
Abstract
Directed evolution offers opportunities to improve promiscuous activities of hydrolases in rounds of diversity generation and high-throughput screening. In this article, we developed and validated a screening platform to improve the perhydrolytic activity of proteases and likely other hydrolases (e.g., lipases or esterases). Key was the development of a highly sensitive fluorescent assay (sensitivity in the µM range) based on 3-carboxy-7-hydroxycoumarin (HCC) formation. HCC is released through an hypobromite-mediated oxidation of 7-(4′-aminophenoxy)-3-carboxycoumarin (APCC), which enables for the first time a continuous measurement of peroxycarboxylic acid formation with a standard deviation of 11% in microtiter plates with a wide pH range window (5–9). As example, subtilisin Carlsberg was subjected to site saturation mutagenesis at position G165, yielding a variant T58A/G165L/L216W with 5.4-fold increased kcat for perhydrolytic activity compared with wild type.
Introduction
Peroxycarboxylic acids (R-CO-O-O-H) are oxidants used for bleaching and disinfection, 1 for instance, in the agricultural industry, food establishments, and medical facilities. 2 In chemical synthesis, peroxycarboxylic acids are employed in the production of epoxides 3 and oxidation of alcohols. 4 Despite their attractive synthesis potential, peroxycarboxylic acids are limited in applications due to hydrolysis in aqueous solutions 5 and their explosive “nature” in concentrated form. 6 In situ generation of peroxycarboxylic acids by hydrolysis of esters or amides in the presence of hydrogen peroxide is an alternative approach for producing active peroxycarboxylic acids when required in applications. 7
Peroxycarboxylic acid production employing hydrolases has been reported for proteases,8,9 lipases, 10 and esterases, 11 which are mainly used in detergent formulations. In a patent application, 8 the reengineering of proteases for peroxycarboxylic acid production was reported for the first time. An esterase from Pseudomonas fluorescens (PFE) has been reengineered to improve the ratio of perhydrolytic to hydrolytic activity up to 2600-fold. 12 Semirational design was used to generate PFE variants (L29P, F57H, F93H, D99E, F125A, F227I) by site-directed mutagenesis. Esterase variants were purified and characterized based on conversion of monochlorodimedone. Total activities for the production of peroxyacetic acid of four selected esterase variants ranged from 5.1 to 13 s−1 compared to 0.12 s−1 of the wild-type esterase. 13
Directed hydrolase evolution offers opportunities to improve promiscuous activities of hydrolases in iterative rounds of diversity generation and high-throughput screening (HTS).14,15 Microtiter plate–based screening systems offer a throughput of at least a few thousand clones per round, which have often proven to reengineer enzyme properties efficiently. 16 Table 1 summarizes methods for determining and quantifying peroxycarboxylic acid concentrations. In general, peroxycarboxylic acids are determined by titration methods ( Table 1 : no. 1–317–19), chromatography methods ( Table 1 : no. 4–620–22), absorbance ( Table 1 : no. 7–1123–27), and fluorescence ( Table 1 : no. 12 28 ) detection. Titration methods imply hydrogen peroxide titration followed by a iodometric determination of the peroxycarboxylic acid concentration17,18 or a direct iodometric determination under acidic conditions. 19 All chromatography methods are limited in throughput and detect peroxycarboxylic acid formation directly in chromatograms ( Table 1 : no. 5 21 ), as well as employ methyl-p-tolyl sulfide ( Table 1 : no. 4 20 ) or 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid; ABTS) after peroxycarboxylic acid isolation and purification ( Table 1 : no. 6 22 ). Sensitivity reaches values below 1 µM. 21 Colorimetric detection systems ( Table 1 : no. 7–1123–27) are routinely used for determining the amount of enzymatically produced peroxycarboxylic acids. Employed chromophores comprise azo-dyes, 23 iodine, 24 or ABTS.25–27 In a liquid system, sensitivity values reached 1.0 µM with a linear detection range from 2.5 to 100 µM, 25 whereas in agar plates, a semi-quantitative detection has been reported (50–500 µM detection range). 23 Only one fluorometric detection system has been reported for peroxycarboxylic acid determination. 28 The latter is based on kojic acid oxidation, which is mediated through hypobromite formation ( Table 1 : no. 10 28 ). So far, activities of purified and semi-purified heme and nonheme haloperoxidase have been determined in cuvettes with the kojic acid oxidation assay. 29 Only the ABTS detection systems ( Table 1 : no. 9 25 and 11 27 ) have been downscaled into 96-well plates and used as endpoint assay for peroxycarboxylic acid quantification.
