
Abstract
Endocannabinoids such as 2-arachidonylglycerol (2-AG) are ligands for cannabinoid receptors that contribute to the transmission and modulation of pain signals. The antinociceptive effect of exogenous 2-AG suggests that inhibition of monoglyceride lipase (MGLL), the enzyme responsible for degrading 2-AG and arresting signaling, may be a target for pain modulation. Here we describe the characterization of MGLL ligands following a high-throughput screening campaign. Ligands were discovered using ThermoFluor, a label-free affinity-based screening tool that measures ligand binding via modulation of protein thermal stability. A kinetic fluorescent assay using the substrate 4-methylcoumarin butyrate was used to counterscreen confirmed HTS positives. A comparison of results from binding and inhibition assays allowed elucidation of compound mechanism of action. We demonstrate the limit of each technology and the benefits of using orthogonal assay techniques in profiling compounds.
Keywords
Introduction
Cannabis has been used as a natural analgesic for centuries, although not without well-documented side effects. Perception of a noxious stimulus (nociception) is a complex sensory phenomenon, dependent on the type and intensity of the stimulus as well as the particular pain pathways activated. Both natural and exogenous cannabinoids can produce antinociception mediated through the cannabinoid receptors CB1 and CB2,1,2 which have been identified both peripherally and in the central nervous systems. Animal studies demonstrate that cannabinoids can be as effective as morphine in suppressing the physical reactions to acute pain.3–5 Unfortunately, the use of exogenous cannabinoids can produce severe motor deficits (including sedation, hypothermia, and catalepsy), complicating the ability to interpret the phenotype of cannabinoid agonists. Effective modulation of pain via the cannabinoid receptors depends on finding pharmacological agents that act peripherally on pain-sensing mechanisms.
A defining characteristic of neurotransmitter systems is rapid onset of signaling, followed by rapid signal inactivation. The endocannabinoid 2-AG is produced on demand by neurons and functions near its site of synthesis; signaling is typically terminated by uptake and enzymatic degradation of the neurotransmitter via monoglyceride lipase (or monoacylglycerol lipase, MGLL; EC 3.1.1.23).6,7 Inhibition of MGLL would increase 2-AG levels, which might in turn prolong its nociceptive effect, enhancing retrograde signaling.4,8 Targeting the cannabinoid receptor system for antinociceptive therapy through inhibition of endocannabinoid hydrolysis may provide for greater selectivity and fewer adverse effects, as the resulting increase in endocannabinoid concentration occurs at the site of action, not systemically.
MGLL belongs to the α/β hydrolase family containing a classical Ser-His-Asp catalytic triad,9,10 similar to that of other lipases. 11 In designing an activity counterscreen for use in profiling hits from a high-throughput screen (HTS), several historical procedures were considered. Ideally, one would use the physiologically relevant substrate 2-AG, but complications arise due to the natural isomerization reaction of 2-AG to 1-AG, yielding a mixture of substrates with mixed kinetic properties. Use of 2-AG could necessitate the use of chromatographic techniques to separate substrate and products.12,13 Radiolabeled substrates are commonly used for monitoring enzyme activity where one can distinguish between the various reaction products but are labor intensive, are low throughput, and may require handling restrictions. Alternatively, fluorescent-based substrate analogues can be exploited to yield a real-time, rate-based assay. 14
Here, we demonstrate that MGLL effectively used several coumarin-based substrates with varying lengths of aliphatic chains, and we employed these in developing a rate-based assay. The coumarin-based assay was exploited to profile screening hits generated using ThermoFluor* technology, a miniaturized, high-throughput thermal stability assay that detects compound binding through perturbations in the free energy of protein stability. ThermoFluor was chosen as the primary assay for HTS to detect all small molecules interacting with MGLL, anticipating a further classification of confirmed screening hits based on their effect on enzyme activity. We present a comparison of compound binding and inhibition, which shows distinct groupings of compounds differentiated by assay limitations and mode of action.
Materials and Methods
Reagents
Unless otherwise stated below, materials were obtained from commercial sources. 4-Methylcoumarin (or 4-methylumbelliferyl)–labeled substrates, as well as coumarin- (umbelliferyl-) arachidonate, were from Sigma-Aldrich (St. Louis, MO).
Enzyme expression and purification
A detailed description of MGLL cloning, expression, and purification was published elsewhere. 15 In brief, the cDNA for MGLL (amino acids 11–313) was cloned from human brain DNA, ligated into the modified pENTR 11cLIC vector (Invitrogen, Carlsbad, CA), and transfected into insect cells using the BaculoDirect Baculovirus Expression System (Invitrogen). The viral stock was propagated for two more amplifications at a low multiplicity of infection (MOI) to render a P2 virus stock and expanded to generate a high-titer P3 stock by infecting Sf9 cells in suspension at a MOI of 0.3, harvesting the virus after 72 h.
Large-scale expression was done in 2-L shake flasks or WAVE bioreactors (GE Healthcare, Piscataway, NJ), infecting Sf9 cells at a density of 1.5 × 106 cells/mL with an MOI of 1. Infected cultures were maintained at 27 °C under constant shaking at 140 rpm. Cells were harvested 65 to 72 h postinfection by centrifugation. Cell viability was determined by Guava ViaCount (Millipore, Billerica, MA) or trypan blue, which was routinely 60% to 80% at time of harvest. Cell pellets were thawed and resuspended in buffer A (50 mM HEPES [pH 7.5], 400 mM NaCl, 5% glycerol, 0.05% β-mercaptoethanol, [1×] Complete EDTA-free protease inhibitor cocktail tablets [Roche, Basel, Switzerland]), homogenized and mechanically lysed with a micro-fluidizer processor (Microfluidics, Newton, MA), and clarified by centrifugation (40 000 g, 1 h). The lysate was loaded on a 1-mL His-Trap FF crude column and ÄKTAxpress system (GE-Healthcare) at 4 °C. MGLL was eluted with five column volumes of buffer A containing 400 mM imidazole. The elution peak was loaded onto a Superdex 200 HR 16/60 (GE Healthcare, Piscataway, NJ) preequilibrated with buffer B (50 mM HEPES [pH 7.5] buffer containing 200 mM NaCl, 2% glycerol, 1 mM dithiothreitol).
Substrate solubility
Stock substrate solutions were dissolved in 100% DMSO at concentrations of 5 or 60 mM. Twelve serial twofold dilutions were created in 100% DMSO in a 384-well polypropylene plate (Greiner Bio-One, Monroe, NC). Substrate dilutions were dispensed (0.35 µL) into a 384 UVStar plate (Greiner Bio-one) using a Cartesian Hummingbird capillary liquid-handling instrument (Genomics Solutions, Ann Arbor, MI). Then, 50 µL buffer C (20 mM PIPES [pH 7.0], 150 mM NaCl) was added and mixed manually using a multichannel pipettor. Absorbance scans of the substrate aqueous dilutions were measured from 230 to 700 nm at 25 °C (Safire 2 ; Tecan, Männedorf, Switzerland). Absorbance at 310 nm and scattering at 600 nm were used to determine the Molar Extinction Coefficient (ϵ310) and solubility of the substrate, respectively.
Rate-based assays
All rate-based assays were performed in black 384-well polypropylene PCR microplates (ABgene, Surrey, UK) in a total volume of 30 µL. The fluorescence change due to substrate cleavage at 37 °C was monitored with excitation and emission wavelengths of 335 and 440 nm (10 nm bandwidth), respectively, measured on a Safire 2 plate reader (Tecan). Assays plates were loaded with substrate dissolved in buffer C (25 µL at 1.2 × final substrate concentration, in 1 × buffer C + 0.003% Triton ”Triton” should be replaced with “Triton X-100”.), followed by enzyme (5 µL of a 6× solution in 1× buffer, final [MGLL] = 10 nM) to initiate the reaction. The initial rate was measured for 3 min while reactions behaved in a zero order manner. Fluorescence was converted to concentration using a coumarin standard curve. Substrate showed an inner-filter effect on product fluorescence above 600 µM (~ 3.0 absorbance units ~ 310 nm). Activity at these concentrations was not used to determine kinetic constants.
