
Abstract
TWEAK (tumor necrosis factor–related weak inducer of apoptosis), a member of the tumor necrosis factor superfamily, acts on cells by binding to its only receptor named Fn14 (fibroblast growth factor–inducible 14). Their engagement activates a number of intracellular signal transduction cascades and consequently leads to cell death, proliferation, migration, or survival depending on the cellular contexts. Studies have indicated that the expression of TWEAK and Fn14 is upregulated in many solid tumors compared with healthy tissues. The activation of TWEAK/Fn14 signaling enhances the proliferation, invasion, and migration of tumor cells. Moreover, the angiogenesis, pro-inflammatory cytokine expression, and epithelial–mesenchymal transitions are promoted upon TWEAK/Fn14 activation. Currently, the tumor necrosis factor receptor–associated factor and nuclear factor kappa B signaling pathways are considered two main downstream pathways activated by TWEAK/Fn14 interaction. In view of these facts, some TWEAK- or Fn14-targeting agents are generated to inhibit the progression of tumors and have achieved initial success in clinical and pre-clinical trials. These agents include monoclonal antibodies, fusion proteins, immunotoxins, and nanoparticles. In addition, some relevant signaling pathways are studied to identify new potential therapeutic targets. Overall, these findings suggest that the TWEAK/Fn14 pathway is critical in the development of tumors, and targeting this signaling is a potential therapeutic approach in future tumor therapy.
Introduction
Tumor necrosis factor (TNF)-related weak inducer of apoptosis (TWEAK), a member of the TNF superfamily, is expressed in various tissues and exerts biological effects including the modulation of cell death (apoptosis or necrosis), proliferation, differentiation and migration, the triggering of angiogenesis, and the induction of inflammatory cytokine expression.1,2 TWEAK functions via binding to fibroblast growth factor–inducible 14 (Fn14), a type I transmembrane protein, its unique receptor reported so far. 2 TWEAK is also the sole ligand binding to Fn14. TWEAK is primarily expressed as a type II transmembrane protein but it can be cleaved by furin to produce a soluble cytokine.1,3 Both membrane-anchored and soluble TWEAK can bind to Fn14.3,4 TWEAK is a glycoprotein with three parts, including the C-terminal extracellular region, transmembrane domain, and N-terminal intracellular region. 5 Fn14 contains the extracellular domain that binds to TWEAK as well as the cytoplasmic tail region essential for signal transduction. 6 The activation of TWEAK–Fn14 pathway is found in injured tissues and a number of other disorders including autoimmune diseases and cancers, triggering multiple downstream signaling pathways to remodel these tissues. 7
TWEAK and Fn14 show low expression in normal tissues and are highly expressed in many tumors and metastases. TWEAK overexpression is found in liver, colorectal, esophageal, bladder, pancreatic, ovarian, and prostate cancers as well as neuroblastoma.8–15 Besides the tumor types mentioned above,11–14,16–18 the increased expression of Fn14 has been also detected in brain glioma, breast cancer, lung cancer, and melanoma.19–28 In addition, Fn14 is highly expressed in the metastases of breast, colorectal, melanoma, non–small cell lung, and prostate cancers.14,23,28–30 Moreover, overexpression of Fn14 is associated with advanced stage of tumors and a worse clinical outcome.12,15–17,19,23,31
Numerous studies suggested that TWEAK/Fn14 signaling regulates several key events that are associated with tumor progression and metastasis, including immune surveillance and angiogenesis.31,32 Its pivotal roles in tumor cells have attracted considerable attention within recent years. Thus, an amount of TWEAK- or Fn14-targeting therapeutic agents is under pre-clinical development and some of them have entered early-phase clinical trials.33–36 In this review, we discuss the roles of TWEAK/Fn14 signaling in tumors and the development of TWEAK- or Fn14-based agents for cancer therapy.
