
Abstract
The TWEAK-Fn14 pathway is upregulated in models of inflammation, autoimmune diseases, and cancer. Both TWEAK and Fn14 show increased expression also in the CNS in response to different stimuli, particularly astrocytes, microglia, and neurons, leading to activation of NF-κB and release of proinflammatory cytokines. Although neutralizing antibodies against these proteins have been shown to have therapeutic efficacy in animal models of inflammation, no small-molecule therapeutics are yet available. Here, we describe the development of a novel homogeneous time-resolved fluorescence (HTRF)–based screening assay together with several counterassays for the identification of small-molecule inhibitors of this protein-protein interaction. Recombinant HIS-TWEAK and Fn14-Fc proteins as well as FLAG-TWEAK and Fn14-FLAG proteins and an anti-Fn14 antibody were used to establish and validate these assays and to screen a library of 60 000 compounds. Two HTRF counterassays with unrelated proteins in the same assay format, an antiaggregation assay and a redox assay, were applied to filter out potential false-positive compounds. The novel assay and associated screening cascade should be useful for the discovery of small-molecule inhibitors of the TWEAK-Fn14 protein interaction.
Introduction
The cytokine TWEAK (tumor necrosis factor–like weak inducer of apoptosis) is a member of the tumor necrosis factor (TNF) superfamily and was first described in 1997. 1 Ligands belonging to this family are type II transmembrane proteins with a small N-terminal cytoplasmic region and a large extracellular part that is cleaved off by furin proteases. TWEAK forms homotrimers in solution 2 and mediates its biological activities mainly by binding to the Fn14 (fibroblast growth factor inducible 14) receptor,3,4 although full-length, membrane-anchored TWEAK is also capable of signaling through Fn14. 5 Fn14 itself is a type I membrane protein with a cysteine-rich extracellular domain required for TWEAK binding while a small cytoplasmic tail contains one TNFR-associated factor (TRAF) binding domain. 6
Binding of TWEAK trimers induces trimerization also of the Fn14 receptor, followed by activation of the proinflammatory NF-κB pathway. 6 In addition, the TWEAK-Fn14 signaling axis regulates proinflammatory TNFα-mediated responses, 7 as well as proangiogenic processes, 8 and therefore seems to be of general importance for wound healing and tissue repair. TWEAK can also stimulate apoptosis in certain cancer cells by inducing TNFα secretion and assembly of a Death-signaling complex containing caspase-8. 9 Continuous Fn14 signaling in chronic tissue injury and inflammation might lead to pathological effects, and involvement of TWEAK-Fn14 in several inflammatory diseases such as rheumatoid arthritis, systemic lupus erythematosus (SLE), and multiple sclerosis has been reported.10,11 Increased expression of TWEAK and Fn14 is found in models of ischemic stroke, 12 whereas Fn14 signaling can promote also growth, survival, and angiogenesis of tumors. 13 Details on the biology of TWEAK and Fn14 and their role in disease are described elsewhere. 14
Nearly all therapeutic approaches of targeting members of the TNF or TNF receptor superfamilies involve neutralizing antibodies against the ligands or receptors, or soluble receptor proteins for competition with the endogenous receptors. Five anti-TNF antibody-based drugs are already on the market, and therapeutic proteins for at least 16 of the other 22 established ligand-receptor pairs are in clinical development. 15 The humanized anti-TWEAK monoclonal antibody RO5458640 (Roche, Basel, Switzerland) is being tested in a phase I clinical trial in patients with advanced solid tumors, whereas a similar antibody from Biogen Idec (Weston, MA) is undergoing a phase I trial for lupus nephritis. In contrast, PDL 192 (Facet Biotech, Redwood City, CA), a humanized monoclonal antibody directed against Fn14, is currently being evaluated in a phase I trial in patients with advanced solid tumors. A soluble Fn14-Fc fusion protein was shown to inhibit TWEAK mitogenic activity on endothelial cells 16 and other cell types. Only very few examples of small-molecule inhibitors for TNF family cytokine-receptor interactions exist, despite the known disadvantages of protein therapeutics often related to drug delivery and cost. For instance, anti-inflammatory compounds are mainly limited to inhibitors of downstream NF-κB, AP1, and JNK/p38 pathways 17 or other less characterized intracellular targets. One reason for the paucity of small organic drugs might be the perceived difficulty of targeting protein-protein interactions with small molecules. 18 Nevertheless, some progress has been made in recent years in disrupting binding of TNF family members to their receptors with compounds, as exemplified by molecules that bind to and destabilize TNFα and CD40L trimers, respectively.19,20
Small-molecule design would benefit from structural studies of the Fn14-TWEAK system. So far, only solution nuclear magnetic resonance (NMR) structures of human apo-Fn14 protein structures are available (PDB codes: 2RPJ, 2EPQ, 2KMZ). No experimental structure of the complex with TWEAK has been determined; therefore, at this time, it is not possible to fully elucidate the binding mode of the two partners. Interestingly, it has been shown that mutations of three charged residues in the Fn14 CRD domain—namely, Asp45, Lys48, and Asp62 and, to a minor extent, Met50—had a deleterious effect on TWEAK binding. This suggests that the residues may define a specific area that is important for complex formation or stability, 21 but we do not know if these amino acids are directly involved in binding to TWEAK or if their mutation triggered structural changes that compromised TWEAK binding. Targeting this area with small molecules may represent a potential strategy to interfere with the FN14-TWEAK complex and therefore for pharmaceutical intervention.
Earlier work on the development of high-throughput screens for this target class used different assay formats, ranging from radioligand binding 22 and DNA-encoded chemical libraries 23 for identification of TNFα inhibitors, homogeneous time-resolved fluorescence (HTRF) energy transfer for inhibitors of the herpes virus HVEM–HVEM-L interaction 24 to AlphaScreen technology for OX40-OX40L interaction inhibitors. 25 Here we report the development of an HTRF assay in a 384-well format for the identification of TWEAK-Fn14 inhibitors and describe the implementation of this assay for screening a library of 60 000 small molecules. In addition, the application and usefulness of several counterassays to filter out false-positive compounds will be discussed.
