
Abstract
Endocannabinoids have been identified to have roles in numerous physiological and pathological processes. Largely due to the association of the effects of Cannabis administration on mental states, the CNS impact of the endocannabinoid system has been the most intensively studied. Here, we provide a brief summary of the endocannabinoid system, comprising the receptors and the multiple endogenous lipid derivatives which activate them, as well as the enzymes which control the levels of these lipid derivatives. We identify pharmacological tools which may be used to interrogate the endocannabinoid system, as well as current and future options to exploit the system in the clinic.
The endocannabinoid system
Our current understanding of the endocannabinoid system (ECS) defines two types of endogenous agonist: esters and amides of fatty acids. The esters and amides are synthesised by two independent enzyme systems; separate hydrolases also terminate their action. The functions of these endocannabinoids are primarily mediated through two G protein-coupled receptors, although there is evidence for activation of TRPV1 vanilloid receptors and PPAR nuclear hormone receptors, as well as through allosteric regulation of other receptors, channels and enzymes (Alexander and Kendall, 2007).
Cannabinoid receptors
In 1990, an orphan G protein-coupled receptor (SKR6) was identified from the cDNA library of rat cerebral cortex (Matsuda et al., 1990) and was identified to mediate the pharmacological effects of (−)-Δ9-tetrahydrocannabinol (Δ9-THC), the major psychoactive constituent of Cannabis. A second cannabinoid receptor was subsequently identified in 1993 from spleen (Munro et al., 1993). These two receptors are referred to as the CB1 and CB2 cannabinoid receptors (Pertwee et al., 2010).
These cannabinoid receptors belong to the family of rhodopsin-like G protein-coupled receptor (GPCR), characterised by binding of the canonical ligand(s) to a binding site in the plane of the plasma membrane made up of the seven transmembrane-spanning domains. Four crystal structures of the CB1 cannabinoid receptors have been described (Hua et al., 2016; Shao et al., 2016). These structures suggested that unusually among the rhodopsin family of GPCR, the extracellular surface of the receptor was also involved in ligand binding. Based on the amino acid sequence, there is relatively poor homology between CB1 and CB2 receptors, with <50% homology for human CB1 and CB2 receptors. CB1 receptors are reasonably well-conserved across species, while CB2 receptors are poorly conserved in comparison (Pertwee et al., 2010). Both CB1 and CB2 cannabinoid receptors couple to the Gi/o family of G proteins to elicit inhibition of adenylyl cyclase activity and enhance extracellular signal-regulated protein kinase activity (Felder et al., 1995). The CB1 receptor, but not the CB2 receptor, has also been shown to activate inwardly rectifying potassium channels and inhibit voltage-gated calcium channels (Felder et al., 1995).
A profound difference between the two receptors is their distribution. Initially, the very high expression of the CB1 receptor in the CNS, reportedly up to 1.8 pmol/mg protein in rat cortical P2 membranes (Devane et al., 1988), lead to the alternative nomenclature of the ‘central cannabinoid receptor’. By contrast, the CB2 receptor was identified initially in the spleen and in cells from the myeloid lineage, leading to the alternative usage of the ‘peripheral cannabinoid receptor’. We know these distinctions to be incorrect; however, since CB1 receptors are found in the periphery (not just on neural elements) and CB2 receptors are also found in the CNS. Indeed, there have been efforts to generate ligands for the CB1 receptor which are ‘peripherally restricted’ in order to avoid CNS side effects (Yu et al., 2010).
CB1 receptors are distributed throughout the brain, with the highest levels in the cerebellum, cerebral cortex, hippocampus and much lower levels in the brainstem (Herkenham et al., 1990). The poor expression in the brainstem has been interpreted as the basis for the lack of respiratory depression effects of Cannabis administration; this is a sharp contrast to the opiates, for which opioid receptors are expressed in the cardiopulmonary centres.
