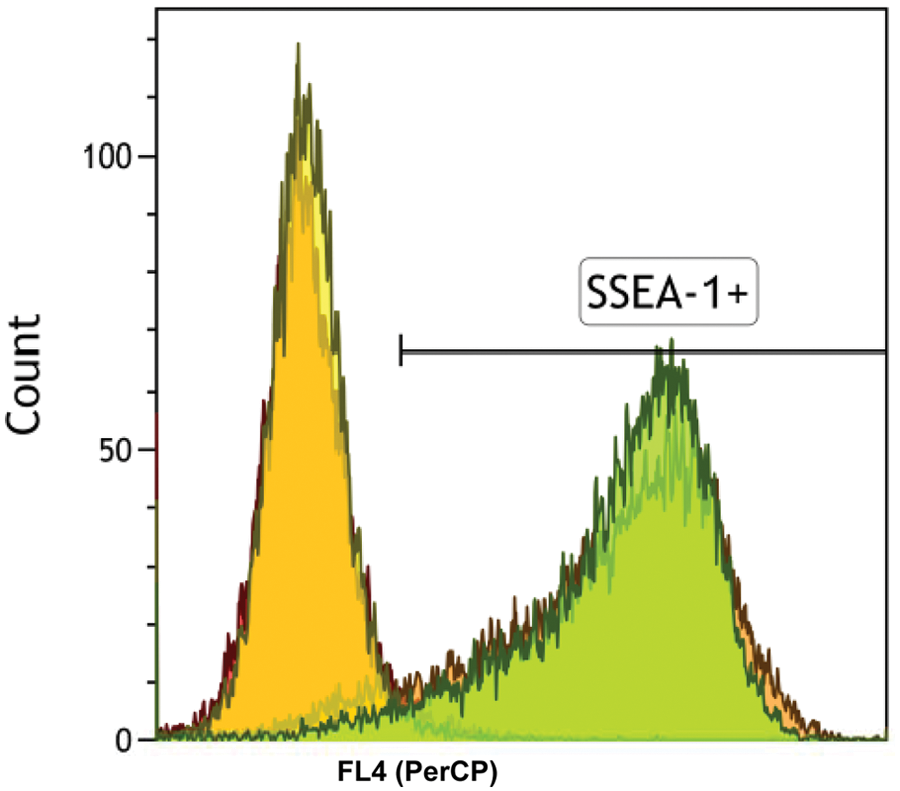
Automated maintenance of embryonic stem cell cultures. Flow cytometry results following staining of embryonic stem cell cultures for SSEA-1 as a marker of pluripotency (gated) or isotype controls. Overlapping peaks illustrate manual and automated cultures.
384-Well microplate format of embryoid body formation
In an effort to increase throughput and decrease variability in EB formation, we automated the addition of embryonic stem cells to 384-well polypropylene round-bottom plates. These plates prevented cell adherence and allowed the embryonic stem cells to aggregate into embryoid bodies at the bottom of the well, in a similar fashion to the hanging drop format of differentiation. Five hundred cells were dispensed into each well in basal differentiation medium using a BioRAPTR FRD workstation, and this method resulted in the formation of a single EB per well in 99.5% of the wells ( Fig. 2A ). The size of the EBs could be altered by varying the starting number of cells and/or time of incubation, but after 5 days, these EBs were ~380 µm in size. Using an ImageXpress Micro high-content imager, we measured the circularity (shape factor) of the EBs formed by this method. The shape factor of the microplate EBs was 0.8 ± 0.1 ( Fig. 2B ), a significant improvement in circularity compared with the hanging drop method (0.5 ± 0.2, p = 1.4e-22; Fig. 2A , B ). In addition, the microplate EBs were more consistent in their shape across wells than hanging drop EBs, with coefficients of variation (CVs) of 12% and 40%, respectively.