Materials and Methods
Reagents
Anhydrous DMSO for preparing compound stock solutions, methyl sulfoxide-d6 (99.9%) as solvent for NMR, and Acetonitrile Chromasolv (>99.9%) as ultra-performance liquid chromatography/mass spectrometry (UPLC/MS) solvent were all obtained from Sigma-Aldrich (Milan, Italy). For the HTRF assay, NaH2PO4, NaF, and bovine serum albumin (BSA) were also obtained from Sigma-Aldrich, whereas Na2HPO4 was purchased from Merck (Milan, Italy). The extracellular domains of human TWEAK coupled to a 6xHIS tag (HIS-TWEAK; cat. 1090-TW) and human Fn14 conjugated to the human Fc domain (Fn14-Fc; cat. 1199-TW), as well as an anti–human Fn14 antibody (anti-hFn14; cat. MAB1199), were all purchased from R&D Systems (Minneapolis, MN); the untagged extracellular domains of human TWEAK (154 amino acid residues; cat. 310-06) and human Fn14 (53 amino acid residues; cat. 310-21) were purchased from Peprotech (London, UK). Eu3+ cryptate, conjugated to anti–human Fc (anti-hFc-K; cat. 61HFCKLA); XL665 conjugated to anti–6XHis antibody (anti-6His-XL665; cat. 61HISXLA); and the counterassay kits for the 6HIS-tag (cat. 62HISPEB) and the human MAB screening (cat. 62HFCPEB) were all purchased from CisBio (Codolet, France). The anti-FLAG M2 antibody (cat. F3165) and the M2 affinity gel (cat. A2220) were obtained from Sigma-Aldrich.
Cloning, Expression, and Purification of Human FLAG-Fn14 and FLAG-TWEAK Extracellular Domains
We used commercial preparations of recombinant Fc-tagged Fn14 (Fn14-Fc) and 6His-tagged TWEAK (HIS-TWEAK) proteins for the development of the primary assay. Untagged TWEAK protein was also purchased and used as a positive control for competing with HIS-TWEAK, which is attached to a fluorophore via an anti-6HIS antibody in the assay. In anticipation of biophysical binding studies with small-molecule inhibitors, we decided to produce the extracellular domains of both TWEAK and Fn14 in our laboratories. The proteins were fused only to small FLAG affinity tags instead of much larger Fc tags and also contained secretion sequences for the expression of the targets into the medium. Schematic representations of the fusion proteins are reported in Figure 1A .
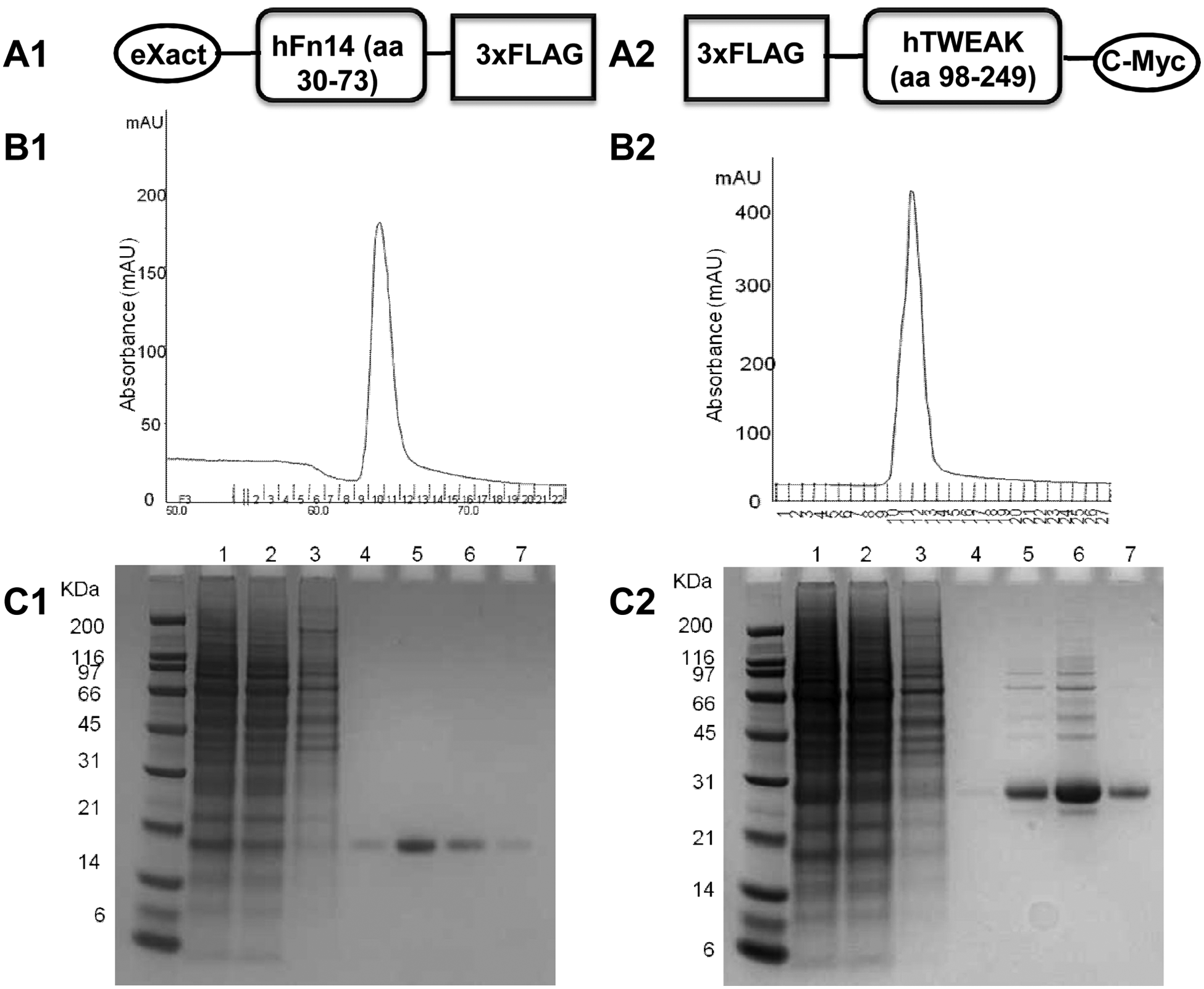
Affinity purification of recombinant Fn14-FLAG and FLAG-TWEAK. (
The human 1000–base pair Fn14 cDNA clone from OriGene (Rockville, MD) was used as the template for PCR amplification using forward primer 5′-GAAGATCTGCG CCAGGCACCGCCCCC-3′ and reverse primer 5′-CGGG ATCCGGCAGGAGGTGCTGCAGC-3′, which contained a BglII or BamHI site, respectively, to facilitate subsequent cloning steps. The PCR product was ligated into a modified p3xFLAG-CMV-13 (Sigma-Aldrich) containing an eXact tag (Bio-Rad, Milan, Italy) and sequenced fully on both strands to ensure an error-free clone. The human TWEAK DNA sequence was amplified by nested PCR using a pool of human cDNAs from kidney, lung, and spleen, according to the TWEAK expression pattern. 26 PCR amplification with external primers (forward primer: 5′-TCCCTCGGG TCCCGGGATGG-3′; reverse primer: 5′-TGCCCAGAGA GCTGTCGAGG-3′) was followed by a second PCR step using internal primers (forward primer: 5′-CCCAAGCTTG GCCGGAAAACACGGGCTCG-3′; reverse primer: 5′-GG GATATCAGGTGAACCTGGAAGAGTCCG-3′) to obtain the sequence of the extracellular domain, which was inserted into a p3XFLAG-Myc-CMV-23 plasmid (Sigma-Aldrich).