Quite rapidly following the molecular identification of the cannabinoid receptors, pharmacological tools were described allowing selective activation or blockade. Ligands for the cannabinoid receptor are often grouped for ease of division into plant-derived cannabinoids (phytocannabinoids), synthetic cannabinoids and endogenous cannabinoids (endocannabinoids) (Mechoulam et al., 2014).
Plant-derived cannabinoid ligands
The most well-described agonist of cannabinoid receptors is THC or Δ9-tetrahydrocannabinol (Figure 1), which was isolated from Cannabis extracts and structurally defined more than 50 years ago (Gaoni and Mechoulam, 1964). Since then, over 500 compounds have been isolated from the plant, of which 105 have been defined as cannabinoids (Husni et al., 2014). Many of these are resorcinol derivatives and include compounds such as cannabidiol, cannabigerol, cannabinol and cannabidivarin, as well as their associated carboxylates, such as Δ9-tetrahydrocannabinolic acid and cannabidiolic acid. Evaluation of the human pharmacology of this abundance has only just begun, although the use of cannabidiol as a potential therapeutic for infantile intractable seizures is currently in advanced clinical trials (O’Connell et al., 2017). The value to the plant of this plethora of apparently unique chemicals is unknown, although there are suggestions that they may have beneficial effects against pathogens.

Structures of some endogenous, plant-derived and synthetic cannabinoids. From the top left, clockwise: AEA: anandamide (also known as N-arachidonoylethanolamine); THC: Δ9-tetrahydrocannabinol; 2AG: 2-arachidonoylglycerol; JWH133; CP55940; metAEA: R-methanandamide. In the centre of the figure is WIN, WIN55212-2.
The Cannabis plant is not the only source of natural ligands which target the ECS. As recently reviewed (Gertsch, 2017), there is the potential for multiple ‘cannabimetic’ agents to be present in the human diet. It has been suggested that these may be broadly beneficial as cardiovascular protective agents, but might also have negative implications in terms of promoting the Western diet and its impact on the expansion of obesity (Gertsch, 2017).
The consequences of a single administration of Cannabis or THC in man are relatively well characterised. A major feature of these effects is their variability, particularly of the psychological consequences, for review, see Hollister (1986). Some of the outward symptoms are more predictable, however. There is a transient increase in heart rate, with a persistent reddening of the conjunctiva. In the ‘right’ context, the perception shortly after consumption of Cannabis preparations is of a ‘high’, or euphoric period, which is following by a ‘dope’; an extended period of drowsiness. Often, this is accompanied by an impairment of short-term memory, as well as poor concentration and cognitive skills. Occasionally, in a manner which may be influenced by dose, cannabinoid combinations and/or environment (and potentially many other undefined factors), negative psychological impact has been reported, including paranoia, panic attacks and psychotic episodes. Few, if any, deaths have been attributed to single, acute Cannabis ingestion.
Although Cannabis is the most commonly used illicit substance in the world, we have less clarity on the effects of long-term administration. Due to the highly lipophilic and high protein binding of cannabinoids, THC can persist for several hours to days depending on the amount consumed and its route of administration, for review, see Hollister (1986). Therefore, abrupt cessation of Cannabis consumption is generally tolerated. An abstinence syndrome usually commences within 48 h of cessation and might last up to 2 weeks, manifesting in the form of craving, anger or aggression, fatigue, anxiety, shakiness, sweating, insomnia, vivid dreams, decreased appetite, irritability and depression.
There are many reasons for the lack of understanding of the impact of long-term, high-dose Cannabis intake. The illegal nature of Cannabis preparations in most of the world is a contributing issue, as well as the natural variation in cannabinoid content meaning that longitudinal and geographical comparisons may suffer from multiple confounding factors.