The roles of TWEAK/Fn14 activation in tumors
Apoptosis and proliferation
TWEAK has been reported to promote the proliferation of normal endothelial cells, keratinocytes infected by human papillomavirus (HPV), and hepatocellular carcinoma cells.8,37,38 Proliferation of endothelial cells is an important step in the formation of blood vessels which is required for tumor growth. 37 HPV16-induced keratinocyte immortalization has been shown to be closely related to epidermis-originated malignancies, and TWEAK/Fn14 activation accompanies HPV16 infection inducing the proliferation of keratinocytes. 38 In addition, TWEAK promotes the proliferation of hepatocellular carcinoma cells in a dose-dependent manner by both autocrine and paracrine ways. 8 However, TWEAK also induces apoptosis of some tumor cell lines such as interferon-γ treated HT-29 (colon adenocarcinoma) and HSC3 (oral squamous cell carcinoma) cells, Kym-1 cells, and PC-3 prostate cancer cells.39–41 As for Kym-1 cells, cell death can be indirectly induced by secretion of endogenous TNF-α and was inhibited by cycloheximide. 40 Also, TWEAK/Fn14 activation promotes androgen-independent apoptosis of PC-3 prostate cancer cells in the presence of inflammatory cytokines (TNF-α/interferon-γ). 41 These results indicate that TWEAK has cytotoxic activity on some human tumor cells, but this function needs co-culturing with sensitizing agents in most cases, suggesting that TWEAK-induced cell death is cytokine dependent. Fn14 can be an anti-tumor target on two sides. Blockade of TWEAK/Fn14 signaling may inhibit tumor cell proliferation. However, Fn14 may serve as a tumor-associated target for sending antibodies, fusion proteins, or immunotoxins to tumor cells without considering its tumor-related functions.
Invasion and migration
Invasion and migration are the most essential characteristics of malignant tumors and are closely related to prognosis of tumors. Many studies have reported that the TWEAK/Fn14 pathway plays an important role in the invasion and migration of tumors (Figure 1). TWEAK/Fn14 signaling stimulates the invasion and migration of glioma cells through the TNF receptor–associated factor 2 (TRAF2), Rho guanosine triphosphatase (GTPase), Rac1, and guanine nucleotide exchange factor (GEF)–GTPase (involving two units: Ect2–Cdc42 and Trio–Rac1) signals.42–44 The Rho GTPase family members RhoA, Rac1, and Cdc42 are pivotal regulators of cell migration and are involved in the induction of lamellipodia and filopodia protrusion as well as the formation of stress fibers. 45 Binding of TWEAK to Fn14 results in the activation of Cdc42 by the Ect2 GEF and the subsequent activation of Rac1 by the Trio GEF, which promotes the migration and invasion of glioma cells. 44 In addition, the activation of the TWEAK/Fn14 pathway promotes invasion of glioblastoma cells. 46 Actually, TWEAK/Fn14 engagement in melanoma cells (B16) inhibits both cell growth and invasion but favors invasion of prostate cancer cells in vitro. 47 Therefore, TWEAK/Fn14 activation exhibits pro-cancer or anti-cancer effect in vitro, depending on the cancer cell lines. The detailed mechanism needs further study.

TWEAK/Fn14 signaling triggers intracellular signaling cascades. The TRAF2 and NF-κB pathways are activated and then lead to downstream cellular responses which are associated with cell invasion and migration as well as angiogenesis. Ect2–Cdc42 and Trio–Rac1 are the two units of GEF–GTPase signaling. MMP-9 and VEGF are increasingly expressed, further inducing tumor metastasis. In addition, tumors produce multiple angiogenic factors, including TWEAK, FGF-2, and VEGF-A. They bind to their respective receptors VEGFR2, FGFR1, and Fn14 on vascular endothelial cells, which overexpress Fn14 under tumor inflammation. In this way, TWEAK combined with FGF-2 and VEGF-A also participates in sprouting angiogenesis.