The recombinant extracellular domains of human Fn14 (Fn14-FLAG, Fn14 amino acids 30–73) and TWEAK (FLAG-TWEAK, TWEAK amino acids 98–249) were expressed using the HEK293 Freestyle system from Invitrogen (Carlsbad, CA). The Fn14-eXact-3xFLAG or TWEAK-3xFLAG-cMyc expression plasmids were transiently transfected into HEK293 cells, which were adapted for suspension growth (FreeStyle 293; Invitrogen) and maintained in FreeStyle HEK293 expression medium (Invitrogen) supplemented with penicillin 100 ug/mL and streptomycin 100 µg/ml. Cells were diluted to a density of 1 × 106 cell/mL, whereas plasmid DNA and Freestyle MAX reagent (Invitrogen) were diluted in OPTI-Pro SFM medium (Invitrogen) separately and incubated in a 1:1 ratio for 15 min before adding the DNA-lipid mixture to the cell suspension. Expression of the two proteins was allowed for 48 h and monitored by analyzing 1 mL of conditioned medium by Nu-PAGE and Western blots, using an anti-Flag antibody. Then the conditioned medium was collected by centrifugation (1200 rpm, 5 min), and the cells were diluted to 1 × 106 cells/mL in fresh 293 expression medium. Recombinant protein production was allowed to continue for another 72 h.
Fn14-FLAG and FLAG-TWEAK were purified from HEK293 conditioned media by affinity chromatography using the anti-FLAG M2 gel settled in a glass column (100 × 15 mm). After washing with loading buffer (20 mM Tris [pH 7.0], 150 mM NaCl), recombinant proteins were eluted with a gradient from 0% to 100% 0.1 M glycine HCl (pH 3.5). Each eluted fraction was analyzed by Nu-PAGE on a 4% to 12% bis-tris acrylamide gel (Invitrogen), followed by Coomassie staining and Western blot analysis with anti-FLAG antibody. Purified proteins were analyzed by capillary electrophoresis to determine their concentration and purity with the Experion Pro260 analysis kit (Bio-Rad).
The 19-KDa and 28-KDa protein bands correspond to the recombinant Fn14-FLAG and FLAG-TWEAK proteins, respectively, and confirm that maximal expression of the two proteins was already achieved after 48 h while protein levels were still high after 96 h (data not shown). Fn14-FLAG and FLAG-TWEAK proteins were then produced in larger quantities under these conditions and purified by affinity chromatography ( Fig. 1B ). Nu-PAGE analysis followed by Coomassie blue gel staining confirmed that both recombinant proteins showed more than 90% purity after the single purification step ( Fig. 1C ). The average yield of the expression and purification protocols was about 0.6 mg/L for Fn14-FLAG and 1.25 mg/L for FLAG-TWEAK, as calculated by comparison with a BSA standard using a capillary electrophoresis system (data not shown).
Selection of Compounds for High-Throughput Screening
A selection approach was applied aimed at sampling the available “drug-like” chemical space in both the Siena Biotech and iNovacia compound collections of around 70 000 and 270 000 unique compounds, respectively. Moreover, to have a better chance of identifying high-quality hits for further drug development, molecular properties were also kept under control. A complex chemoinformatics protocol was built within Pipeline Pilot (version 8.0.1.) from Accelrys (San Diego, CA) to perform the compound selection. A “drug-likeness” filter was used to remove molecules with unsuitable calculated physicochemical properties as defined by the widely applied Lipinski’s rule of five 27 and Veber’s rule. 28 Moreover, to avoid complex stereoisomer mixtures, numbers of chiral centers for the molecules was limited to three. Molecules with experimentally determined aqueous solubility <20 µM were removed, whereas for molecules without experimental data, an in-house generated insolubility model 29 was applied to filtering out compounds predicted to have aqueous solubility <40 µM. Molecules with groups expected to make them highly reactive, interferent, or promiscuous 30 were also discarded. Finally, molecules bearing more than four aromatic rings were removed as lipophilic and essentially flat compounds are often associated with low aqueous solubility and poor development potential. 31 After applying these filters, about 30 000 compounds from the Siena Biotech collection were selected for the screen. Most of these compounds were also commercially available.
Then, structurally diverse compounds from those already present in the Siena Biotech subset were selected from the wider iNovacia collection. Molecules with a Tanimoto similarity >0.5 with respect to Siena Biotech compounds were discarded. Then, a genetic algorithm based on multiobjective Pareto optimization was used, resulting in multiple solutions with best possible trade-offs among the optimized properties. Optimization was carried out to maximize structural diversities, measured using ECFP6 fingerprint descriptors, both within the selected iNovacia subset as well as compared with the Siena Biotech collection. The Pareto method provided about 5000 compounds, which were in turn used as input for the last step of the selection, aimed at choosing not only singletons but small analogue series consisting of five to six analogues for each selected compound, leading to a total of about 30 000 compounds. This set constituted the iNovacia selection.
Peptides were designed as follows: ADLDKSMDS ASSRARPHSDF, corresponding to the Fn14 amino acid sequence, which includes all the four key residues involved in binding to TWEAK 21 ; ADLDKSMG and ARPHSDF, which are fractions of the previous peptide; and the cyclic peptide cyclo(DLDKSMGGDG), which was designed to include the four key residues and locate them in a similar 3D conformation as depicted in the NMR solution structure for the Fn14 protein available at the time (PDB code: 2RPJ). 32 The peptides were synthesized by CASLO (Lyngby, Denmark) and added to the screening library. Then, 10 µL of a 10-mM DMSO solution of each compound was added to 1 of 352 wells of a 384-well plate, leaving the last two columns empty for controls.
Assay Protocols for the TWEAK-Fn14 Interaction Assay
Two different assay formats for the HTRF-based TWEAK-Fn14 interaction assay were developed due to the use of two dissimilar liquid-handling robots at the two companies, for high-throughput screening (HTS) and confirmation assays. Three hundred and eighty-four-well low volume microplates from Corning-Costar (Corning, NY; cat. 3676) with nonbinding surface were used in both cases. For the HTS primary screen, 5-mM DMSO compound solutions were added as either 20 nL per well for the primary screen or 400 nL per well with serial DMSO dilutions for concentration response curve (CRC) assays, using an ATS-100AV acoustic dispenser from EDC Biosystems (Fremont, CA). Alternatively, CRC analysis was performed by adding 5 µL to each well of compound solutions prediluted in assay buffer (10 mM sodium phosphate [pH 7.5] with 1 mg/mL BSA) using a Freedom Evo liquid-handling robot from Tecan (Männedorf, Switzerland).