Brightfield images of automated and manual embryoid bodies (EBs). (
Cardiomyocyte formation
The EBs formed through the microplate method were transferred to 96-well gelatin-coated tissue culture–treated plates using a Biomek FXP Multichannel Pipettor. During an additional 3 days of adherent differentiation, cardiomyocytes were identified through the observation of spontaneously contracting regions of cells (
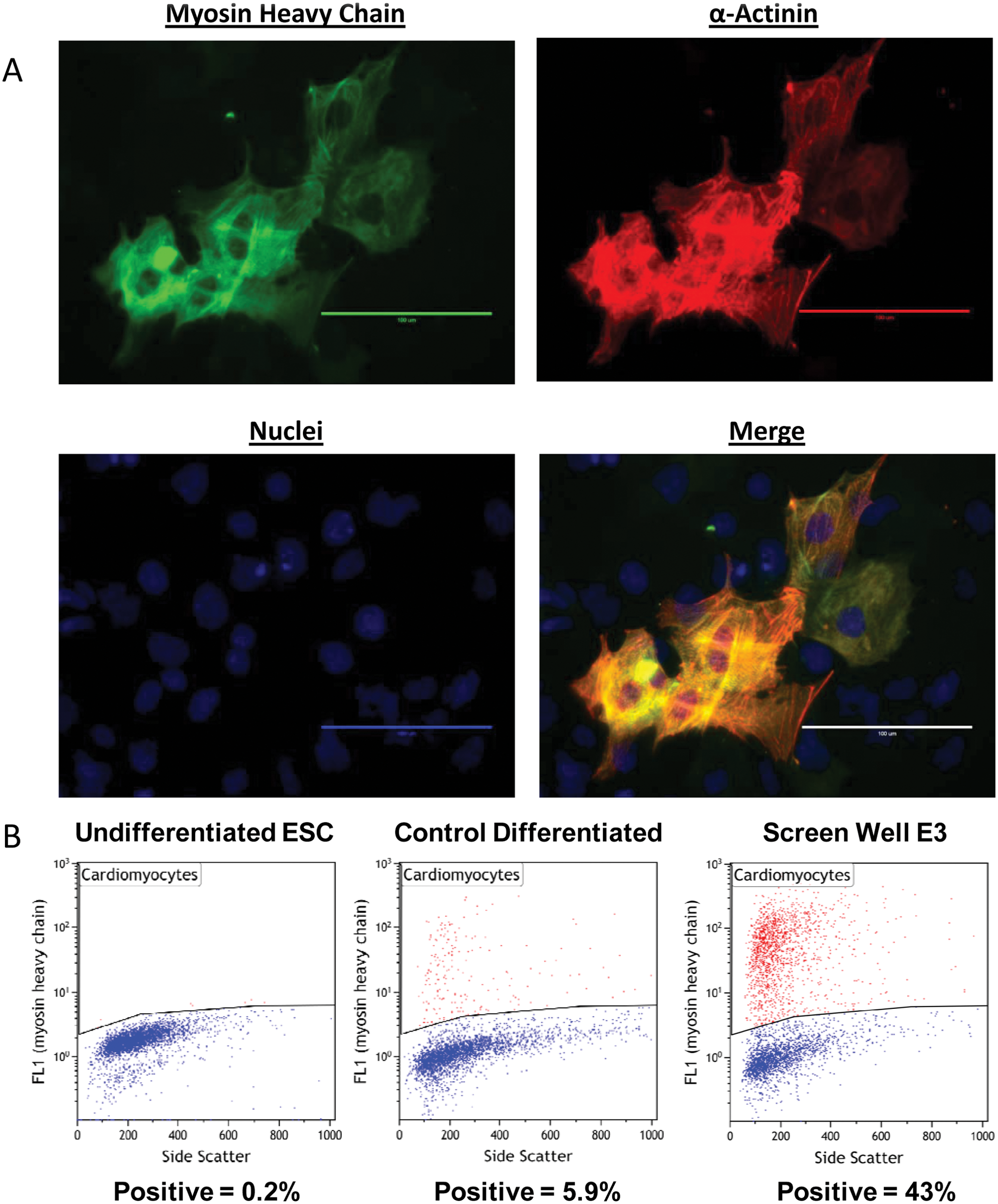
Analysis of cardiomyocytes following optimized automated embryonic stem cell differentiation. (
Cardiomyocyte optimization
By having a single embryoid body per well, we were able to use the automated microplate format of differentiation to screen for conditions that would enhance cardiomyocyte formation. We used two successive DOE-derived screens to test combinations of various factors ( Table 1 ) that were thought to promote cardiomyocyte formation. Concentrations and time of addition were also varied in addition to different combinations of factors. Differentiated cells were analyzed for cardiomyocytes by flow cytometry as above (myosin heavy chain positive). Screen results identified conditions that produced differentiated cell cultures that were >40% cardiomyocytes ( Fig. 3B , “Screen Well E3”).