Synthetic cannabinoids
Around the time of the identification of a brain receptor for THC (Devane et al., 1988), a number of medicinal chemistry programmes were initiated in order to attempt to mimic some of the beneficial aspects of Cannabis/THC, such as pain relief (see below), without the negative aspects (the ‘high’, memory loss and dissociation from the environment). Many high-potency agonists were identified, including CP55940 and R-(+)-WIN55212-2 (Figure 1). This allowed the division of agonists into three chemical classes: classical (similar to THC, Figure 1), bicyclic (similar to CP55940, Figure 1) and aminoalkylindole cannabinoids (similar to R-(+)-WIN55212-2, Figure 1). Not only were many of these synthetic cannabinoids more potent than THC, they also displayed greater efficacy. As a result, the effects in man were very similar to the effects of higher dose THC/Cannabis and so none of these agents reached the clinic. That is, with one exception. Nabilone is a structural analogue of THC and has been licenced since 1985 for treating chemotherapy-associated nausea and vomiting. Many of the early compounds show limited selectivity between CB1 and CB2 receptors (Figure 2). More recently, agonists with good selectivity (Figure 2) for CB1 receptors (e.g. methanandamide, Figure 1) and CB2 receptors (e.g. JWH133, Figure 1) (Abadji et al., 1994; Huffman et al., 1999).

Selectivity of some endogenous, plant-derived and synthetic cannabinoids. Illustrated are calculated occupancies of CB1 and CB2 cannabinoid receptors expressed as a % of total at ligand concentrations of 100 nM. Indicated are groups of endocannabinoids, AEA and 2AG (Felder et al., 1995; Mechoulam et al., 1995), CB1-selective agonists, ACEA (N-arachidonoyl-2’-chloroethylamine) and methanandamide (Hillard et al., 1999; Lin et al., 1998), non-selective agonists, THC, CP55940, HU210, WIN55212-2 (Felder et al., 1995; Showalter et al., 1996), CB2-selective agonists, JWH133, JWH015 and HU308 (Hanus et al., 1999; Huffman et al., 1999; Showalter et al., 1996).
In Figure 2, we have indicated, based on published receptor affinities (see the legend to Figure 2 for references), the relative occupancies of human CB1 and CB2 cannabinoid receptors at 100 nM concentrations of a number of cannabinoid ligands. While the predicted occupancies of the two receptors is low (<20%) for the endocannabinoids anandamide and 2-arachidonoylglycerol, it is difficult to compare realistic estimates of ambient extracellular endocannabinoids due to the hydrophobicity of the compounds. It may be that local levels upon stimulation of the ECS (see below) may be in excess of those levels. For many investigations, both in vitro and in vivo, ‘non-selective agonists’ such as THC, CP55940 and R-(+)-WIN55212-2 (Figure 1) are used to define a role for cannabinoid receptors in general. At 100 nM, it can be seen that THC has equivalent occupancies at CB1 and CB2 receptors, albeit lower than the same concentration of CP55940. At this same concentration, R-(+)-WIN55212-2 displays almost twice the occupancy at CB2 receptors compared to CB1 receptors (Figure 2). This highlights the mild CB2-selectivity of R-(+)-WIN55212-2, a feature which is often overlooked.
The roles of either CB1 or CB2 cannabinoid receptors can be indicated through the use of more selective agonists. The figure highlights that ACEA and methanandamide provide >80% occupancy of the CB1 receptor at 100 nM, with less than 10% occupancy at CB2 receptors, illustrating the utility of this concentration of these ligands for selective CB1 receptor activation (Figure 2). Similarly, for the CB2 receptor, 100 nM concentrations of JWH133, JWH015 or HU308 provide substantial (>80%) occupancy of CB2 receptors, with much less CB1 occupancy (Figure 2).