In each well, the following protein and antibody solutions were sequentially added: 5 µL of a 2-nM solution of HIS-TWEAK protein (final concentration 0.5 nM) and 5 µL of a 1-nM Fn14-Fc solution (final concentration 0.25 nM), both prediluted in assay buffer, and 10 µL of the HTRF detection reagents (anti-hFc-K and anti-6His-XL665), reconstituted according to the CisBio protocol and premixed 1:400 in HTRF buffer (10 mM sodium phosphate [pH 7.5] plus 100 mM NaF with 1 mg/mL BSA). After 3 h of incubation at room temperature (RT), HTRF signals were measured using either a Victor 2V plate reader from PerkinElmer (Waltham, MA) or a GeniusPro plate reader from Tecan, using 340 nm as excitation wavelength, a 620-nm filter for the Europium donor fluorescence, and a 665-nm filter for the XL665 acceptor fluorescence detection. HTRF signals were calculated as the HTRF ratio (ratio of fluorescence measured at 665 nm and 620 nm) × 104 (thereby using the signal at 620 nm as an internal standard). Data were normalized to the mean HTRF ratio value of negative control (only DMSO added = 100% or C100%) and positive control (1 µM untagged human TWEAK = 0% or C0%). Compound activity was calculated as the percentage of residual HTRF ratio using the following equation: [(HTRF ratioCmpd – C0%)]/[(C100% – C0%)] × 100. A hit threshold of 80% was used for the primary screen (i.e., compounds with a residual activity below 80% were considered primary actives). The Z′ factor was calculated using the following equation: Z′ = 1 – 3 × [(SD100%) + (SD0%)]/abs[C100% − C0%], where SD100% and SD0% are the standard deviation values of negative and positive control, respectively. 33 Concentration response curves were fitted with the four-parameter logistic nonlinear regression model using the IDBS XLfit software.
HTRF Counterassays
The HTRF-based 6-HIS tag counterassay was performed according to a protocol from CisBio. The following reagents were added sequentially into a 384-well low-volume plate from Corning-Costar (cat. 3824): 10 µL control or serially diluted compound solution, 5 µL anti-6HIS-cryptate, and 5 µL 6HIS peptide-XL665 from the assay kit. The plates were incubated at 2–8 °C for 2 h before reading the HTRF signal. The HTRF-based human MAB counterassay was also performed according to CisBio instructions. Applying the same 384-well low-volume plate, the following reagents were sequentially added into the well: 10 µL control or serially diluted compound solution, 5 µL human IgG-XL665, and 5 µL anti–human Fc-cryptate from the assay kit. Reactions were incubated for 18 h at RT before reading the HTRF signals.
Counterassay for Aggregate Formation with Dynamic Light Scattering
The 10-mM stock solutions of compounds in DMSO were sonicated for 10 min and appeared clear when inspected directly after sonication. Aliquots were then further diluted in DMSO to yield 0.5-mM and 2.5-mM compound solutions in DMSO. Special care was taken to avoid exposure to water vapor when working with the DMSO stock solutions. The 10-µM and 50-µM solutions of the compounds in 10 mM sodium phosphate (pH 7.5), 100 mM sodium fluoride (without BSA) were prepared by dilution of the 0.5-mM and 2.5-mM DMSO stock solutions, respectively. The DMSO content of the aqueous solutions used in the measurements was 2%. The DMSO stock solution and the two buffer solutions were monitored for aggregate formation. Dynamic light-scattering (DLS) experiments were carried out on a DynaPro MS800 instrument from Wyatt Corp. (Santa Barbara, CA) equipped with a 248-channel Multi-Tau USB correlator and a temperature control unit. The control of the instrument, data collection, and evaluation were carried out by DYNAMICS software (Dynamics Software, Veenendaal, the Netherlands). Solutions of the compounds were loaded into 12-µL quartz cuvettes, and the measurements were carried out at 25 °C. The data acquisition time was set to 10 s. The result of the measurement represented the average of 10 acquisitions. All compound solutions were monitored for at least 20 min.
Counterassay for Redox-Active Compounds
The assay was used in a slightly modified form as described. 34 To increase sensitivity, a concentration of 1 mM dithiothreitol (DTT) was applied in the assay instead of 50 µM. A solution of DTT and 5 µM resazurin in 50 mM NaCl, 50 mM HEPES (pH 7.5) was mixed with increasing concentrations of the compound, incubated for 60 min at RT, and analyzed on a Victor 2V plate reader.
Analysis of Purity, Identity, Solubility, and Chemical Stability of Hit Compounds
Compound solutions of 10 mM in DMSO were diluted to a final compound concentration of 100 µM in phosphate-buffered saline (PBS; 1% v/v DMSO, 10 mM Na2HPO4, 140 mM NaCl, 2.7 mM KCl, 1.8 mM KH2PO4, pH 7.4). Two samples were prepared: one for immediate analysis and a second sample stored at 37 °C for 24 h. Analysis was performed on a reversed-phase high-performance liquid chromatography (HPLC) system with UV and electrospray ionization–mass spectrometry (ESI-MS) detection. Purity was defined as the relative area of the sample peak at 220 nm, whereas the molecular identity of the compound was determined by the presence of a molecular ion in the MS of the sample chromatogram. Chemical stability was defined as the ratio of the relative area of the sample peak after 24 h to that of the 0-h sample in the chromatogram. For solubility measurements, samples were diluted to a compound concentration of 250 µM in 50 mM phosphate buffer (pH 7.0) with a residual DMSO concentration of 2.5% v/v. Subsequently, the samples were agitated for 1 h and filtered. Two standards of 25 µM and 250 µM were prepared in a 50:50 mix of acetonitrile and water. Analysis was performed on a reversed-phase HPLC with UV detection. Solubility was determined as the ratio of sample peaks to the standard curve.
Results
Assay Principle
We have developed a novel sensitive in vitro protein-protein interaction assay using HTRF technology for the identification of small molecules capable of interfering with the binding between TWEAK and Fn14. The assay is based on two antibodies that are coupled to the HTRF reagents europium cryptate (for fluorescence emission) and XL665 (as acceptor molecule) and bind to HIS-TWEAK and Fn14-Fc by means of their 6His and Fc tag, respectively (
Optimization of Assay Reagents, Kd Determination, and DMSO Tolerance
Initial time course experiments were conducted with varying concentrations of HIS-TWEAK (1–10 nM) and Fn14-Fc (0.1–0.5 nM) to establish the time necessary for the reaction to reach equilibrium. Controls, in which one of the components was excluded, were performed to ensure that all components were indeed required for the time-dependent specific increase of the HTRF signal and that the background signal was stable over time (data not shown). With 0.25 nM of Fn14-Fc and 1 nM HIS-TWEAK, a threefold increase in signal over background could be observed. Using 3 nM HIS-TWEAK instead of 1 nM, a fivefold signal increase was seen ( Fig. 2A ). In both cases, the estimated time to reach equilibrium was about 3 h at room temperature, whereas the signal itself was stable for longer periods.