Recently, it was found that Fn14 overexpression strengthens the invasive and metastatic potential of non–small cell lung adenocarcinoma cells by the upregulating integrin α6. 48 In addition, Fn14 overexpression also increases the expression of matrix metalloproteinase (MMP)-1 genes, which is related to malignant transformation of cells. 48 Furthermore, TWEAK upregulates the messenger RNA (mRNA) level of MMP-9, which favors the invasiveness of glioma cells. 49 Actually, Fn14 overexpression enhances androgen-independent progression of prostate cancer through MMP-9 and also correlates with poor treatment outcome. 16 Meanwhile, the silencing of Fn14 expression by small interfering RNA (siRNA) can decrease invasive capacity of tumor cells. 17 In contrast, increasing the Fn14 levels through adenoviral vectors enhances the invasive activity.17,19 Therefore, TWEAK/Fn14 activation promotes cell invasion in certain tumor types.
Angiogenesis
Tumor cells can stimulate the division, proliferation, and migration of vascular endothelial cells through releasing a variety of angiogenic growth factors. The formation of new blood vessels from the pre-existing vasculatures plays a key role in primary tumor growth and metastasis.50–52 TWEAK is produced in the tumor microenvironment and can act on vascular endothelial cells to promote angiogenesis in rat corneas and HEK293 xenograft tumor. 10 Fibroblast growth factor (FGF)-2 and vascular endothelial growth factor (VEGF)-A are two factors contributing to angiogenesis. Knockdown of TWEAK or Fn14 strongly reduced VEGF expression in human urothelial carcinoma cells. 12 TWEAK/Fn14 interaction upregulates VEGF expression through the nuclear factor (NF)-κB pathway. 53 In turn, the expression of FGF-2 and VEGF-A induces the Fn14 gene expression in vascular endothelial cells. 54 These findings demonstrated that FGF-2, VEGF-A, and TWEAK might cooperate during tumor angiogenesis via binding to their respective receptors on the endothelial cell surface (Figure 1).
Inflammation
The initiation and promotion of tumors are often tightly linked to chronic inflammatory processes and associated with repetitive tissue damage as well as the corresponding repair processes.55–57 TWEAK is involved in tumor inflammation by activating the NF-κB pathway and inducing pro-inflammatory cytokine expression. 6 For example, TWEAK stimulated adjacent endothelial cells of tumors to express elevated levels of TNF-α, interleukin (IL)-8, monocyte chemotactic protein-1, intercellular adhesion molecule-1, and E-selectin.37,58 These cytokines, chemokines, and adhesion molecules together contribute to the transendothelial migration of leukocytes, which is an important process of inflammatory response. 59 TWEAK may also act on the tumor-associated innate immune cells and stimulate additional production of growth factors, cytokines, chemokines, and angiogenic factors during the development of tumors. 6 Thus, inhibition of TWEAK/Fn14 signaling could reduce the response of inflammation and inhibit tumor progression.
Morphogenesis of cells
Morphogenesis is one of the fundamental aspects of epithelial–mesenchymal transition (EMT), which tightly correlates with tumor progression. TWEAK can stimulate a branching morphogenic phenotype in Eph4 mammary epithelial cells, and blocking Fn14 expression can inhibit this morphogenesis. 60 E-cadherin is one of the administrators of the epithelial phenotype. The downregulation of E-cadherin is considered to be the critical step in EMT. 61 TWEAK induces phenotypic changes of proximal tubular cells, accompanied by loss of E-cadherin and epithelial integrity. 62 This phenotype was also found in epithelial cells after the treatment of the pro-oncogenic factor hepatocyte growth factor (HGF). 63 HGF is a ligand of the epithelial tyrosine kinase receptor c-Met, which functions in mesenchymal formation during epithelial morphogenesis. 63 Importantly, HGF/Met activation significantly increased the Fn14 mRNA and protein expression in vitro. 30 Another signal transduction route the Rho/Rac GTPase engages in the EMT. 64 TWEAK–Fn14 signaling axis stimulates the migration and invasion of glioma cells through a multi-Rho GTPase pathway that includes Cdc42 and Rac1. 44 Therefore, we suggest that TWEAK/Fn14 signaling may trigger morphogenesis of cells. By understanding this process, we may be able to prevent it. But further studies are needed to understand the molecular mechanism that drives morphogenesis and to provide promising targets for the prevention of tumor progression.