Methods for Determination and Quantification of Peroxycarboxylic Acids
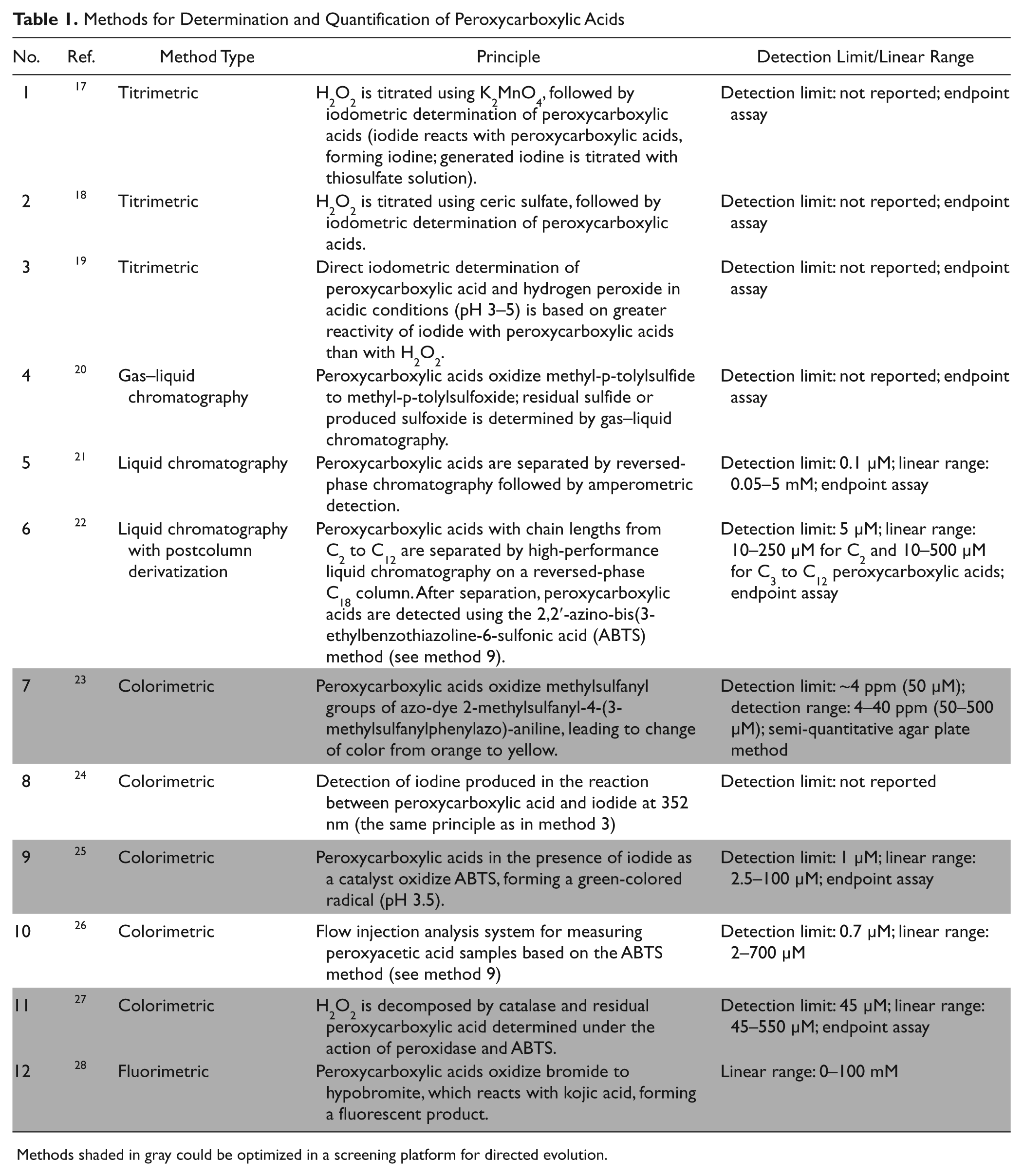
Methods shaded in gray could be optimized in a screening platform for directed evolution.