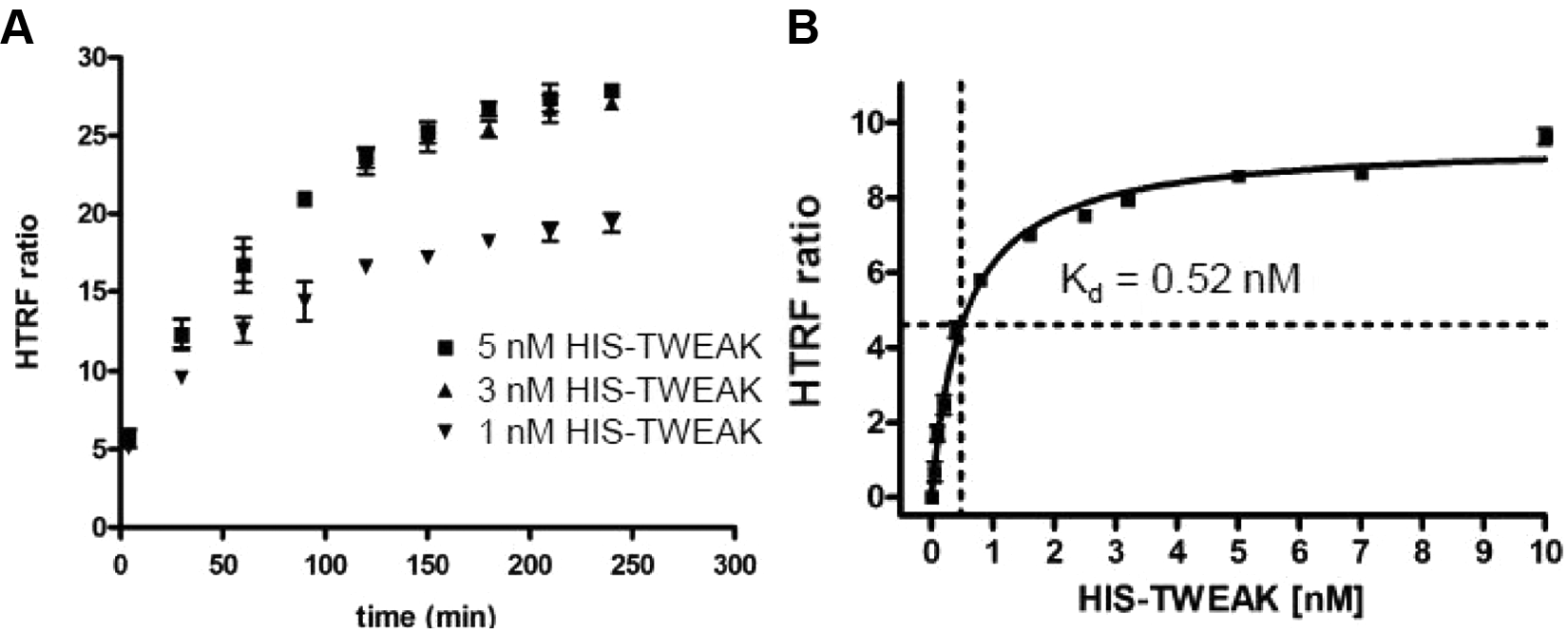
(
Although CisBio protocols recommend using HTRF detection reagents at a dilution of 1:400, we found that it was possible to use 1:800 dilution with no loss in signal-to-background (S/B) values (data not shown). Thus, to minimize reagent consumption, 1:800 dilutions were used in subsequent experiments. The DMSO tolerance of the assay was investigated by dispensing 0 to 800 nL DMSO into wells of an assay plate with a final assay volume of 20 µL per well. In total, 1 µM of untagged TWEAK extracellular domain served as control for total inhibition of the binding, whereas assay buffer was used as negative control. No effects on the signal up to 4% final DMSO concentration were observed (data not shown). Performing a single-point screen at a compound concentration of 10 µM in 0.1% DMSO and concentration response curves with 200 µM as the highest compound concentration in 2% DMSO were therefore possible without effects on the overall signal.
To determine optimal reagent concentrations for a sensitive assay, the dissociation constant (Kd) was measured by incubating increasing concentrations of HIS-TWEAK ligand (from 0–10 nM) in the presence of 0.25 nM Fn14-Fc for 3 h at room temperature. A Kd of 0.5 nM was estimated for HIS-TWEAK ( Fig. 2B ). Our aim was to screen at or below the Kd to achieve optimal sensitivity for detection of even weak small-molecule inhibitors of this protein-protein interaction. At the same time, a minimal S/B window between 2 and 3 was estimated to be necessary to ensure acceptable assay quality of the HTS, requiring protein concentrations in the low nanomolar range. Hence, final assay concentrations were set to 0.5 nM HIS-TWEAK and 0.25 nM Fn14-Fc with an incubation time of 3 h at room temperature for reaching equilibrium.
Investigation of Reference Compounds for the HTS
Several compounds were tested in CRCs in the assay to identify a suitable positive control for the HTS. The compound SPD304 has been reported to bind to TWEAK-related TNFα trimers, stabilizing an intermediate dimer that is no longer able to interact effectively with the TNFα receptor, with an IC50 value of 22 µM. 19 The compound was tested for similar effects in the TWEAK-Fn14 interaction assay, but no significant inhibition up to 200 µM was observed (data not shown). The cyclic peptide CWDDGWSFC has been shown to bind specifically to TWEAK, interfering with its binding to CD163, 35 but no inhibition of binding to Fn14 up to 200 µM peptide concentration was observed (data not shown). The same applied to the three linear peptides ADLDKSMDSASSRARPHSDF, ADLDKSMG, and ARPHSDF. The compound suramin is known to bind to and antagonize several proteins, among which are also the two trimeric cytokine ligands OX4024 and TNFα 36 and their respective receptors. However, suramin was not compatible with the assay format due to interference with the fluorescent signal and therefore could not be tested (data not shown).