The signaling pathways activated by TWEAK/Fn14 engagement
TRAF signaling pathway
Fn14 acts on cells through connecting adaptor molecules with the downstream signal transduction molecules. The TRAF family is a group of important adaptor molecules, which are associated with Fn14 and downstream signaling pathways. 65 It has been identified that TRAF1, 2, 3, and 5 bind to the Fn14 cytoplasmic tail.65–67 Among them, TRAF2 is an anti-apoptotic protein and binding to Fn14 results in the activation of the NF-κB pathway, which then induces multiple cellular responses. 68 Cellular inhibitor of apoptosis protein-1 (cIAP1) sensitizes cells to TNF-α and activates the NF-κB signals. TWEAK/Fn14 function involves a cIAP1–TRAF2 complex. TWEAK/Fn14 engagement promotes the lysosomal degradation of cIAP1–TRAF2 and further sensitizes immortalized tumor cells to TNF-α-induced death, whereas primary cells remain resistant. 69 Such discrepancy between these two types of cells may be because TNF-α regulates cell cycle by binding to either of the two receptors, TNF receptor 1 (TNFR1) and TNFR2.70,71 TNFR1 may induce cell death, whereas TNFR2 promotes cell proliferation. 72 The formation of cIAP1–TRAF2 complex limits Fn14 signaling and protects tumor cells from TWEAK-induced TNF-α sensitization. 69 Our previous study showed that HPV16 infection induces proliferation of keratinocytes through TWEAK/Fn14 interaction, possibly by activating Ras and TRAF2 and upregulating TNFR2 expression. 38 In addition, TWEAK stimulates Src homology 3 domain–containing guanine nucleotide exchange factor (SGEF) recruiting to the Fn14 cytoplasmic tail via TRAF2 partner. 42 Mutation of the Fn14–TRAF domain site or depletion of TRAF2 expression by siRNA oligonucleotide could block SGEF recruitment to Fn14 and inhibit SGEF activity and subsequent tumor cell migration. 42 This suggested that the Fn14–TRAF2–TNFR axis is involved in the apoptosis and proliferation of tumor cells (Figure 2).

Fn14–TRAF2–TNFR axis regulates the apoptosis and proliferation of tumor cells. TWEAK/Fn14 interaction activates the TRAF2 signaling pathway. Then, Fn14 recruits a cIAP1–TRAF2 complex. cIAP1 sensitizes cells to TNF-α. TNF-α generates different effects by binding to two receptors: TNFR1 and TNFR2. Fn14–TRAF2–TNFR1 may play a role in the apoptosis of cells, and Fn14–TRAF2–TNFR2 may be responsible for cell proliferation.