Compounds were dissolved in 100% DMSO to 10 mM, and 11-point dilutions series (1:2 dilutions, also in 100% DMSO) were generated. Compounds (45 nL) were predispensed from compound dilution plates into assay plates using the Hummingbird. These predispensed plates were stable for months (data not shown) but were typically used within hours. Plates were assayed as above, after diluting compound with 25 µL of 1.2× substrate and initiating reactions with 5 µL 6× MGLL in buffer C. The final concentration of MGLL in enzymatic assays ranged from 0.5 to 40 nM but was typically 2 nM in compound inhibition assays. Final compound concentrations ranged from 0.015 to 15 µM, except when assayed at a single concentration (noted below).
The kinetic constants for MGLL enzymatic activity and inhibition were obtained from a fit of data to equation (1).
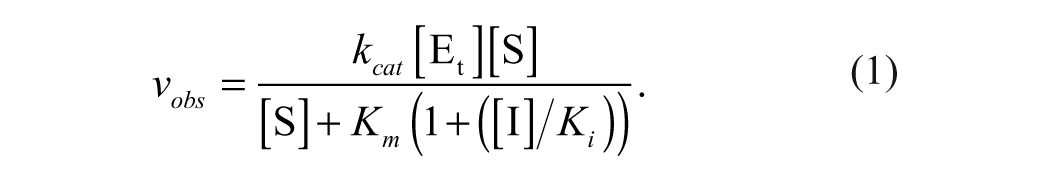
vobs is the observed rate of enzyme activity at each substrate concentration, [S]; kcat is the catalytic constant; [Et] is the total active enzyme concentration (where Vmax = [Et]kcat); Km is the apparent substrate dissociation constant; [I] is the inhibitor concentration; and Ki is the inhibitor dissociation constant. Intrinsic kinetic constants, kcat and Km, were determined in the absence of inhibit or from the initial linear rates of 11 substrate concentrations ranging from 0 to 600 µM. Inhibitor dissociation constants, Ki, were determined by varying both substrate and inhibitor concentrations.
The IC50 values for the experimental compounds were determined from the fit of equation (2) to the dose-response plot of the fractional activity as a function of inhibitor concentration.
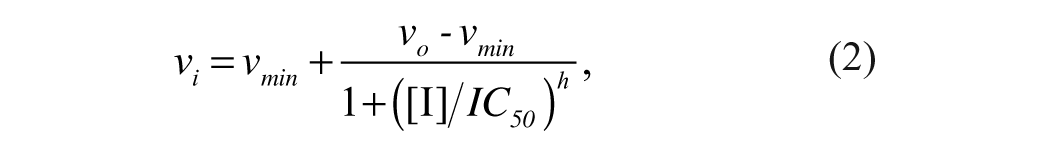
where vi is the fractional activity of the enzyme at inhibitor concentration [I], vo is the enzyme activity at zero inhibitor concentration, vmin is the minimum enzyme activity at saturating inhibitor concentrations, IC50 is the inhibitor concentration that gives 50% activity, and h is the Hill coefficient.
For inhibitors that have been determined to be kinetically competitive with the substrate, the IC50 is related to Ki, Km, and [S] by equation (3).
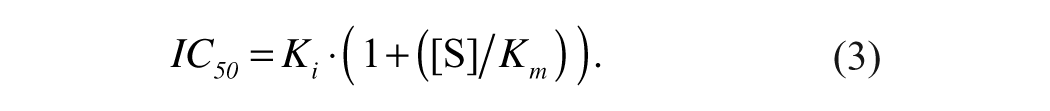
The IC50 approaches Ki at [S] << Km. At [S] = Km, IC50 = 2 · Ki. The substrate concentration (10 µM) used for compound analysis was 14-fold below the Km (134 ± 5 µM, determined below). Percent inhibition values were determined by comparing the enzyme rate using 4 µM compound (vi) with the enzyme rate in the absence of compound (v0).
ThermoFluor assay
Plate-based protein thermal stability of MGLL was assayed by ThermoFluor using instruments available from Johnson & Johnson Pharmaceutical Research & Development, L.L.C. (Exton, PA). Details of the ThermoFluor-based protein stability assay are described elsewhere.16–19 Briefly, protein unfolding is induced by increasing temperature and detected by monitoring the fluorescent probe, 1,8-ANS, which exhibits a change in fluorescence associated with protein unfolding due to noncovalent, nonspecific dye association with hydrophobic protein residues buried in the native state.
For ligand-binding studies, compounds (45 nL) were dispensed from the same compound dilution plates described above into black 384-well polypropylene PCR (ABgene) assay plates using a Cartesian Hummingbird. Protein at a concentration of 0.05 mg/mL in 50 mM PIPES (pH 7), 200 mM NaCl, 100 µM 1,8-ANS, and 0.001% Tween-20 was added to assay plates (3 µL) containing predispensed compound. Wells were overlaid with silicone oil (1 µL, type DC 200; Fluka, St. Louis, MO) to prevent evaporation during thermal cycling. Final compound concentrations varied from 150 to 0.15 µM, except when assayed at a single concentration (noted below). Assay plates were heated at a rate of 1 °C/min for all experiments over a temperature range sufficient to measure protein unfolding. Fluorescence was measured by continuous illumination with UV light (Hamamatsu LC6; Hamamatsu, Bridgewater, NJ) supplied via fiber optics and filtered through a custom band-pass filter (380–400 nm; >6 OD cutoff; Omega Optical, Battleboro, VT). Fluorescence emission was detected by measuring light intensity using a CCD camera (Sensys, Roper Scientific, Trenton, NJ) filtered to detect emission at 500 ± 25 nm (Omega Optical, Battleboro, VT), resulting in simultaneous and independent readings of all 384 wells. One or more images were collected at each temperature, and the sum (or average of the sums) of the pixel intensity associated with a given well versus temperature was fit to standard equations, yielding the Tm, as described in the Data Analysis section.