Additives for Screens
Quantitative PCR was used to assess expression of additional cardiomyocyte genes to further confirm the enhancement of cardiomyocyte differentiation. Under basal differentiation conditions, Nkx2.5 and cardiac actin were upregulated 220- to 250-fold compared with undifferentiated embryonic stem cells ( Fig. 4A , “Control”), further illustrating that the automated microplate format was conducive to cardiomyocyte formation. However, differentiating cells under one of our optimized conditions increased Nkx2.5 3850-fold and cardiac actin 1750-fold compared with undifferentiated embryonic stem cells ( Fig. 4A , “Optimal”). This 7- to 17-fold increase over basal differentiation (cardiac actin, p = 1.14e-6; Nkx2.5, p = 0.0013) is similar in magnitude to the 7-fold increase in positively stained cells observed by flow cytometry (43% vs. 6%), supporting an increase in cardiomyocyte formation. The size of contracting regions was also increased under optimal conditions ( Fig. 4B ), although this increase was not quantified.
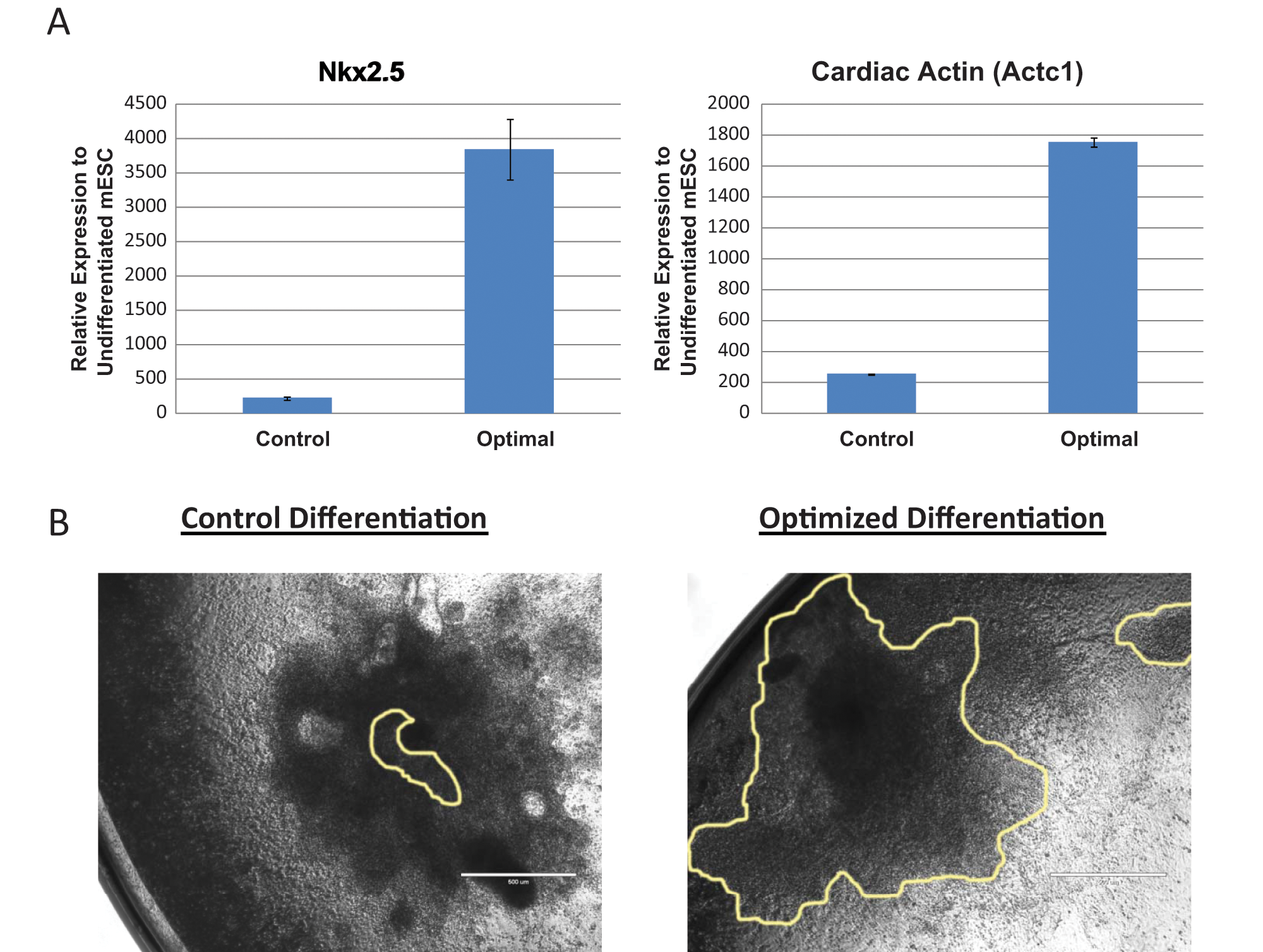
Validation of optimized cardiomyocyte differentiation. (
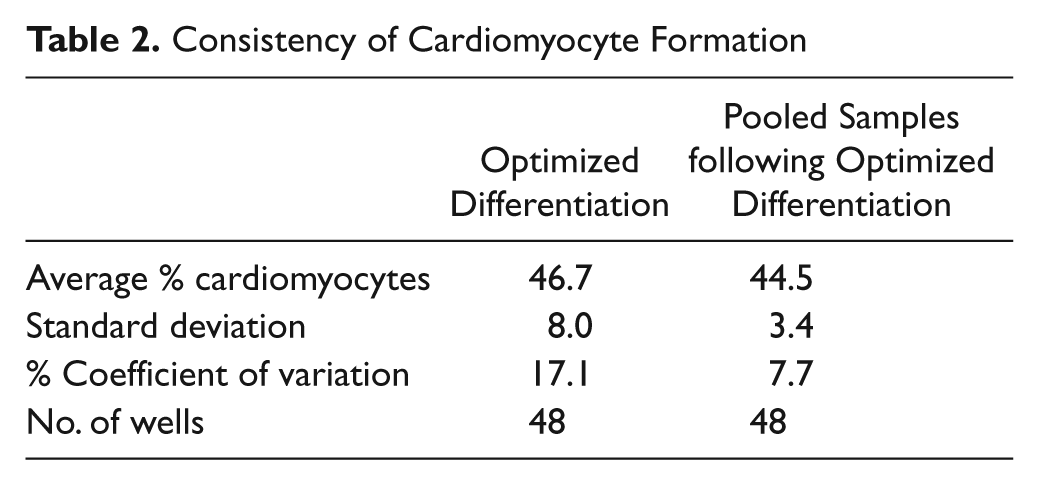
Consistency of cardiomyocyte formation
To determine the consistency of cardiomyocyte formation under optimized differentiation conditions, we analyzed 48 wells of optimally differentiated embryoid bodies by flow cytometry. The resulting differentiated cells showed an average of 46.7% cardiomyocytes with 17.1% variance ( Table 2 ). By comparison, 48 wells of optimally differentiated cells that were trypsinized, pooled, redistributed into 48 wells, and then analyzed by flow cytometry showed 7.7% variance. This suggests that there is <10% biological variability in this highly complex system.
Consistency of Cardiomyocyte Formation
Discussion
Two major barriers to the use of differentiated embryonic stem cells in drug discovery remain the labor-intensive methods and resulting low yield of cell types of interest from current differentiation protocols. Although embryoid body formation offers more “embryo-like” differentiation conditions, 27 the delicate procedures and inconsistent resulting lineages leave much to be desired. We have described a system that can automate and optimize differentiation processes that begin with EB formation. By adapting the hanging drop method to a 384-well microplate format amenable to screening (single EB/well), we were able to enhance the formation of cardiomyocytes to more than 40% of the total differentiated cells.