NF-κB signaling pathway
The NF-κB pathway, inducing the production of cytokines, chemokines, adhesion molecules, and metalloproteases, is a critical pathway linking inflammation with cancer development and progression.73,74 TWEAK/Fn14 signaling activates both classical and alternative NF-κB pathways. 75 The activation of classical NF-κB pathway is characterized by phosphorylation of inhibitor of NF-κB (IκB) and nuclear translocation of p50/p65 heterodimers while the alternative NF-κB pathway is activated through the stabilization of NF-κB-inducing kinase and the translocation of p100 and p52/RelB heterodimers into the nucleus. 76 The activation of non-canonical NF-κB pathway and NF-κB-inducing kinase is responsible for TWEAK-stimulated invasion of glioblastoma cells. 46 Actually, TWEAK/Fn14 engagement activates non-canonical NF-κB signaling in both melanoma and prostate cancer cells but produces two different results (inhibition or stimulation of cell invasiveness). 47 Mutation of TWEAK at Tyr (176) does not disrupt TWEAK trimerization but fails to induce Fn14-mediated NF-κB signaling. 77 Fn14 deletion mutant in human breast cancer cells is associated with abrogation of NF-κB activation as well as tumor invasion in vitro. 24 TWEAK-induced NF-κB activation is partly mediated by TRAF2 and TRAF5.65,76 In turn, the increased presence of NF-κB signaling in tumor tissues induces the expression of Fn14 and nuclear p65. 14
In fact, Fn14 promotes tumor cell growth and drug resistance via upregulating Bcl-xl and Bcl-W expression upon NF-κB transcriptional activity.26,78 In non–small cell lung cancer, TWEAK–Fn14 signaling promotes tumor cell survival through myeloid cell leukemia sequence 1 (Mcl-1). 79 TWEAK stimulates Mcl-1 expression via NF-κB activity and confers protection against cisplatin- and radiation-induced cell death; depletion of Mcl-1 impaired colony formation in vitro and TWEAK-induced cell survival. 79 Thus, both the TWEAK/Fn14 signaling axis and Mcl-1 demonstrated their potential as therapeutic targets for non–small cell lung cancer. In addition, TWEAK/Fn14 activation can upregulate VEGF and SGEF expression through the NF-κB pathway to promote cancer cell metastasis.53,79 TWEAK also upregulates non-canonical NF-κB-dependent MMP-9 expression to promote glioma cell invasion. 46 Therefore, NF-κB-dependent gene expression plays an important role in TWEAK/Fn14 signaling to affect many aspects of tumor biology.
TWEAK and Fn14 as biomarkers and therapeutic targets
Since TWEAK/Fn14 signaling correlates closely with multiple biological activities of tumors, TWEAK and Fn14 may serve as potential biomarkers that indicate prognosis and prediction of cancers. Fn14 is overexpressed in primary breast carcinomas and is sensitive with high specificity to predict brain metastasis. 80 The treatment of mice with brain metastasis of breast carcinoma improves survival and downregulates TWEAK and Fn14 expression in tumors. 80 Moreover, Fn14 protein is related to human glioma tumor grade. 81 Fn14 expression is also an independent prognostic factor in gastric cancer, non–small cell lung cancer, and hepatocellular carcinoma.82–84 Furthermore, low serum levels of TWEAK are related to locoregional failure in patients with squamous cell carcinoma in head and neck, and high CD136/TWEAK expression ratio in tumors is also related to poor prognosis. 85
In view of their high tumor-associated expression and multiple action, TWEAK and Fn14 are two attractive targets in treating tumors. Therefore, a number of agents targeting TWEAK or Fn14 have been developed in recent decade (Table 1). They exert anti-tumor effect through three approaches: neutralization of soluble TWEAK,33–35 blockade of Fn14 signals,21,87–89 and killing the Fn14-positive tumor cells directly.22,28,90,91
TWEAK- or Fn14-targeting therapeutic agents to treat cancers.