Thermal stability profiles obtained using ThermoFluor were fit to equation (1) to determine the Tm.16,17
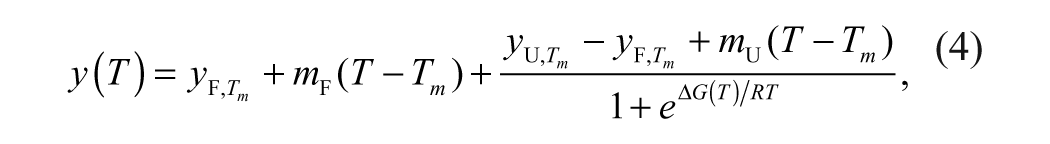
where
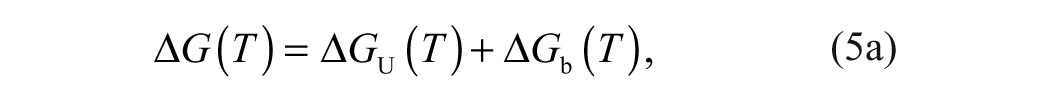
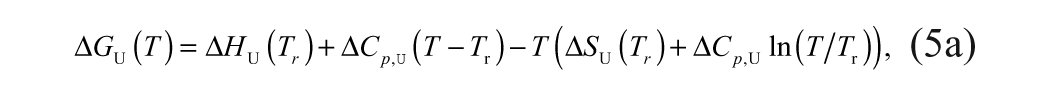
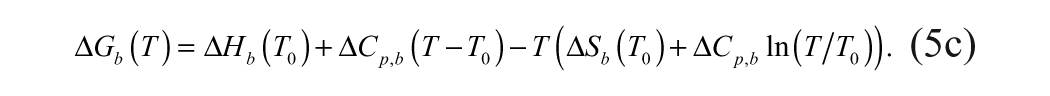
Binding affinities are determined from a ligand concentration-dependent increase in the Tm of MGLL. The expression of total ligand concentration, Lt, and total protein concentration, Pt, needed to raise the Tm to a given value is
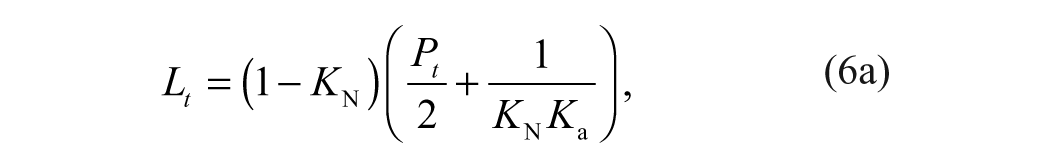
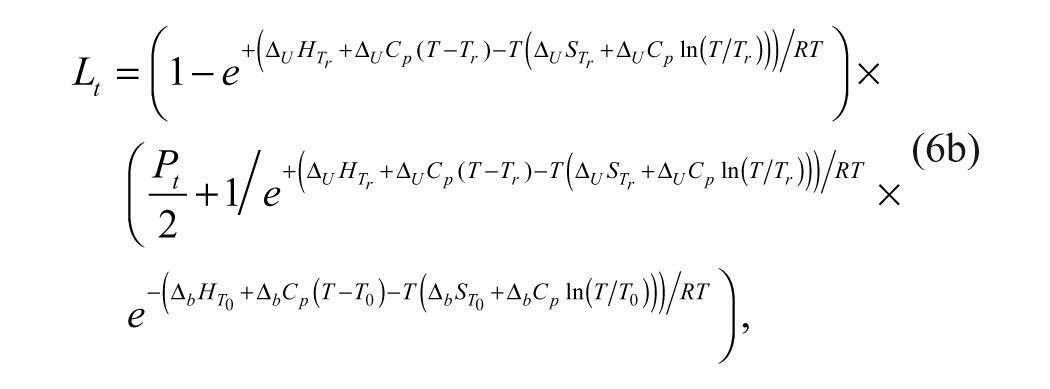
where KN is the equilibrium constant for folding (equivalent to the inverse of the unfolding equilibrium constant, KU), and Ka is the equilibrium constant for ligand association (equivalent to the inverse of the dissociation constant, KD). Equation (6b) is transcendental with respect to temperature and thus cannot be solved explicitly for Tm as a function of total ligand concentration, Lt.16,17 Therefore, estimates of ligand binding require an inversion of the dependent and independent variables.
Results
Solubility of aliphatic substrates
The natural substrate for MGLL is 2-AG ( Fig. 1A ). Five different 4-methylcoumarin (4-MC-) and coumarin (C-) based acyl-substrates with various aliphatic chain lengths were selected based on their commercial availability and were explored as a mimetic of 2-AG in MGLL activity assays ( Fig. 1B ). The substrates selected for MGLL assays also have been used previously for the analysis of lipase activity in other systems.20–22 Prior to testing for enzymatic hydrolysis, substrate solubility was measured spectrophotometrically. Substrate absorbance was measured as a function of concentration, monitoring both the absorbance in DMSO at the peak wavelength ( Fig. 2A ) and the absorbance at longer, nonabsorbing wavelengths (600 nm, to assay solubility from light scattering). The absorbance of 4-methylcoumarin butyrate (4MC-B) substrate exhibited peak absorbance at 285 nm and 310 nm, with ϵ310 nm = 6000 M−1cm−1 in DMSO ( Fig. 2A , inset), and insignificant scattering at 600 nm ( Fig. 2C ), consistent with solubility >400 µM in buffer C.
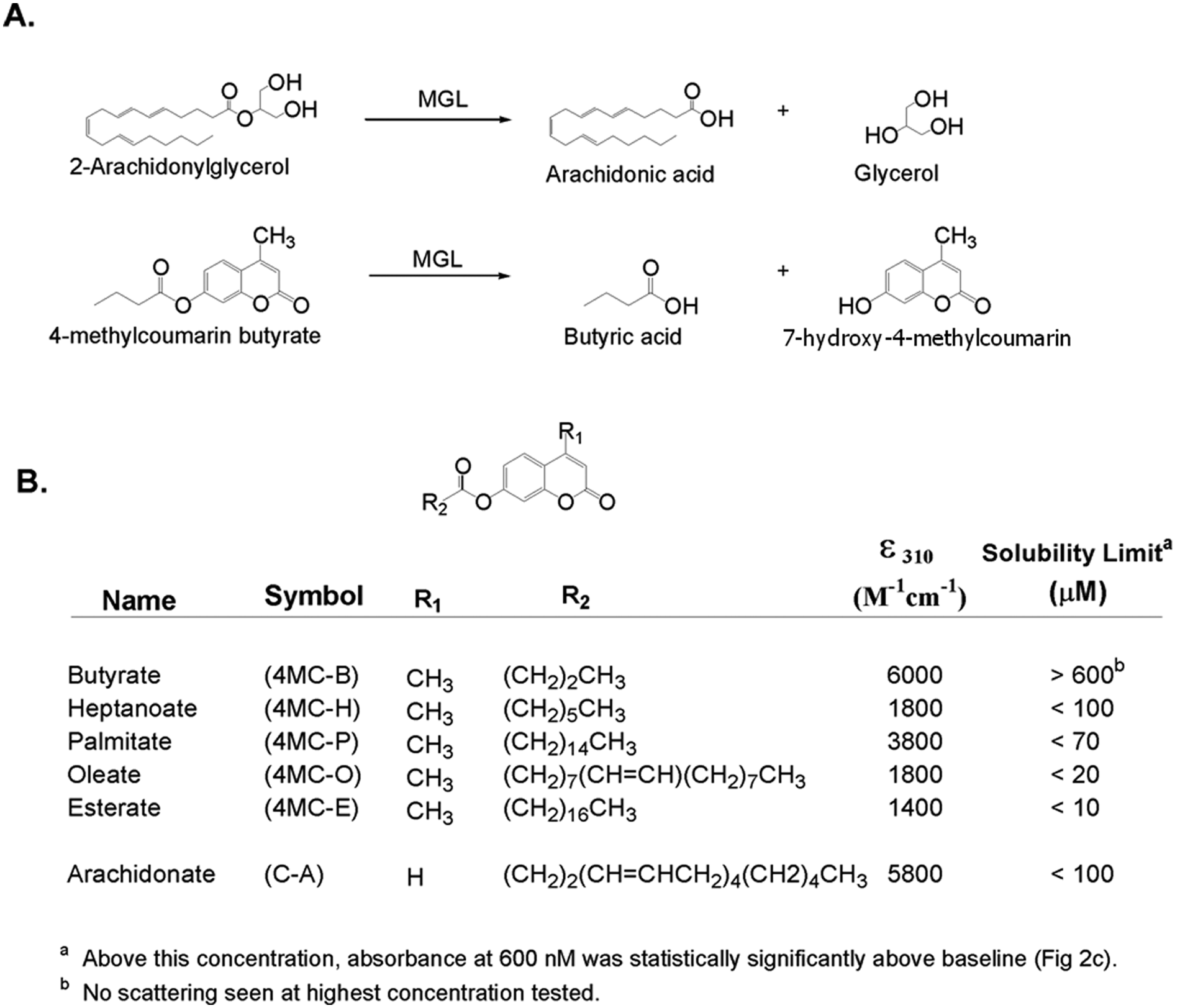
Monoglyceride lipase (MGLL) substrates. (
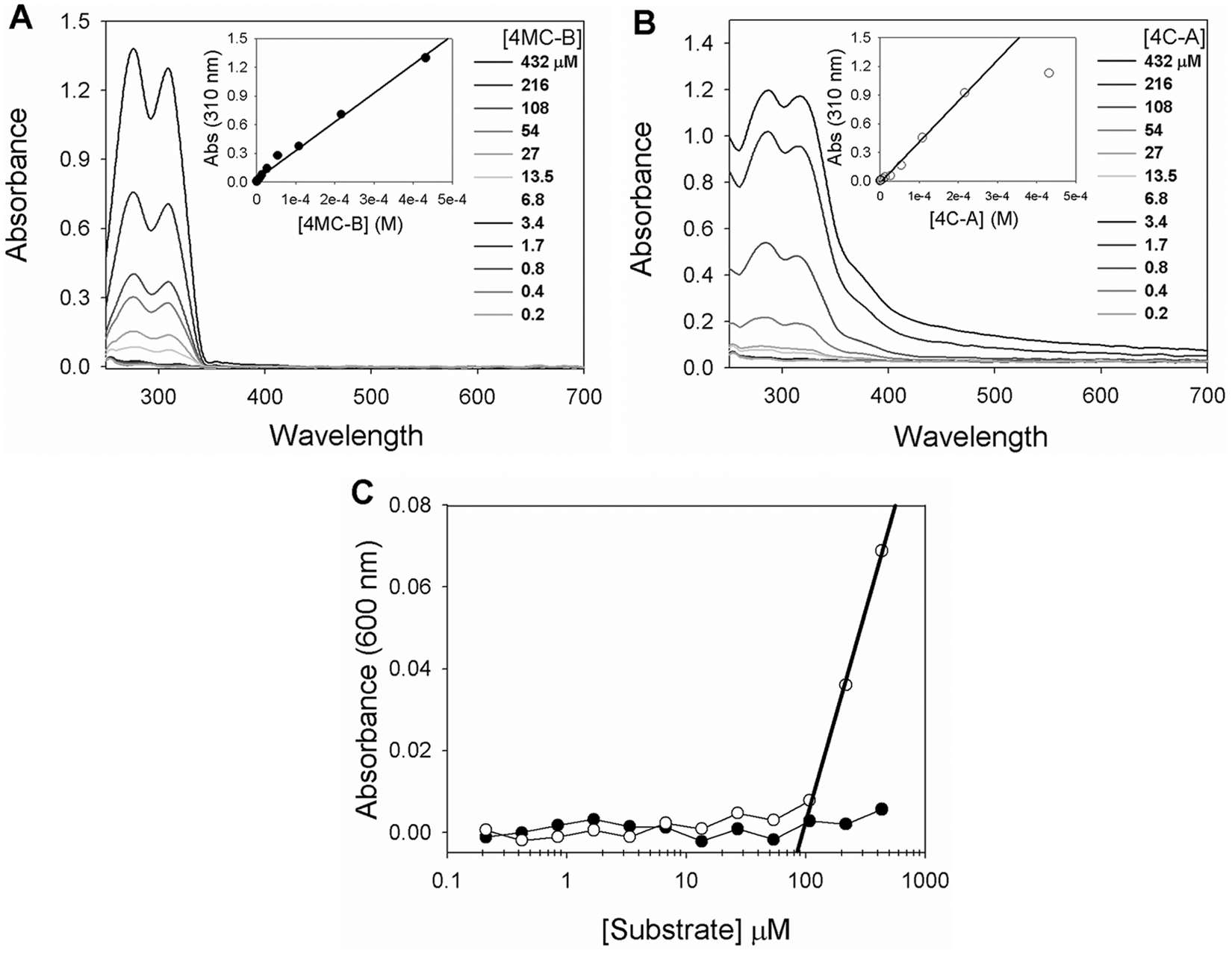
UV/Vis analysis of substrates. Absorbance scans of (