One key to the optimization screen was the consistency of the basal EB formation and differentiation. This consistency begins by maintaining embryonic stem cells in a pluripotent state while minimizing well-to-well and plate-to-plate variability, one of the main advantages of automation. The plate-based differentiation protocol described here provided a process that enabled high-throughput studies, and these studies require low variability across plates to ensure a robust signal-to-noise ratio. The consistency of differentiation was even more pronounced with our process when compared with hanging drop protocols. Over numerous experiments, the percentage of cardiomyocytes in the basal differentiation conditions was 6% to 10% (data not shown). However, changing reagent lots (i.e., recombinant proteins, serum) could affect the screen results or consistency between experiments.
Although a 40% cardiomyocyte yield is a significant enhancement over basal differentiation, this proof-of-principle study was not exhaustive, and further improvements can likely be made. Using defined medium could give more control over any serum effects and identify additional reagents that influence cardiomyocyte differentiation. Also, early additives were not removed in this study, but media changes were possible with this system (data not shown). As the time of exposure to various proteins or compounds has been shown to affect cardiomyocyte differentiation, this advantage over the hanging drop method could be exploited to further enhance cardiomyocyte formation.
Although this cardiomyocyte percentage may be sufficient for some drug studies, other assays may require further purification of cardiomyocytes from noncardiomyocytes. This could include sorting cells based on external, lineage-specific markers such as SIRPA 28 or by the transgenic expression of drug resistance cassettes from lineage-specific promoters. Regardless, an increased yield in the desired cell type will lower the starting number of embryonic stem cells required to achieve the desired output. As a reference, differentiating the initial 500 embryonic stem cells using our optimized differentiation method resulted in >10 000 cardiomyocytes on day 8 (data not shown). In addition, this low starting number of cells would allow the plating of ~8000 EB wells from a single well of a six-well plate, potentially yielding >80e6 cardiomyocytes. Obviously, experiments of this magnitude could not be observed visually, but cardiomyocyte contraction could be monitored in an automated fashion such as by using high-content imaging with indicator dyes to measure calcium flux.
This method also provides significant cost savings beyond increasing cell yield. Although the optimization screen ended in 96-well plates, the high-cost reagents (i.e., recombinant proteins) were added only during the initial 384-well stage. This smaller well volume significantly reduced reagent expenses, whereas the higher density plate (vs. 96-well low-attachment plates) helped reduce labware costs. The fact that the maximal cardiomyocyte percentage was reached after only 8 days saves both time and money with less media changes required as compared with longer differentiation protocols. For differentiation protocols of this length, the attachment phase may be performed in 384-well tissue culture plates (data not shown), thereby further reducing the footprint of a given experiment.
Further adjustments to this method will be required for utilization with human embryonic or induced pluripotent stem (iPS) cells. These cells are less amenable to embryoid body formation, but a number of methods have been shown to enhance this process.29,30 We have begun testing human iPS cells in our microplate format and have achieved the formation of a single EB per well (data not shown). However, these EBs have been inconsistent in their eventual formation of cardiomyocytes, and the lack of consistency inhibits our ability to perform screens to enhance cardiomyocyte differentiation. Work is ongoing to develop a consistent baseline of cardiomyocyte formation with these cells.
Our data suggest that this automated 384-well microplate method is a significant improvement over current methods. Automation significantly lowers the effort required for current microplate methods, and the use of 384-well plates further reduces the space and cost per experiment. In addition, our method provides superior consistency in EB formation and cardiomyocyte differentiation as well as a format more amenable to screening when compared with hanging drops or static suspension methods. With additional enhancements, the automated microplate method of embryonic stem cell differentiation may help move embryonic stem cell–derived lineages further into drug screening.
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
*
Beckman Coulter, the stylized logo, Biomek, BioRAPTR, FRD, and Vi-Cell are trademarks of Beckman Coulter, Inc. and are registered in the USPTO. Biomek and BioRAPTR workstations and AAO for BioRAPTR software are for laboratory use only and not for use in diagnostic procedures. Gallios flow cytometer and Kaluza software are for research use only and not for use in diagnostic procedures.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.