In this report, we describe a validated microtiter plate–based HTS system (96-/384-well format) for the directed evolution of proteases into peroxycarboxylic acid producers. Key was the development of a highly sensitive fluorescent assay (µM range) based on 3-carboxy-7-hydroxycoumarin (HCC) formation. HCC is released through a hypobromite-mediated oxidation of 7-(4′-aminophenoxy)-3-carboxycoumarin (APCC), which enables for the first time a continuous measurement of peroxycarboxylic acid formation. In addition, the APCC detection system can be employed in expression media without purification steps in a pH range from 5 to 9.
Materials and Methods
Materials
All chemicals were of analytical reagent grade or higher quality and purchased from Fluka, Sigma-Aldrich (Steinheim, Germany) or AppliChem (Darmstadt, Germany), with the exception of APCC (see below APCC synthesis). The peroxyacetic acid (PAA) solution was purchased from Sigma-Aldrich (32% PAA). All enzymes were purchased from New England Biolabs GmbH (Frankfurt, Germany) and Fermentas GmbH (St. Leon-Rot, Germany).
Thermal cycler (Mastercycler gradient; Eppendorf, Hamburg, Germany) and thin-wall PCR tubes (Multi-ultra tubes; 0.2 mL; Carl Roth GmbH, Karlsruhe, Germany) were used in all PCRs. The PCR volume was always 50 µL. The amount of DNA in cloning experiments was quantified using a NanoDrop photometer (ND-1000; NanoDrop Technologies, Wilmington, DE). An Infinite M1000 (Tecan Group AG, Zürich, Switzerland) plate reader was used for detection of fluorescence, and Sunrise (Tecan Group AG) was used for absorbance detection.
Subtilisin Carlsberg and subtilisin Carlsberg T58A/L216W genes were kindly provided by Henkel AG & Co. KGaA (Düsseldorf, Germany).
APCC Synthesis
APCC synthesis was performed as reported 30 with an overall yield of 6%. Structure of APCC was confirmed by 1 H-NMR analysis (Bruker AMX 300; Bruker BioSpin, Rheinstetten, Germany).
General Reaction Conditions
Standard reactions were performed in 96-well microtiter plates (polystyrene black flat 96-well no. 655076; Greiner Bio-One GmbH, Frickenhausen, Germany) in sodium phosphate buffer (100 mM, pH 7.5; final volume of 100 µL) with the following composition: PAA solution (0.05–100 µM), varied concentrations of sodium bromide (10, 50, 100, or 400 mM), and APCC (0.5 mM). Finally, the following conditions with varied hydrogen peroxide concentrations in sodium phosphate buffer (100 mM, pH 7.5; final volume of 100 µL) were employed: PAA standard solutions (0.05–100 µM), sodium bromide (100 mM), APCC (0.5 mM), and varied concentrations of hydrogen peroxide (1, 10, 30, or 100 mM). APCC conversions were measured by fluorescence intensity (382 nm [ex.] and 445 nm [em.], gain 100; 30 °C) using an Infinite M1000 microtiter plate reader. Reported values are the average of three measurements with average deviations from the mean values (10-min incubation; 30 °C). All prepared PAA solutions were used within 1 h.
A final calibration curve, fluorescence build up versus peroxycarboxylic acid formation, was recorded in 96-/384-well microtiter plates (polystyrene black flat 96-/384-well no. 655076/no. 781076; Greiner Bio-One GmbH) in sodium phosphate buffer (100 mM, pH 7.5) containing a PAA standard solution (0.05–100 µM), sodium bromide (100 mM), and APCC (0.5 mM) in a final volume of 100 µL/40 µL.
Stability of HCC in the presence of peroxyacetic acid and hydrogen peroxide was determined to ensure a reliable determination of peroxycarboxylic acid production (see Results section). Fluorescence data were analyzed using GraphPad Prism software (GraphPad Software, San Diego, CA).