The coumarin-arachidonate (C-A) substrate was characterized by a similar absorbance at 285 nm and 310 nm, and ϵ310 nm = 5800 M−1cm−1 ( Fig. 2B ), but displayed significant scatter at 600 nm above 100 µM ( Fig. 2C ), the apparent solubility limit of this substrate. The 4-methylcoumarin esterate (4MC-E), 4-methylcoumarin oleate (4MC-O), and 4-methylcoumarin palmitate (4MC-P) substrates had significantly lower apparent extinction coefficients, consistent with impure material or incomplete dissolution. Consistent with the latter interpretation, these substrates showed significant light scattering at 600 nm (even at 10 mM in 100% DMSO), with solubility limits decreasing as a function of increasing aliphatic chain length ( Fig. 1B ). For reasons of solubility and selectivity, unless otherwise stated, all subsequent kinetic assays used 4MC-B due to its higher solubility.
4MC-B kinetic constants and solvent effect on MGLL activity
Steady-state rates for the hydrolysis of 4MC-B were measured under initial-rate conditions ( Fig. 3A ). The rate of product formation was determined within 3 min of reaction initiation, where less than 5% of the substrate was hydrolyzed. A plot showing the rate of product formation as a function of substrate concentration is hyperbolic and was fit to the Henri-Michaelis-Menten equation ( Fig. 3B ), yielding Km = 134 ± 5 µM, kcat = 31 ± 4 s−1, and kcat/Km = 1.4 ± 0.1 × 107 M−1 min−1.
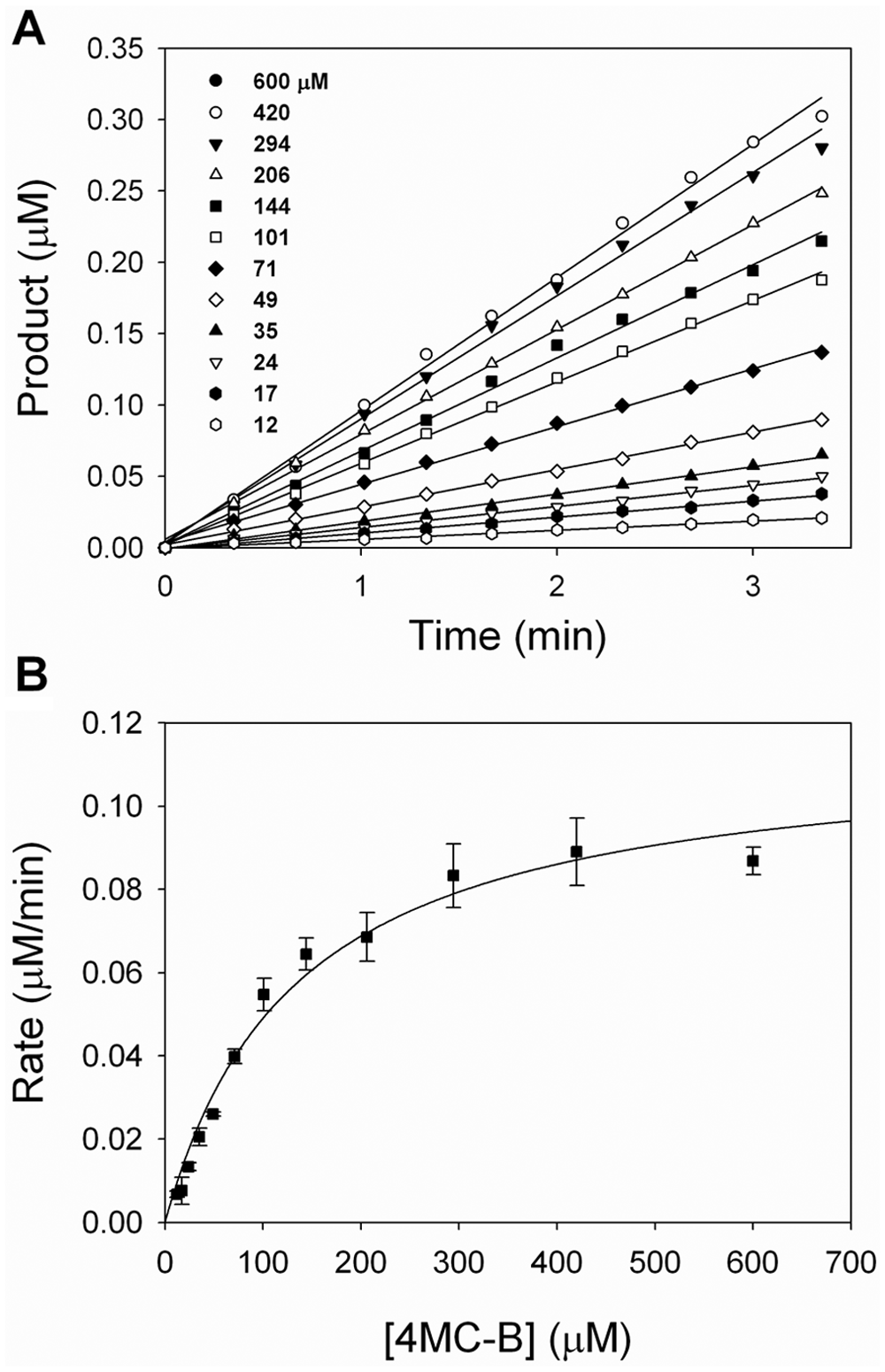
Kinetic constants for 4-methylcoumarin butyrate (4MC-B) hydrolysis by monoglyceride lipase (MGLL). (
IC 50 assay for inhibition screening
The MGLL activity assay was further evaluated using an active site inhibitor ( Fig. 4A ) that was identified as a possible tool compound during the HTS campaign described below. 15 Progress curves for the hydrolysis of 4MC-B in the presence of the tool compound ( Fig. 4B ) are consistent with rapid, reversible inhibition. A plot of the initial reaction rates as a function of inhibitor concentration ( Fig. 4B , inset) was fit to equation (2), giving IC50 = 0.1 ± 0.02 µM, with a Hill slope of h = 1. The Ki value for this compound, determined by varying both substrate and inhibitor concentrations ( Fig. 4C ), was described well by a global fit to a competitive binding model using equation (1), yielding Ki = 0.2 ± 0.03 µM.