During the 1990s, selective antagonists for both CB1 and CB2 receptors were generated by Sanofi in France (Rinaldi-Carmona et al., 1994, 1998). Rimonabant (SR141716A) and SR144528 (Figure 3) are CB1- and CB2-selective antagonists (Figure 4), respectively, and have also been described as inverse agonists, where they have the opposite effect to agonists. Equally common in their use are the antagonists/inverse agonists AM251 and AM630 to define CB1 and CB2 receptors, respectively (Figure 3). Classical antagonists are inert and act by blocking the effects of the agonist, and so any effects of the antagonists are entirely agonist dependent. In comparison, inverse agonists are effective in the absence of agonists, thought to act by stabilising a more inert form of the receptor than a tonically active basal state. A problem with the cannabinoid world is that it is very difficult to be sure that the endogenous agonists (see below) are absent (Howlett et al., 2011), and that a control situation measured in the absence of added agonists is indeed a ‘basal’ state.

Structures of CB1- (AM251 and rimonabant) and CB2-selective (AM630 and SR144528) antagonists.
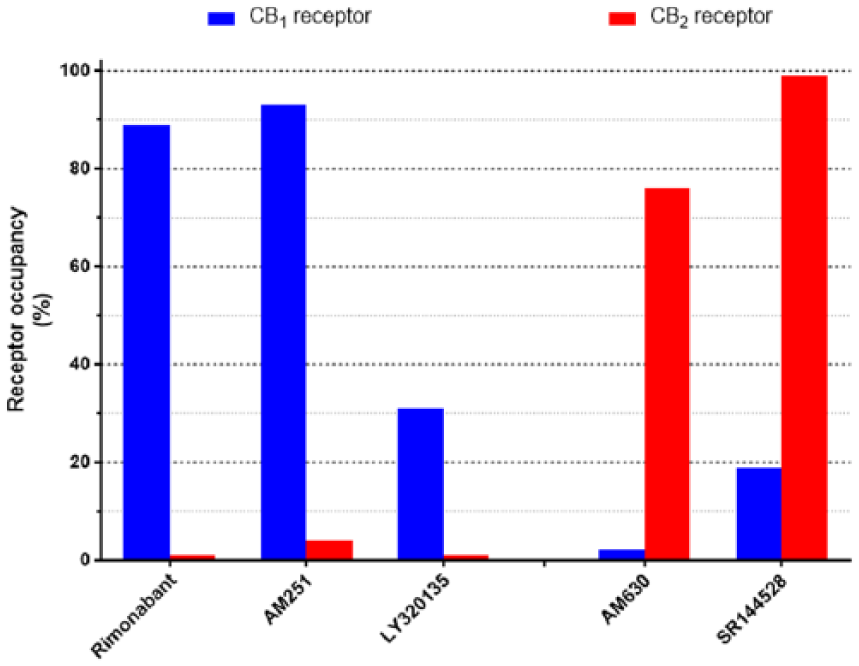
Selectivity of cannabinoid antagonists. Illustrated are calculated occupancies of CB1 and CB2 cannabinoid receptors expressed as a % of total at ligand concentrations of 100 nM. Indicated are three CB1-selective antagonists, rimonabant, AM251 and LY320135 (Felder et al., 1995, 1998; Lan et al., 1999) and two CB2-selective antagonists, AM630 and SR144528 (Rinaldi-Carmona et al., 1998; Ross et al., 1999).
In Figure 4, we have indicated, based on published receptor affinities (see the legend to Figure 4 for references), the relative occupancies of human CB1 and CB2 cannabinoid receptors at 100 nM concentrations of antagonists/inverse agonists. For the CB1-selective antagonists rimonabant and AM251, 100 nM concentrations represent a useful level of discrimination with ~90% CB1 occupancy and less than 5% CB2 receptor occupancy (Figure 4). The structurally distinct CB1 receptor antagonist LY320135, on the other hand, has a lower CB1 receptor occupancy at this concentration, although with a very much lower occupancy at the CB2 receptor. AM630 and SR144528 at 100 nM also exhibit good discrimination, for the CB2 receptor (Figure 4).