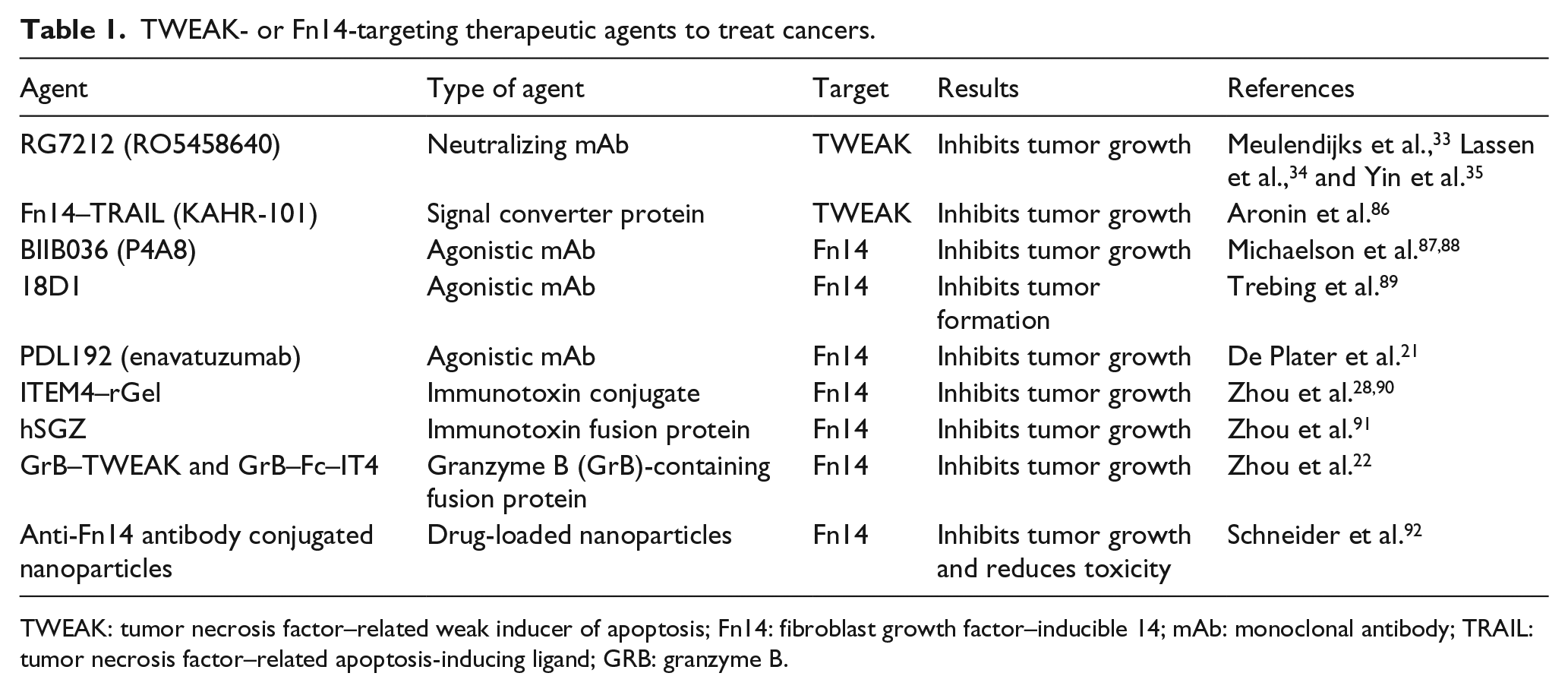
TWEAK: tumor necrosis factor–related weak inducer of apoptosis; Fn14: fibroblast growth factor–inducible 14; mAb: monoclonal antibody; TRAIL: tumor necrosis factor–related apoptosis-inducing ligand; GRB: granzyme B.
RG7212, a neutralizing monoclonal antibody (mAb) targeting TWEAK, can inhibit TWEAK/Fn14 signaling and suppress the growth of solid tumors in patients.33,34 In pre-clinical trials, RG7212 blocked TWEAK/Fn14 signaling, NF-κB activation, and cytokine secretion in cultured tumor cells and even inhibited tumor growth in murine models. 35 Phase I studies were conducted in recent years, showing that RG7212 possesses excellent tolerability and favorable pharmacokinetics as administered systemically.33,34 In colon carcinoma–bearing mice, anti-TWEAK antibody (P2D10) can reduce 5-fluorouracil-induced diarrhea without affecting the anti-tumor effect of 5-fluorouracil. 93
BIIB036, PDL192, and 18D1 are three Fn14-specific mAbs. They may diminish tumor growth in vivo and by various ways, including blockade of Fn14, inhibition of TWEAK–Fn14 engagement, and inducing antibody-dependent cellular cytotoxicity (ADCC).21,36,87–89,94 BIIB036 and PDL192 have demonstrated impressive efficacy in vivo that is, at least in part, mediated by ADCC and activation of NF-κB signaling.21,36,87,88,94 In addition, BIIB036 activates the NF-κB pathway, stimulates the production of IL-8, and induces death of human cancer cells.36,88 The 18D1 antibody inhibits metastatic colony formation of RENCA (murine renal cell carcinoma) cells predominantly via ADCC. 89 In a Phase I clinical trial, PDL192 showed significant drug toxicity in liver and pancreas in the presence of an anti-tumor dose. 36 Therefore, more clinical research is necessary to determine the safety and effectiveness of these Fn14 mAbs.