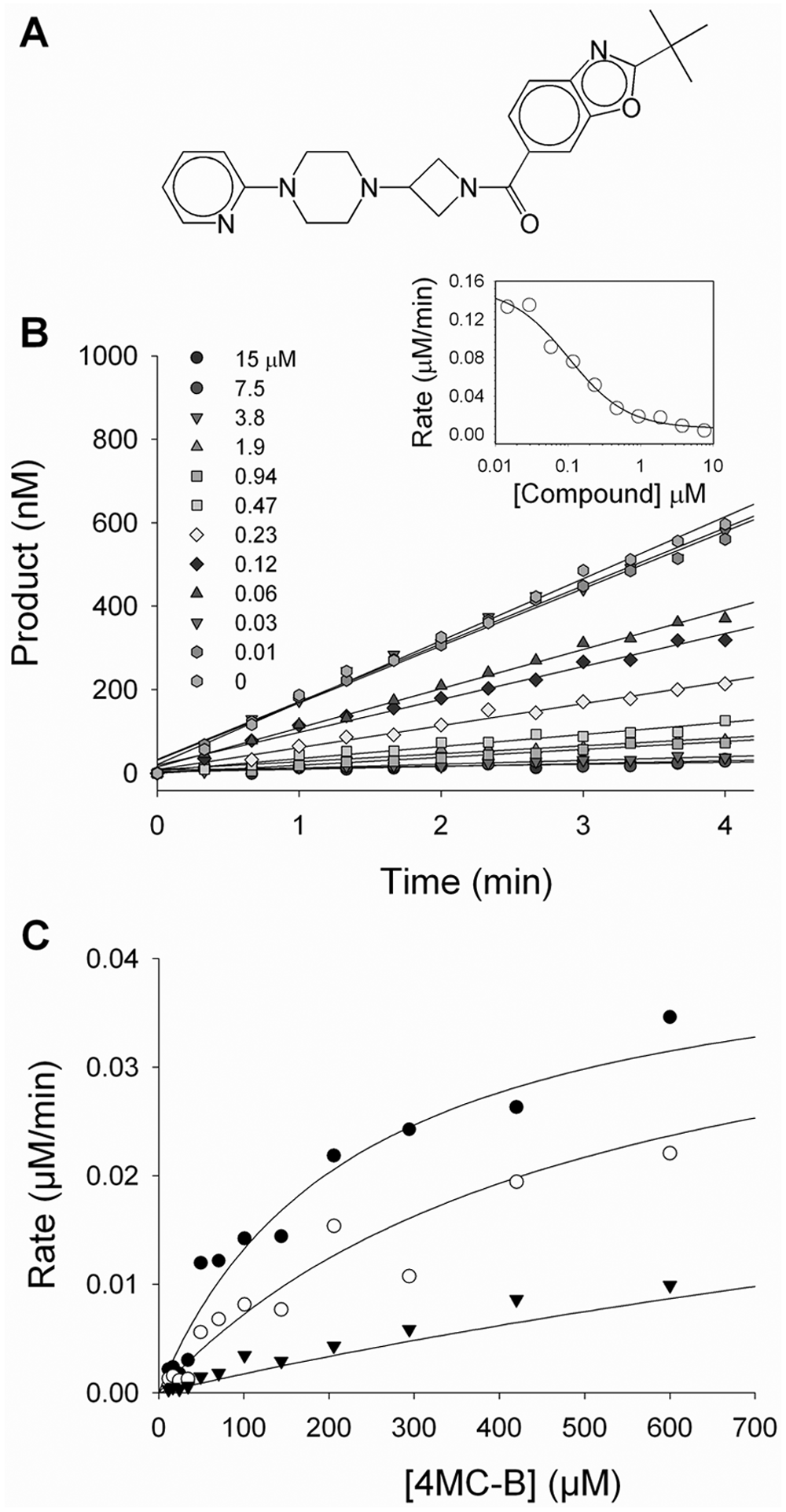
Inhibition of monoglyceride lipase (MGLL) by a tool compound. (
High-throughput screen
An HTS campaign using ThermoFluor was performed testing compound libraries at (nominally) 22 µM. 17 Figure 5A illustrates the typical thermal stability transitions for the unfolding of MGLL in the absence and presence of several ligands. Each unfolding transition is characterized by a distinct Tm, the midpoint temperature of unfolding defined by equation (4). Ligand-binding affinities are determined from a series of thermal unfolding curves, 17 as shown for several ligands ( Fig. 5B ). The Tm of MGLL changes in proportion to both the concentration and affinity of the ligand, defined by equation (6b). For all examples, a shift in ΔTm continues to occur well above the concentration of MGLL (0.7 µM) used in the assays. The magnitude of ΔTm (the increase in MGLL thermal stability) resulting from ligand binding is a function of both compound concentration and the dissociation constant for compound binding, KD.16–18,23 For HTS assays, where all compounds are assayed at a single concentration, ΔTm is directly proportional to binding affinity.