Abuse of Cannabis and synthetic cannabinoids
Cannabis ‘abuse’ has been recognised for some time; laws criminalising the possession of Cannabis preparations were passed in the United Kingdom and United States since 1928 and 1937, respectively. In many countries worldwide, attempts to circumvent the law lead to the availability of particular synthetic cannabinoids, often labelled as ‘legal highs’ or ‘designer drugs’. Many of these were based on a series of compounds synthesised originally as pharmacological tool compounds by John Huffman in the United States, with codified names such as JWH018 (Showalter et al., 1996). Similarly, HU210, a high-potency synthetic cannabinoid was synthesised in the labs of Raphael Mechoulam (1988) in Israel to investigate cannabinoid effects, but has since been used illicitly. These illegal synthetic cannabinoids are marketed with names such as K2, spice or black mamba. Although many countries have introduced legislation specifically aimed at preventing the synthesis and consumption of synthetic cannabinoids, there is a persistent issue with abuse of these synthetic cannabinoids. Reports in the mainstream media have suggested 130 suspected overdoses in 1 week in July 2016 in New York (http://edition.cnn.com/2016/07/17/health/synthetic-drug-k2-schumer-legislation/). The abuse of these synthetic cannabinoids appears to be rife among the homeless and incarcerated, presumably as a mechanism to ‘fill the time’. In 2016, over 30 individuals were suggested to have become intoxicated using a synthetic cannabinoid termed AMB-FUMINICA (sold as AK-47 24-Karat Gold) (Adams et al., 2017), leading to a popular media description of a ‘zombie outbreak’. Causes of death associated with these synthetic cannabinoids have been attributed to a mixture of CNS and cardiac events. The toxicokinetics of these agents is only just being addressed; often the compounds are sprayed onto plant material and smoked, giving rise to complex metabolites (Thomas et al., 2017). It remains to be established whether the lethal effects of these synthetic cannabinoids is due to ‘on-target’ effects via CB1 or CB2 cannabinoid receptors, either independently or as the result of combination with other substances/targets. Given the relative paucity of toxicity observed previously in the many studies using these agents in animal models, it seems more likely that an ‘off-target’ effect is responsible, through an as yet undefined receptor or ion channel, for example.
Endogenous cannabinoid ligands
As identified above, endogenous cannabinoids fall into two classes, esters and amides. The first putative endocannabinoid to be identified was named anandamide, also known as N-arachidonoylethanolamine (AEA, ananda is the Sanskrit word for bliss and amide describes the chemical bond in its structure – Figure 1), which was initially detected in porcine brain (Devane et al., 1992). AEA is a partial agonist at both CB1 and CB2 receptors (Mackie et al., 1993). A second endocannabinoid, 2-arachidonoylglycerol (2AG, Figure 1), was identified in short order, both of these are lower affinity than many of the synthetic agonists (Figure 1). There are differences in the relative efficacy of these two endocannabinoids in that 2AG appears to be a full agonist, while AEA appears to have partial agonist properties at both receptors (Mechoulam et al., 1995; Sugiura et al., 1995). The significance of this differential efficacy has been interpreted to be that signalling through the cannabinoid receptors is complicated; with a dependence not only on the absolute concentration of a single agonist, but with a dependence on the individual agonist concentrations and also on their concentrations relative to each other (Gonsiorek et al., 2000).
Turnover of the two endocannabinoids is, for the main, independent of each other (Figure 5). Although alternative pathways have been suggested, the best understood pathway of AEA biosynthesis involves a minor phospholipid component, N-arachidonoylphosphatidylethanolamine, generated by an N-acyltransferase (Natarajan et al., 1982). A selective N-acylphosphatidylethanolamine phospholipase D (NAPE-PLD) activity is able to generate phosphatidic acid and AEA (Figure 5) (Schmid et al., 1983). Termination of AEA signalling is mediated by hydrolytic enzymes, where fatty acid amide hydrolase (FAAH) appears to be a primary regulator of AEA (and other N-acylethanolamine) levels inside the cell (Schmid et al., 1985). N-Acylethanolamine acid amidase (NAAA) is a lysosomal enzyme, particularly associated with macrophages (Ueda et al., 1999), which is also able to hydrolyse N-acylethanolamines, including AEA. FAAH2, in contrast to FAAH and NAAA, is not found in murids, but is found in human (Wei et al., 2006), and is also able to hydrolyse AEA and other N-acylamides, with a physical association with intracellular lipid droplets (Kaczocha et al., 2010). The differential tissue and subcellular locations of these enzymes is presumed to regulate AEA (and other N-acylamides) in a differential manner.