Cloning of Subtilisin Carlsberg and Variant T58A/L216W Genes into the Shuttle Vector pHY300PLK
Subtilisin Carlsberg and its variant T58A/L216W (along with its pre-pro-sequence and promoter) were cloned into pHY300PLK shuttle vector (Takara Bio, Inc., Shiga, Japan). Both genes were amplified (95 °C for 30 s, one cycle; 95 °C for 30 s, 57 °C for 30 s, and 72 °C for 90 s, 24 cycles; 72 °C for 3 min, one cycle) using primers (5′-GATGGATCCCCGGGACCTCTTTC-3′ and 5′-TAGT CTAGATTATTGAGCGGCAG-CTTCG-3′), Pfu DNA polymerase (1.5 U), dNTP mix (0.20 mM), and 25 ng plasmid template. The amplification product was purified using the QIAquick PCR Purification Kit (Qiagen GmbH, Hilden, Germany), followed by digestion using BamHI (20 U) and XbaI (20 U) restriction enzymes, and again purification (QIAquick PCR Purification Kit). The (shuttle) cloning vector pHY300PLK was also digested using BamHI (20 U) and XbaI (20 U) restriction enzymes and purified using the QIAquick PCR Purification Kit. Digested subtilisin Carlsberg genes (wild type and variant T58A/L216W) and vector pHY300PLK were ligated using T4 DNA ligase (1 U), resulting in pHY300Car (wild type) and pHY300Per (variant T58A/L216W). Plasmid constructs were subsequently transformed into Escherichia coli DH5α, isolated, and sequenced. pHY300Car and pHY300Per were transformed subsequently into Bacillus subtilis DB104.
Site Saturation Mutagenesis at Position G165
Site saturation mutagenesis of the subtilisin Carlsberg variant T58A/L216W at position G165 was performed as described. 31 The following oligonucleotides were used for mutagenesis: 5′-GAAACACGAATACAATTNNKTATCC TGCGAAATACGA-3′ and 5′-TCGTATTTCGCAGGATA MNNAATTGTATTCGTGTTTC-3′. PCR conditions: First step: 98 °C for 30 s, one cycle; 98 °C for 10 s, 55 °C for 30 s, and 72 °C for 3 min, four cycles. Second step: 98 °C for 30 s, one cycle; 98 °C for 10 s, 55 °C for 30 s, and 72 °C for 3 min, 14 cycles. Third step: 72 °C for 3 min, one cycle. PCR mix contained furthermore Phusion High-Fidelity DNA polymerase (1 U; New England Biolabs GmbH), dNTP mix (0.20 mM), and both primers (25 µM each) together with DNA template (pHY300Per; 10 ng). Following the PCR, DpnI (40 U; New England Biolabs GmbH) was supplemented, and the mixture was incubated (4 h; 37 °C). PCR products were purified using a QIAquick PCR Purification Kit and transformed into E. coli DH5α. Obtained clones were pooled and plasmids isolated and transformed into B. subtilis DB104 for protease expression.
Cultivation and Expression in 96-Well Microtiter Plates
Colonies grown on LB agar plates containing tetracycline (15 µg/mL) were transferred into 96-well microtiter plates (flat-bottom, polystyrene plates; Greiner Bio-One GmbH), containing LB medium (150 µL) supplemented with tetracycline (15 µg/mL). After 24 h of cultivation in a microtiter plate shaker (Multitron II, Infors GmbH, Einsbach, Germany; 37 °C; 900 rpm; 70% humidity), a replicator tool (EnzyScreen BV, Leiden, the Netherlands) was used to inoculate an expression 96-well microtiter plate in a total volume of 200 µL LB media supplemented with potassium phosphate buffer (90 mM, pH 7.6) and 15 µg/mL tetracycline. Master plates were stored at −80 °C after addition of glycerol (final concentration 20 vol %). The expression plates were cultivated (24 h; 37 °C; 900 rpm; Multitron II, Infors GmbH) and the cell biomass separated from cell culture supernatant by centrifugation (Eppendorf 5810R, 3200 g). Expressed protease variants in the cell culture supernatants were used for activity determination.