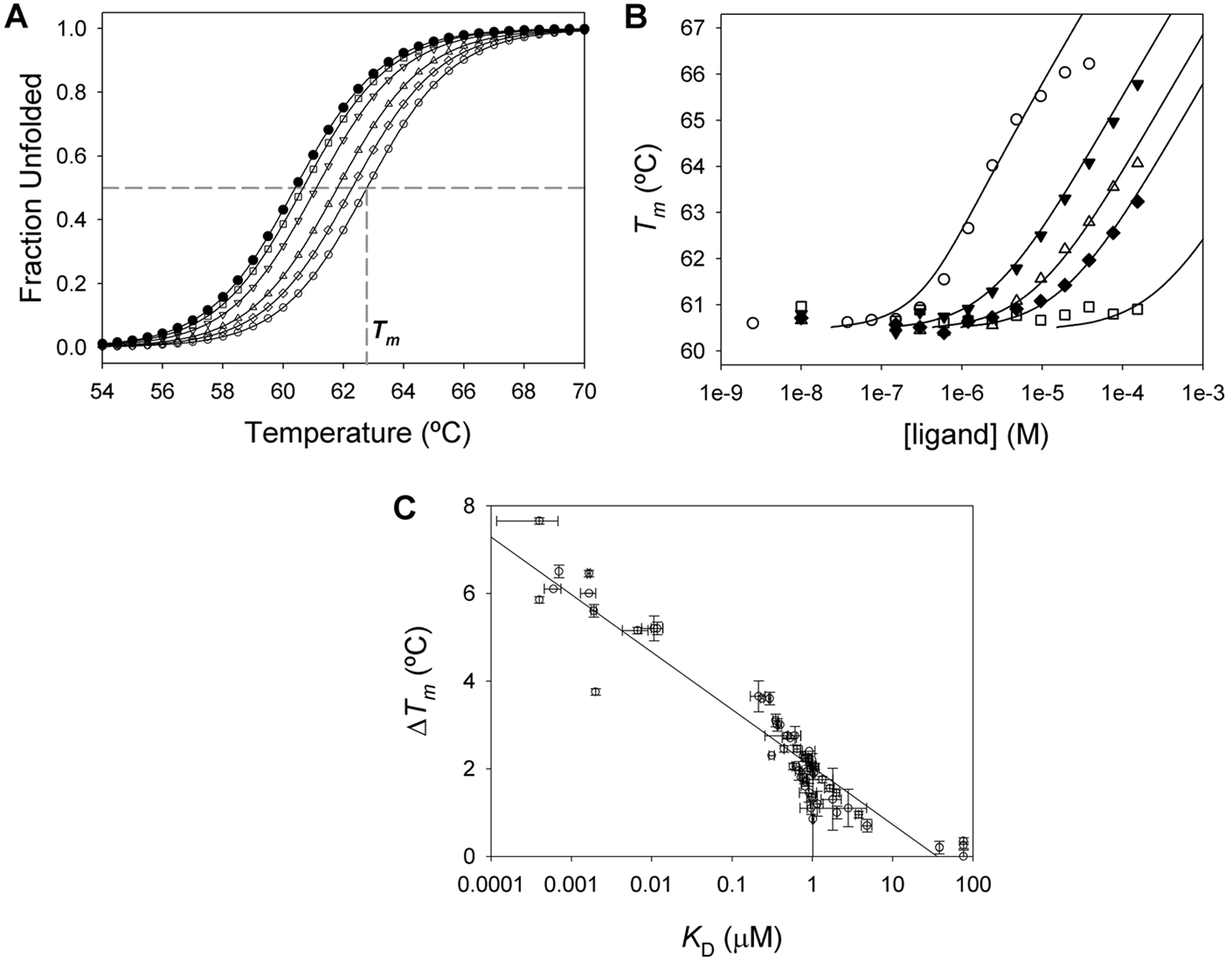
Compound affinity by ThermoFluor. (
Among the example compounds, one weak binding inhibitor ( Fig. 5B , squares) is representative of ligands with a binding affinity near the upper concentration tested (~120 µM) and is more accurately assayed by increasing the ligand concentration. 16 In contrast, the highest affinity compound shown for comparison has a KD = 0.5 µM ( Fig. 5B , open circles), an affinity below the concentration of MGLL. For compounds with even tighter binding affinity, the shape of the curve takes on a sigmoidal appearance until the protein is saturated; these tight-binding profiles still allow for precise quantitation of the binding constant.17,23 That same representative compound shows another common attribute of thermal shift assays, the effect of compound solubility limit on the concentration-response curve; the highest two to three concentrations of this compound are above its solubility limit (~5 µM by light scattering). The protein stability responds only to the amount of soluble ligand present in solution.
The compound ranking obtained from single-point stability-perturbation measurements correlates to that obtained from concentration-response curves. The ΔTm determined from a single ligand concentration has a linear dependence with respect to the log of the binding constant ( Fig. 5C ). As shown for a number of HTS hits, there is a strong correlation between ΔTm and log(KD), particularly when ΔTm > 1 °C, or roughly five to six times the standard deviation of the ligand-free Tm value for MGLL. For those compounds that generate a small ΔTm < 1 °C at the nominal concentration used in screening, there is a poor correlation between ΔTm and log(KD). 16
Correlation between functional and binding activities
ThermoFluor screening positives were tested for their effect on enzyme activity using the 4-MC-B enzyme activity assay at a single concentration of compound to yield a percent inhibition. A correlation of confirmed positives relates % activity of MGLL versus ΔTm ( Fig. 6A ). Most of the compounds that were discovered due to an effect on protein stability also affected the enzymatic activity, although this is not necessarily guaranteed (e.g., if a compound binds outside the protein active site). Compounds giving ΔTm < 1 °C generally exhibited less than 50% inhibition, whereas compounds giving a ΔTm > 1 °C generally exhibited greater than 50% inhibition.
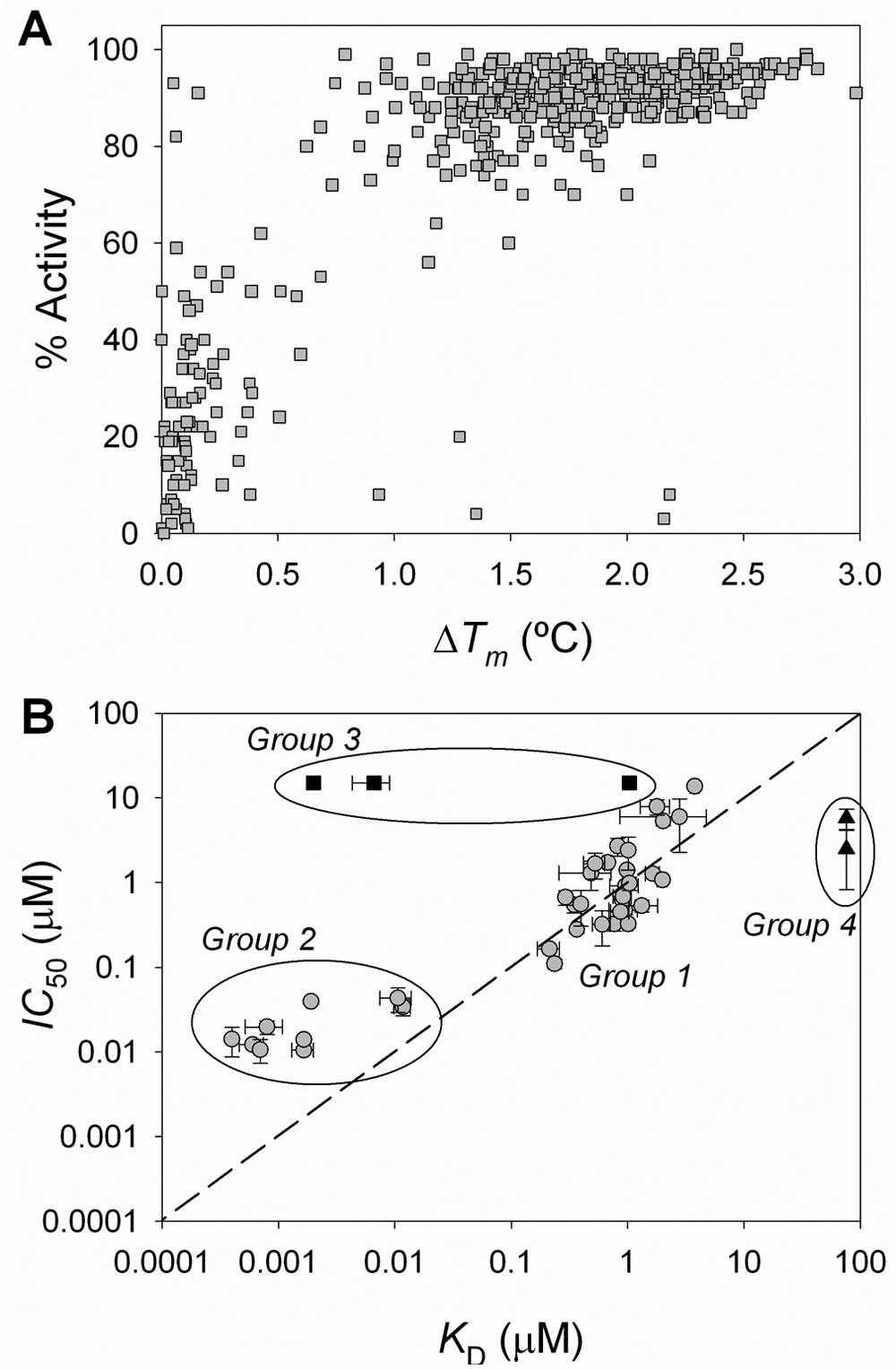
Correlation of compound functional inhibition (enzymatic activity) versus compound binding (by ThermoFluor). (
Mechanism-of-action indications from orthogonal assay correlations
Compounds that were identified from the ThermoFluor-based HTS were further evaluated as MGLL inhibitors by measuring binding affinity, effect on enzyme activity, quality control (QC)/concentration analyses, and solubility (as in Fig. 2 ). The same source of compounds was used in all measurements to minimize differences in sample concentration or preparation when comparing activity in binding and enzyme assays. A plot of IC50 versus KD clearly shows a clustering of compounds into four separate groups ( Fig. 6B ). Most compounds showed good correlation between ThermoFluor KD and IC50 values (group 1). The variation between observed IC50 and KD values is larger than is typically observed for sample replicates using the same technologies. Although measurements were made on the same compound source plate, they were not assayed within the same time frame, which may contribute to the observed variability.