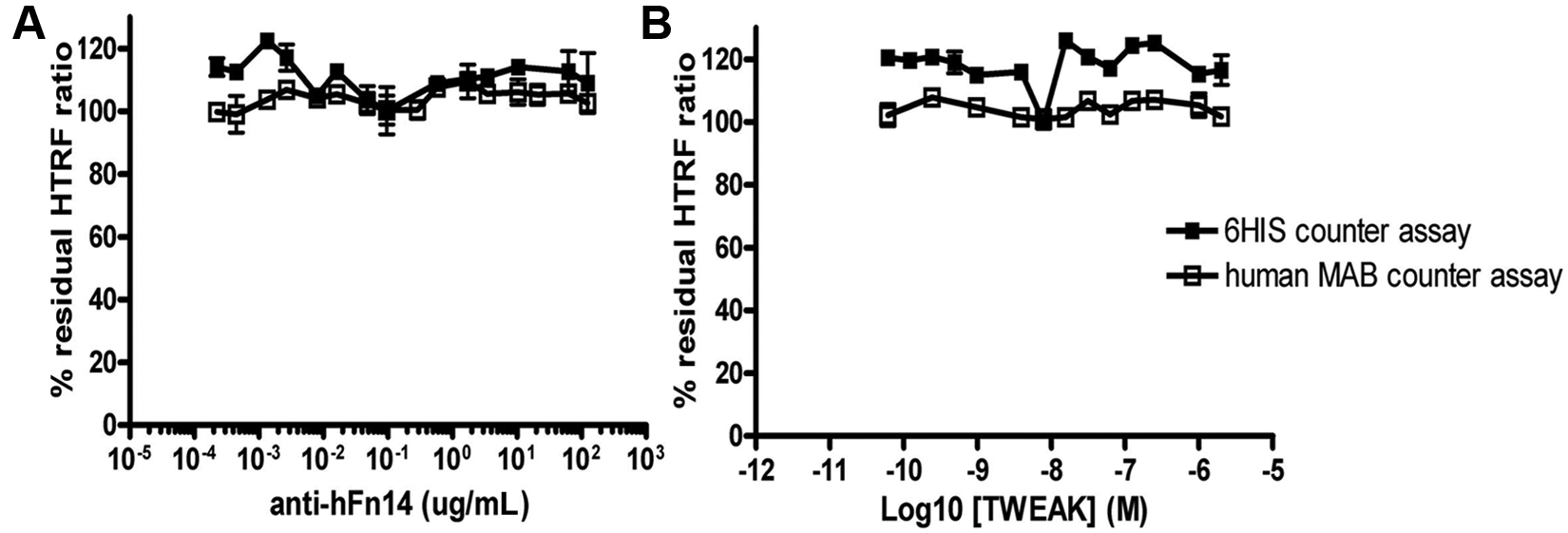
We then investigated unlabeled human TWEAK as well as a neutralizing anti–human Fn14 antibody and found that both proteins competed with the tagged HIS-TWEAK and Fn14-Fc proteins. Whereas the commercial untagged TWEAK preparation gave a biphasic inhibition curve ( Fig. 3A ), FLAG-TWEAK protein, which was later produced and purified in our laboratories, did not show such behavior; a monophasic inhibition curve with an IC50 value of 210 nM ( Fig. 3B ) was obtained. The in-house produced Fn14-FLAG protein ( Fig. 3C ) and an anti-Fn14 antibody ( Fig. 3D ) were also able to interfere in the binding reaction with the HIS-TWEAK and Fn14-Fc proteins. As expected, both the untagged commercial TWEAK, the FLAG-tagged proteins produced in-house, and the anti-Fn14 antibody were inactive when tested in the HTRF counterassays ( Fig. 4 ; data not shown for Fn14-FLAG). Because of the biphasic inhibition behavior of the commercial untagged TWEAK preparation, we decided to use both the untagged TWEAK at 1 µM and the anti-Fn14 antibody at 100 µg/mL as controls in the HTS. Untagged TWEAK was used to define the 0% of the residual (100% inhibition of binding) HTRF signal in the HTS, whereas the antibody served as an additional quality control (QC) during the screen.
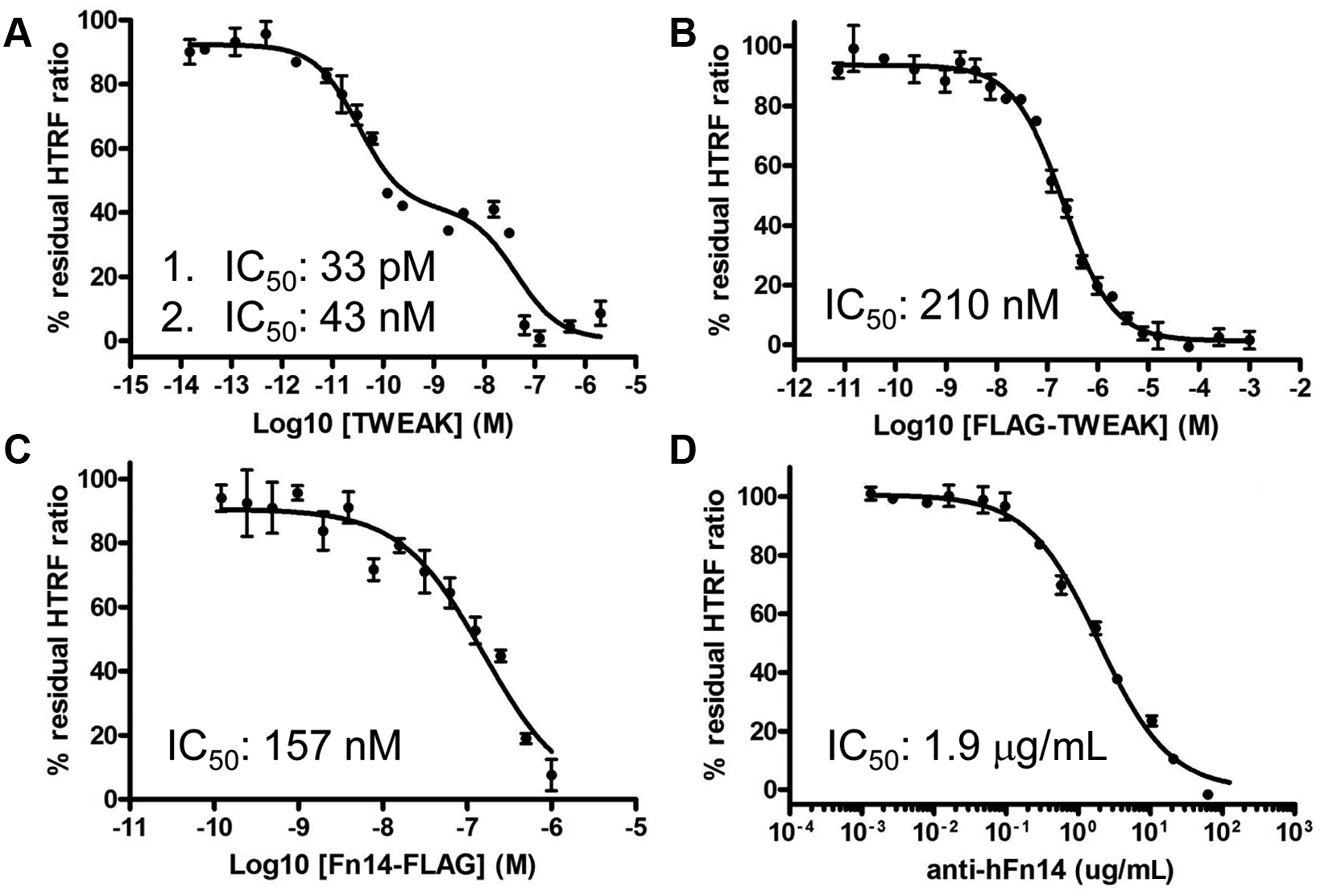
Concentration response analysis of reference compounds tested in the HIS-TWEAK–Fn14-Fc interaction assay. Although a commercial untagged TWEAK preparation gave a biphasic inhibition curve with IC50 values of 0.03 and 43 nM (
Anti-hFn14 antibody (
Assay Validation and HTS
First, a prescreen was performed with the aim of evaluating assay performance and reproducibility. A total of five plates were tested under the conditions of the forthcoming HTS. The results from the prescreen were satisfactory and resulted in a Z′ value above 0.5 for all plates and an average value of 0.7 (
Our approach for finding novel inhibitors of the TWEAK-Fn14 protein interaction was to screen a diverse library of approximately 60 000 small molecules. The 60 000 compounds were screened at a single concentration of 10 µM in the HTS. The average Z′ value of the screen was 0.66. Based on the overall activity distribution of all screened compounds, a hit threshold in terms of residual HTRF ratio was set to 80%. A total of 1033 compounds (1.7% hit rate) showed a residual HTRF ratio of ≤80%. However, inspection of the donor and acceptor signals showed that for the vast majority of these compounds, the decrease of the HTRF ratio was due to an increase in donor signal rather than an expected decrease in acceptor signal. To filter out those compounds that affect both fluorescence excitation and emission and thus are not compatible with the assay format, all 1033 compounds were rerun and only compounds showing both a residual HTRF ratio of ≤80% and a decrease in the HTRF acceptor signal of at least 15% without any significant change in the donor signal were considered primary hits. Fifteen primary hit compounds (0.025% hit rate) with a residual HTRF ratio ≤80% and a residual acceptor signal at 665 nm ≤85% were identified by this procedure.