Anti-Fn14 antibody conjugated nanoparticles were developed to improve efficacy and reduce toxicity. They selectively target Fn14-positive cells and rapidly penetrate brain tissues without significant off-target effects. 92 Although an amount of work is needed in this field, the early success achieved seems promising for the future applications of this technology in delivering chemotherapy drugs to tumor cells with lowest cell toxicity.
Granzyme B (GrB)–TWEAK and GrB–Fc–IT4 are the two highly effective constructs that have the pro-apoptotic serine protease GrB as cytotoxic payload. GrB–TWEAK is composed of the human Fn14-binding domain as the targeting moiety and GrB–Fc–IT4 is constructed from an Fn14-targeting humanized single-chain antibody. 22 They showed high affinity and selective cytotoxicity against Fn14-expressing triple-negative breast cancer lines and inhibited tumor growth in vivo. 22 They are completely human constructs and are not likely to induce immune responses in patients.
ITEM4–rGel and hSGZ are two kinds of Fn14-targeting immunotoxins. ITEM4–rGel, obtained from Escherichia coli cells, is made of the ITEM4 mAb conjugated to a recombinant form of gelonin (rGel).28,90,91 hSGZ is a humanized, bivalent single-chain protein. 28 Both agents are highly cytotoxic to Fn14-positive cells in vitro and have showed significant growth inhibition but no obvious toxicity in cancer xenograft models.28,90 They induce cell death through different mechanisms and the effects appear to be dependent on cell type.28,90 Thus, these two Fn14-targeting immunotoxins have excellent efficacy and safety in pre-clinical studies.
Fn14–TRAIL (TNF-related apoptosis-inducing ligand) is a multi-functional fusion protein composed of the Fn14 extracellular domain and the TRAIL that interacted with TRAIL receptor to induce apoptosis in malignant cells. 86 This fused protein was developed to inhibit TWEAK/Fn14 signaling and to promote TRAIL signaling. Fn14–TRAIL could suppress the growth of hepatocellular carcinoma both in vitro and in vivo. 86 Moreover, it is well tolerated by mice without affecting liver histology. 86 Fn14–TRAIL can be oligomerized by TWEAK, forming a stable complex that exhibits higher efficiency in inducing apoptosis of malignant lymphoblasts. 95 Therefore, fusion proteins with more than one function can be used for targeting TWEAK/Fn14 pathway in the future cancer therapy.
Since TWEAK/Fn14 activation functions in tumor cells through the NF-κB pathway,46,47,53,75,96 small molecule inhibitors were discovered to disrupt the TWEAK–Fn14–NF-κB signaling axis. 97 By using a cell-based drug-screening assay, aurintricarboxylic acid was identified to suppress TWEAK–Fn14–NF-κB dependent signaling but not TNF-α–TNFR–NF-κB driven signaling. 97 Aurintricarboxylic acid can inhibit chemotactic migration and invasion of glioma cells upon TWEAK stimulation, without affecting cell viability or Fn14 expression. 97
Conclusion
The TWEAK/Fn14 signaling pathway participates in the pathogenesis of tumors. It exhibits an important role in the growth, invasion, and migration of tumor cells. Also, TWEAK/Fn14 activation triggers downstream signals to regulate several key events which are associated with the inflammation, angiogenesis, and EMT of tumors. Fn14 can serve as a marker for tumor progression. Moreover, some TWEAK and Fn14-targeting agents have already been tested in pre-clinical trials and showed effective outcomes. However, much more pre-clinical research is still needed. It is anticipated that safe and effective therapeutics will be applied in clinical practice.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by the National Natural Science Foundation of China (Projects No. 81472876 and 81630081).