In group 2 ( Fig. 6B ), the IC50s of compounds appear not to correlate with the KD values and give an apparent underestimation of compound potency in the activity assay relative to the binding assay. This was due to the sensitivity limit inherent to the enzymatic assay protocol, which was limited by the enzyme concentration (40 nM). Thus, all compounds with KD < 40 nM only require 20 nM compound to achieve 50% inhibition of 40 nM MGLL, whereas KD values are well determined for very tight-binding inhibitors, although the protein concentration (1 µM) is ~100-fold greater than the affinity. The IC50 curves for these compounds yielded Hill coefficients greater than 1, consistent with activity being limited by mass action.
An example of both a group 1 compound (fit with an IC50 = 5.2 · 10−7 M and h = 1) and a group 2 compound (either fit with a Hill coefficient fixed at h = 1 [IC50 = 1.6 · 10−8 M] or varied, giving IC50 = 1.7 · 10−8 M and h = 1.7) is compared in Figure 7A . The residuals for the group 2 compound clearly show that h = 1 inadequately describes the data. The concentration of MGLL had been reduced to 25 nM for these assays; compounds with an effective inhibition lower than approximately half the enzyme concentration are best described by the quadratic Morrison equation for tight-binding inhibitors. 24 Using the quadratic Morrison equation, the fit and residuals of the group 2 compound were indistinguishable from the fit where IC50 and h were varied (not shown). In general, additional replicates at varying [MGLL] at lower concentrations could help quantitate IC50 values in this tight-binding regime. However, their affinities are readily distinguished from ThermoFluor-based KD measurements.
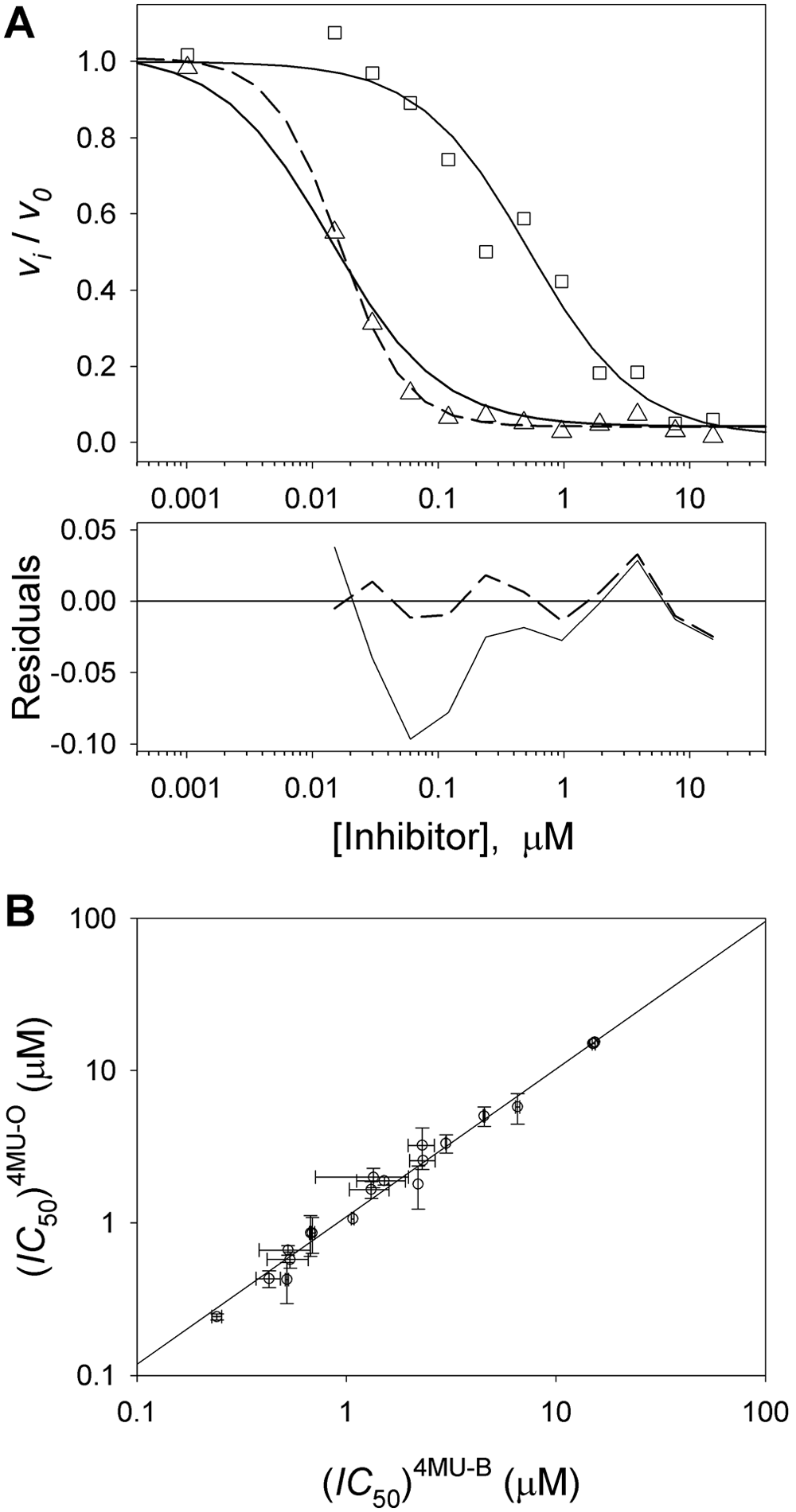
IC50 estimates and comparison of IC50s measured for short or long substrate: (
Compounds in group 3 ( Fig. 6B ) reproducibly showed binding affinity by ThermoFluor (with KD values <1 µM) yet had no effect on enzyme activity at concentrations up to 15 µM. To test for direct binding interactions, these same compounds were assayed using isothermal titration calorimetry (data not shown) and confirmed a lack of binding of group 3 compounds at 25 °C or 37 °C (two temperatures were chosen in case ΔHb ~0 kcal/mole at one of these temperatures). In this case, the binding observed using ThermoFluor is either due to compound binding preferentially at elevated temperatures or because compound binding is only observed in the presence of the ANS dye present in ThermoFluor experiments. In support of the former hypothesis, these same compounds also increased the protein thermal stability when monitoring protein stability via changes in tryptophan fluorescence (data not shown). For other compounds associated with other HTS programs, the latter hypothesis has also been confirmed through the use of ThermoFluor using a different reporter dye. Regardless, these compounds were not pursued further.