Physicochemical Properties of Hits
The 15 primary hits of commercial origin were analyzed in terms of molecular identity, purity, chemical stability, and aqueous solubility. Three compounds did not give any UV trace, and therefore purity and/or identity could not be established. One compound failed our purity criterion of ≥80%. The remaining 11 compounds were all chemically stable and of sufficient solubility and purity for further analysis (data not shown). However, because of the low absolute number of hits, all 15 compounds were progressed to concentration response analysis.
Hit Confirmation in Concentration Response and Counterassays
Hit confirmation was performed by testing the ability of the 15 primary hits to inhibit HIS-TWEAK–Fn14-Fc binding in a concentration-dependent manner. All compounds were also tested in the two HTRF counterassays to remove false-positive compounds interfering with the HTRF assay reagents rather than with the HIS-TWEAK–Fn14-Fc protein-protein interaction. Only 5 of the 15 tested compounds showed clear CRCs without showing any significant effects in either of the two counterassays ( Table 1 ). All four peptides were found to be inactive in the HIS-TWEAK–Fn14-Fc interaction assay (data not shown). Two of the five active compounds were potent but partial inhibitors (compounds A and E) with IC50 values of 2.2 and 0.9 µM, respectively. Two other compounds were fairly weak inhibitors (compounds C and D), whereas only compound B showed both full inhibition and, with an IC50 value of 1.8 µM, significant potency in the HIS-TWEAK–Fn14-Fc interaction assay.
Concentration Response Curves of the Five Confirmed Hit Compounds in the HIS-TWEAK–Fn14-Fc Interaction Assay (■), the 6His Counterassay (□), and the Human MAB Counterassay (△)
Two compounds showed potent but partial inhibition in the HIS-TWEAK–Fn14-Fc interaction assay, whereas two other compounds were only weak inhibitors. The IC50 value for compound D was not determined. Only compound B demonstrated both full and potent inhibition. None of the five compounds showed activity in the two homogeneous time-resolved fluorescence counterassays. HTRF, homogeneous time-resolved fluorescence; ND, not determined.
Aggregation Potential and Redox Activity of Hits
Other assays were applied to make sure that the hit compounds inhibited binding of HIS-TWEAK to Fn14-Fc in a specific and biologically relevant manner. First, DLS was performed with the aim of identifying aggregating and therefore potentially promiscuous inhibitors. 37 Although none of the compounds appeared to be completely soluble at 10 and 50 µM, no compound formed well-defined aggregates in solution that would have been characteristic of promiscuous inhibitors (data not shown). Small aggregates detected in the solutions were polydisperse and most probably can be attributed to limited solubility of the compound.
Next, all hit compounds were resynthesized and tested again in the HIS-TWEAK–Fn14-Fc interaction assay. We noticed that compound B completely lost activity in the HIS-TWEAK–Fn14-Fc interaction assay when the resynthesized batch was used ( Fig. 5A ) and decided to further investigate the reason for this behavior. We speculated that impurities in the commercial batch might have been responsible for the activity measured in the HTS and in the first concentration response assay. Therefore, an additional counterassay was applied that measured the oxidation potential of compounds in the presence of resazurin and DTT. 34 Four hit compounds were not active in the redox assay (data not shown), but we found that the original batch of compound B displayed a clear concentration-dependent activity, whereas the resynthesized batch was inactive ( Fig. 5B ). This strongly indicates that compound B is a false positive.