A schematic overview of the endogenous cannabinoid system. Illustrated are the three major phases of endocannabinoid turnover: synthesis, hydrolysis and transformation. The two major synthetic enzymes are diacylglycerol lipase (DAGL) and N-acylphosphatidylethanolamine phospholipase D (NAPE-PLD). 2-Arachidonoylglycerol (2AG) hydrolysis may be mediated via monoacylglycerol lipase (MAGL), αβ hydrolase 12 (ABHD12) or αβ hydrolase 6 (ABHD6). In parallel, anandamide (AEA) can be hydrolysed by fatty acid amide hydrolase (FAAH), FAAH2 or N-acylethanolamine acid amidase (NAAA). Both 2AG and AEA can be transformed into apparently independently bioactive metabolites through cyclooxygenase-2 (COX-2), lipoxygenase (LOX) or epoxygenase/cytochrome P450 (EPOX/CYP).
2AG biosynthesis appears to be predominantly as a result of the action of the two isoforms of diacylglycerol lipase (DAGL, Figure 5) (Bisogno et al., 2003), while DAG itself accumulates following activation of phospholipase C, which can occur following activation of particular GPCRs or growth factor receptors. In the brain, monoacylglycerol lipase (MAGL) activity is responsible for the majority of 2AG hydrolysis, while αβ hydrolase 6 (ABHD6) and αβ hydrolase 12 (ABHD12) appear to have more minor roles (Blankman et al., 2007). As MAGL is predominantly cytosolic (albeit associated reversibly with membranes) and ABHD6 is through to be membrane-bound, there is a potential for differential subcellular regulation of 2AG (and other monoacylglycerols) levels.
NAPE-PLD has been suggested to have a presynaptic location, while DAGL is suggested to be perisynaptic, associated with postjunctional dendritic spines (Yoshida et al., 2006).
One area of convergence of the ester and amide lineages of endocannabinoid turnover is oxidative metabolism via cyclooxygenase-2, lipoxygenase and epoxygenase (cytochrome P450) metabolism (Figure 5) (Urquhart et al., 2015). Not only is arachidonic acid (AA, the product of 2AG and AEA hydrolysis, Figure 5) a substrate for these enzymes, but both AEA and 2AG, as congeners of AA, are also substrates. The products of oxidative metabolism of AA and their actions are reasonably well-described, so while it is apparent that the oxidative metabolites of AEA and 2AG are bioactive, their precise molecular mechanism(s) of action are unknown.
Selective inhibitors have been described for many, although not all of these enzymes. Thus, selective high-potency inhibitors for MAGL, ABHD6, FAAH/FAAH2, NAAA and DAGL, but not NAPE-PLD or ABHD12, have been used both in vitro and in vivo to attempt to understand the roles of these enzymes in physiology and pathology.
Therapeutic potential of targetting the ECS
The broad distribution of the ECS lends itself to exploration of multiple areas of human diseases and disorders; for review, see Alexander (2016). As mentioned above, nabilone has been used for decades as an anti-emetic, while one of the isomers of THC has itself been used for anti-emetic purposes and for stimulation of appetite in patients with terminal illnesses, such as HIV/AIDS. A plant extract, which contains a mixture of cannabinoids, predominantly THC and cannabidiol, termed Sativex/Nabiximols, is available for the treatment of multiple sclerosis and cancer-related pain. Cannabidiol itself is in trials to treat childhood seizure syndromes.