Group 4 compounds ( Fig. 6B ) displayed IC50 values in the 5- to 10-µM range but showed no or little apparent binding by ThermoFluor at concentrations >70 µM in concentration-response studies. Such behavior might be expected for compounds that could quench product fluorescence in the enzyme assay or that are thermally unstable. Either of these artifacts can make it difficult to determine a binding constant in ThermoFluor precisely, especially for weak binding compounds (KD > 5 µM). Regardless, the highest priority would be given to compounds in groups 1 and 2 as potential lead molecules in a subsequent drug discovery lead optimization program for MGLL inhibition.
Discussion
Our strategy for discovery of potent monoacylglycerol lipase inhibitors was to first conduct a high-throughput screen employing a ThermoFluor-based binding assay applied to MGLL and a proprietary compound library. Second, it was necessary to develop a rate-based fluorescent assay for monoacylglycerol lipase to confirm functional activity of compounds discovered via the binding HTS assay. Validation included both single-point and concentration-response curves for both ThermoFluor and enzyme activity. Furthermore, compounds that showed a similar rank order across both assays were selected for compound development activities. The detailed comparison of affinities from the binding assay, KDs, and from the enzymatic assay, IC50s, was used to develop additional insights into the mechanism of ligand action and to eliminate false-positive compounds from either method.
During assay development, proper determination of substrate kinetic constants is critical in that they form the basis for selecting concentrations of enzyme and substrate in screening assays and for addressing inhibitor mechanism-of-action for compounds identified from HTS. Although the products of monoacylglycerol hydrolysis, arachidonic acid and glycerol, can be monitored through liquid chromatography/mass spectrometry (LC/MS) and radioactive methods, such methods are cumbersome for large-scale screening of compound libraries. The incorporation of an optical probe attached to the substrate allows for spectroscopic differentiation of substrates and products and importantly lends itself to assay miniaturization. A rate-based kinetic assay, in lieu of an end-point assay, minimizes contributions from spectroscopic artifacts (e.g., compound absorbance or fluorescence) that can give rise to false-positive or false-negative results.
Another challenge commonly encountered for enzymes involved in fatty acid metabolism is intrinsic to the reactants and products of enzymatic reactions. For MGLL, the low solubility of substrates with aliphatic chains longer than butyrate made it difficult to determine their kinetic constants. Generally, it is important to ensure that surrogates of the natural enzyme substrate are functionally relevant in comparison to the native reactions, as exemplified here by the challenge of substantially truncated acyl chain lengths employed out of the necessity to improve substrate solubility. Although solubility limits of substrates prohibited quantitation of kinetic constants for all but 4MC-B, a comparison of initial rate data for all substrates as dilute solutions confirmed comparable initial velocities (data not shown). Initial rates versus total substrate concentration for these substrates yielded apparent hyperbolic curves, where substrate concentrations had reached their respective solubility limits (data not shown). Minimally, initial velocity data provided supporting evidence that substrate utilization is comparable for the 4MC-B substrate in comparison to substrates with longer acyl chains.
Another concern is associated with the use of the short aliphatic chain substrate as a surrogate for the natural, longer aliphatic chain for MGLL activity. Truncated substrates could potentially yield different structure-activity relationships (SARs) than one with a longer aliphatic chain. Therefore, a small number of compounds were assayed for MGLL inhibition using the larger 4MC-O substrate, 10-fold below the solubility limit of this substrate and well below the Km of 4MC-B. A correlation plot of IC50 values determined using 4MC-O and 4MC-B displayed excellent correlation between inhibitors of the oleate- versus butyrate-based fluorescent substrate ( Fig. 7B ). These data validate routine use of the shorter substrate to monitor the SAR of MGLL inhibitors.
Recently, several studies of MGLL have been published that use a similar approach of labeling alkyl chains with absorbance- or fluorescent-based spectrophotometric dyes. Muccioli et al. 25 employed the chromogenic moiety, 4-nitrophenol, in several alkyl esters that were successfully hydrolyzed by MGLL in absorbance-based assays. Similarly, Wang and coworkers 26 used 7-hydroxycoumarinyl-arachidonate in an HTS campaign for the discovery of competitive MGLL inhibitors. These studies benefit from the same approach that was undertaken in our current research, incorporating spectroscopic probes into various alkyl chains of different length and degree of saturation. However, our approach has been to rank MGLL ligands via the orthogonal techniques of binding and activity assays followed in parallel. Using a common source plate for assays, normalizing for compound concentration, and including solubility and QC measurements all serve to maximize our capacity to discern how small but significant chemical changes can affect compound mechanism of action.
It is common in ThermoFluor-based HTS to discover ligands that bind to a target protein outside of the active site; these may have no effect on enzyme activity or, conversely, may be allosteric modulators of activity. Likewise, it is common in activity-based assays to identify ligands that appear to affect enzymatic activity but do so in a manner that is not consistent with competitive substrate binding, as is the case for chelating or denaturing agents. A distinct benefit of using a ThermoFluor stability assay for the initial HTS is that compound affinities from the single-concentration measurements are well resolved based on ΔTm (for compounds where ΔTm > 1 °C), whereas a similar single-point enzyme screen ranks numerous compounds equivalently as giving ~100% inhibition. Relative compound activity cannot be discriminated using enzyme inhibition assays without dose-response studies.
A comparison of ThermoFluor-derived binding constants, KDs, and kinetic inhibition of 4MC-B hydrolysis, IC50s, allowed for classification of compounds into four groups ( Fig. 6B ). Most compounds were in group 1 and were essentially equivalent in both binding and activity assays. The subset of compounds in group 2 were more potent in the binding assay as compared with the enzyme assay because affinities fell below the enzyme concentration employed in the activity assay. These highly active compounds are often difficult to discriminate when probed exclusively by an enzymatic assay, when further dilutions of the enzyme may not be practical in efforts to extend the lower limit of quantitation. A unique feature of the thermal shift assay is its utility with respect to quantifying binding affinities of extremely high-affinity ligands, in the range of sub-nanomolar KD values. The compounds in both groups 3 and 4 were only active in either the binding assay or the enzymatic assay, respectively. Regardless of the nature of these results, whether artifacts or legitimately active, pragmatically only compounds that are confirmed to bind and affect MGLL function were selected for further studies in the drug discovery program.
The combination of a binding assay, both initially for HTS and subsequently for affinity estimates, and a spectroscopic enzymatic assay, employing a soluble surrogate of the native reaction, were key tools in the discovery of MGLL inhibitors. The strength of combining parallel, orthogonal techniques at the stage of in vitro discovery bolsters our ability to prioritize compounds that have the best chance of showing activity in the next stages of discovery. This combination of approaches highlights the strengths and limitations of either approach when undertaken alone but importantly provides an efficient means of eliminating compounds with inconsistent activity without the need for further evaluation. Finally, this work is a step forward in expediting the identification of inhibitors against MGLL, a key factor in the future development of antinociception therapeutics targeting the cannabinoid system.
Footnotes
Acknowledgements
The authors thank Mark E. McDonnell and Mark J. Macielag at Janssen Research & Development, LLC, Spring House, Pennsylvania, for synthesis of the MGLL tool compound.
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
The research represents work done in the course of employment; there was no financial support above and beyond normal compensation for employees.
*
The ThermoFluor assay was developed by 3-Dimensional Pharmaceuticals, Inc., which has been merged into Johnson & Johnson Pharmaceutical Research & Development, L.L.C. ThermoFluor is a trademark registered in the United States and certain other countries and is protected under U.S. patents US6020141, US6036920, US6214293, US6232085, US6268158, US6268218, US6291191, US6291192, and US6303322.