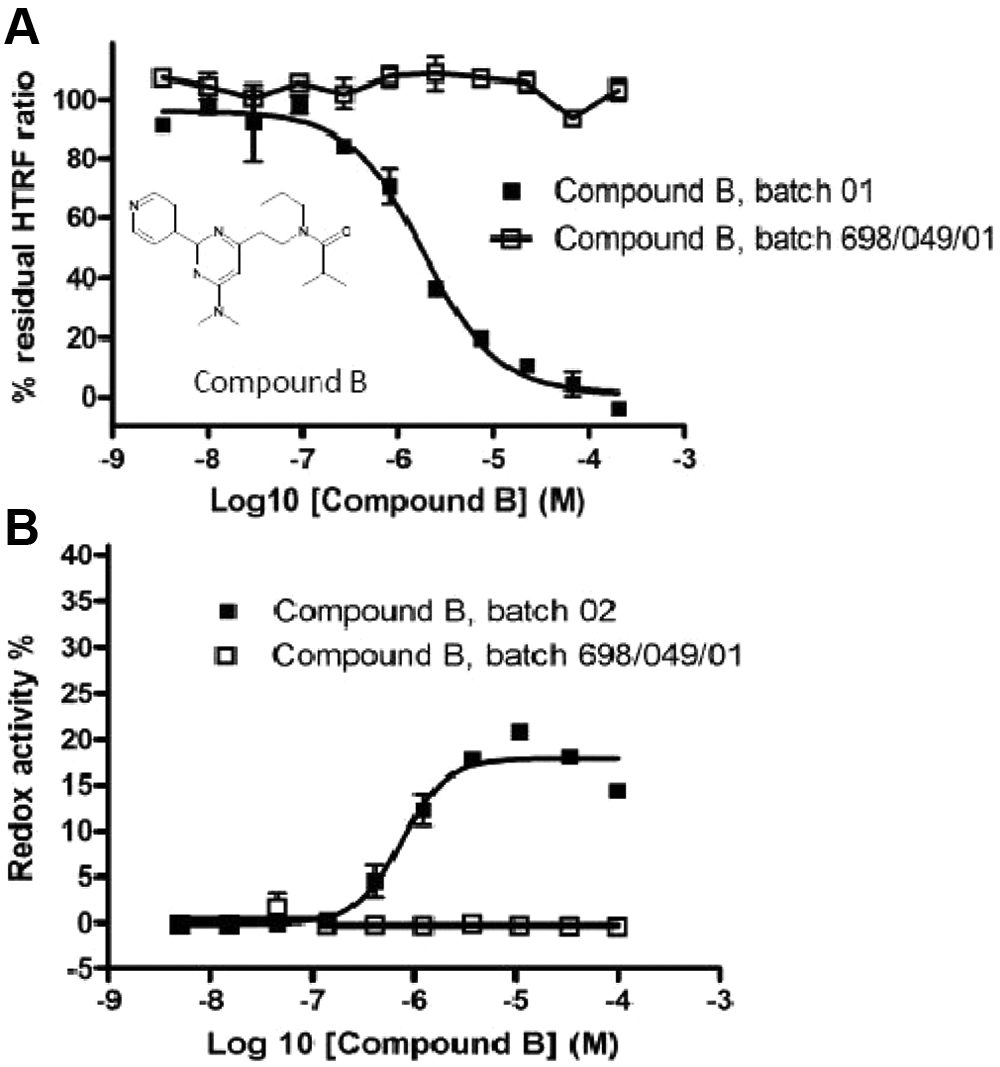
Concentration response analysis of a commercial source (batch 01 and batch 02) and a resynthesized source (batch 698/0497/01) of compound B in the HIS-TWEAK–Fn14-Fc interaction assay (
Discussion
In the past years, Fn14 from the TNF receptor superfamily and its ligand TWEAK have emerged as a signaling axis that is prominently featured in (patho)-physiological remodeling of tissues. TWEAK signals through upregulated Fn14 and seems to direct many different outcomes depending on the cellular context, including a long-lasting production of cytokines, chemokines, and matrix metalloproteinases by noncanonical NF-κB activation 38 as well as induction of apoptosis, the latter via cross-regulation of TNFα.7,39 TWEAK-Fn14 signaling can therefore promote chronic inflammation, pathological angiogenesis, and cell death, and targeting of this pathway may be a promising approach for many diseases with inflammatory components.
So far, only protein-based therapeutics are in development for directly inhibiting the interaction between TWEAK and Fn14, even though small organic molecules could offer advantages such as penetration of the blood-brain barrier for neuroinflammatory diseases. To this end, we have developed an HTS assay based on recombinant human HIS-TWEAK and Fn14-Fc and demonstrated that it can be used for screening large compound libraries. The assay uses the proteins at physiological concentrations,18 and their Kd of 0.5 nM in our assay is similar to affinities of 0.8 to 2.4 nM reported elsewhere.3,16,31 We deliberately chose to work with very low protein concentrations in order to increase the chances of finding small-molecule inhibitors. This is a difference compared with published HTS assays for other members of the TNF superfamily. For instance, whereas only 0.25 nM of Fn14-Fc receptor and 0.5 nM of HIS-TWEAK ligand were used in our HTS, a similar assay for inhibitors of the OX40-OX40L interaction based on AlphaScreen technology used 10 nM of receptor and 100 nM of ligand. 24
We also implemented a number of counterassays to ensure that only inhibitors specific for HIS-TWEAK or Fn14-Fc were selected for further development. The need for these filters became evident in the fact that only a third of all primary hits (5 of 15) displayed no activity in the two HTRF counterassays, indicating that some of the other compounds interfered either in the binding between the Fc part of the Fn14-Fc fusion protein and the anti-Fc antibody or in the recognition of the HIS tag by the anti-6HIS antibody. Although we did not identify aggregating compounds as a potential source of false positives in our list of primary hits, we found that a redox-active impurity was the cause of a concentration-dependent activity in the HTS assay for compound B because a resynthesized batch of the same compound was inactive in both the HIS-TWEAK–Fn14-Fc interaction assay and the redox counterassay. This redox-active impurity possibly acts by oxidizing cysteine residues in the Fn14-Fc domain, leading to a change or disruption of the 3D structure and a concomitant loss of its ability to bind HIS-TWEAK.
Further characterization of specific small-molecule inhibitors and confirmation of binding to either TWEAK or Fn14 will require biophysical interaction analysis by means of surface plasmon resonance (SPR), isothermal titration calorimetry, or NMR technology. To this end, we were successful in expressing and purifying both proteins, each of them containing only a small FLAG tag, which is unlikely to interfere with subsequent analysis. A comparison of our FLAG-TWEAK preparation with untagged protein from a commercial source underscores the importance of homogeneous and nonaggregated material for these studies. FLAG-TWEAK protein generated in our laboratories showed the expected monophasic inhibition when competing with labeled HIS-TWEAK in the HTS assay, whereas a commercial TWEAK preparation resulted in a biphasic curve. The reason for the behavior of the commercial protein is unknown, but it might possibly be caused by protein aggregates or precipitates of different sizes in that sample. Homogeneous and nonaggregating proteins at µM concentrations are also of utmost importance for successful crystallization for future structural studies.
We found only four primary hits that were not active in any of the four counterassays. These compounds were also either fairly weak or only partially active. The overall low hit rate of 0.007% indicates a general difficulty of finding potent small-molecule inhibitors of strong protein-protein interactions such as the TWEAK-Fn14 pair. Although this may be related to the chemical space we explored in terms of chemical scaffolds we had at our disposal, it could also suggest an opportunity to overcome common drug-likeness rules such as those that were applied to filter our screening set. This would gain access to a portion of the chemical space where bigger and/or more lipophilic compounds could be more efficient in covering sufficient protein surface area to disrupt protein-protein interactions. However, the development of such compounds into drugs might not be trivial due to low specificity or pharmacokinetics issues.
Another possibility would be the design of molecules that specifically target the protein-protein interaction area by applying computer-aided drug design techniques to experimental structures of the complex that are not available at the moment. Future crystal structures of the TWEAK-Fn14 complex and of the individual proteins in complex with small-molecule inhibitors of the protein-protein interaction will be extremely helpful in the rational design of high-affinity protein-protein interaction inhibitors. 32 Structure-based drug design, possibly combined with a rescreen of a significantly larger compound set to find additional chemical starting points, should lead to the development of novel anti-inflammatory or antiangiogenic compounds based on targeting the TWEAK-Fn14 pathway.
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work has been supported by a grant from the European Commission FP6 (ADIT, contract no. LSHB-CT-2005-511977).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.