There are a further range of indications for which cannabinoid-related drugs may hold promise. From studies of Cannabis use, the most promising treatment appears to be for pain relief. As mentioned above, there is a lack of clarity about the most responsive indications due to the inherent variability of the natural product.
The CB1 antagonist rimonabant was licenced for less than 2 years in Europe, but not the United States, for treating obesity/metabolic disorder. It was successful, inasmuch as there were substantial and sustained reductions in weight and waist circumference. However, withdrawal of the drug was prompted by frequent reports of depression and suicidal ideation. The mechanism of rimonabant action was thought to involve both central and peripheral sites. Centrally, rimonabant was proposed to target the reward pathway, which is consistent with the observation that palatable food intake is stimulated by cannabinoid agonists and reduced by cannabinoid antagonists. A secondary indication for rimonabant was lined up for assisting in smoking cessation, which again is thought to be mediated by the central reward pathway.
Satiety, metabolic disorder and obesity remain areas of interest, despite the failure of rimonabant, although the current trend appears to be for peripherally restricted antagonists, driven largely by academic, rather than industrial, studies. Glaucoma is an area where cannabinoids have been administered in the past to reduce intraocular pressure and which shows promise for future exploitation (Cairns et al., 2016). Further areas of intense interest for cannabinoid therapeutic potential include mental/psychological disorders, such as schizophrenia, attention-deficit hyperactive disorder, stress and anxiety, as well as neurodegenerative disorders, cancer, inflammatory and immune disorders, bone and cardiovascular disorders.
Targetting the hydrolase enzymes is an attractive alternative to supplementing with a cannabinoid receptor agonist. As the latter results in global activation of receptors irrespective of the level of endogenous tone, this has been used as a rationalisation for the lack of success of full agonists in the clinic. Using enzyme inhibitors, however, leads to an amplification of endocannabinoid tone ensuring that those pathways at which cannabinoid receptors are being activated are enhanced without altering inactive pathways. There has, however, been limited application of ECS enzyme-targetting agents in human; one such investigation of a selective FAAH inhibitor in knee osteoarthritis was discontinued through lack of efficacy (Huggins et al., 2012). A further amplificatory mechanism which shows potential is the use of positive allosteric modulators (Abood, 2015), although these are some distance from therapeutic exploitation in the cannabinoid system.
Cannabis in the clinic
Many countries and some states in the United States have allowed Cannabis preparations to become available for medicinal usage. As indicated above, the most evidence-based reasons for prescribing these are for pain and seizures, but there are trials ongoing associated with relief from anxiety and post-traumatic stress disorder, for example. While within a clinical trial, reproducibility levels are likely to be relatively high, there is likely to be more variability between trials. This variability emanates from multiple factors, including natural inter-individual variation in pharmacokinetics, but a prominent variation lies in the composition of the Cannabis preparation itself. As indicated above, there may be as many as 105 cannabinoids in the Cannabis plant and these will likely vary in their dosage dependent on the plant cultivar, growth conditions, the part of the plant harvested, the method of harvesting, storage, refinement, preparation and administration. This complicates the interpretation of such studies. Even with good quality control mechanisms and standardisation of Cannabis preparations, there are issues about how a suitable placebo might be constructed and administered for a rigorous clinical trial.
Despite the problems with standardisation, there are potential ethical reasons to support the wider availability of Cannabis; this is in the area of ‘harm reduction’. The argument proposes that although Cannabis usage long term may not be ‘safe’, it may be less damaging than some alternatives, such as the synthetic cannabinoids and opioids (Lucas, 2017).
Conclusion
Even though the human relationship with Cannabis has been ongoing for millenia, our knowledge of the complexity of the ECS is still limited, meaning that we have yet to exploit naturally occurring and synthetic agents which target this system to their full potential.
Footnotes
Acknowledgements
The authors thank the University of Philadelphia in Jordan for the scholarship for Wafa Hourani.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship and/or publication of this article.