Screening Procedure
The supernatant from the expression plate was incubated (2 h; room temperature [RT]) under conditions at which catalase activity was suppressed (3-amino-1,2,4-triazole [50 mM] and hydrogen peroxide [30 mM]) according to Tephly et al. 32 After inhibition, the reaction mixture was diluted five times with sodium phosphate buffer (100 mM, pH 7.5) and used for activity determination as described below in 96- and 384-well formats.
384-Well plate-format APCC assay for detection of perhydrolytic activity
Methyl-butyrate conversion was initiated by addition of “incubated” supernatant to the reaction mixture containing hydrogen peroxide (31.3 mM), sodium bromide (100 mM), methyl-butyrate (100 mM), APCC (0.5 mM), and sodium dodecyl sulfate (SDS; 0.9 mM) in sodium phosphate buffer (100 mM, pH 7.5; reaction volume 40 µL). Perhydrolytic activity was monitored as the increase in fluorescence intensity per minute (measured at 382 nm [ex.] and 445 nm [em.]; gain 84; 5-min reaction time; 30 °C) using an Infinite M1000 microtiter plate reader. Standard deviation of perhydrolytic activity for 96 clones was calculated from the change of fluorescence intensity per minute (ΔFI/min).
96-Well plate format for detection of proteolytic activity
Screening was performed in 96-well microtiter plates by addition of catalase-inhibited supernatant to an N-succinyl-Ala-Ala-Pro-Phe-p-nitroanilide solution (suc-AAPF-pNA, 0.22 mM) in sodium phosphate buffer (100 mM, pH 7.5; final volume 100 µL). The proteolytic activity was monitored as the increase in absorbance per minute at 405 nm (5-min reaction time; 30 °C).
Expression and Purification of Subtilisin Carlsberg and Variants
Shaking flasks (1 L) containing LB medium (250 mL) supplemented with tetracycline (15 µg/mL) were inoculated with a 1:250 dilution of overnight culture (B. subtilis DB104) harboring pHY300Car or variants. After 60 h of expression, cell biomass was removed by centrifugation (Sorvall 4 °C; 8000 g; 30 min). The resulting supernatant was filtered using a glass fiber filter (#30; Schleicher & Schuell Microscience, Dassel, Germany) and using a polyethersulfone (PES) membrane filter (0.2 µm; Sartorius, Göttingen, Germany). The supernatant was concentrated to 10 mL using a Millipore ultra-filtration stirred cell with a 10-kDa cutoff (regenerated cellulose ultra-filtration membrane; Millipore, Schwalbach, Germany). The concentrated supernatant was diluted up to 200 mL in HEPES buffer (10 mM, pH 7.0) and concentrated to 10 mL. Subtilisin Carlsberg and variants were purified by anion and subsequent cation exchange chromatography. The concentrated supernatant was loaded onto a Toyopearl Super Q 650c (TOSOH Bioscience GmbH, Stuttgart, Germany) anion exchange column (ID/length 15 × 125 mm) preequilibrated with running buffer 1 (HEPES, 10 mM, pH 7.0). The flow rate and pressure were managed by an ÄKTA prime system (Amersham, GE Healthcare Europe, Freiburg, Germany) using the Unicorn software package. Subtilisin Carlsberg and variants did not bind to the anion exchange column in contrast to many impurities and were collected in the flow-through (buffer 1; flow rate of 3 mL/min). Collected subtilisin Carlsberg and variants were loaded on a Toyopearl Super SP-650c (TOSOH Bioscience GmbH) cation exchange column (ID/length 15 × 125 mm) that was preequilibrated with running buffer 2 (HEPES, 20 mM, pH 7.0). Subtilisin Carlsberg and variants were eluted with 4 % NaCl in running buffer 2 (flow rate 1.5 mL/min). Purified samples were concentrated (Amicon ultra-4 centrifugal filter device [Millipore]; 10-kDa cutoff membrane), the total protein content determined (BCA assay kit; Pierce, Born, Germany), and the homogeneity analyzed by an Experion Pro260 Analysis Kit (Bio-Rad, Munich, Germany).
Characterization of Subtilisin Carlsberg Wild Type and Variants
The kcat and KM values were determined from initial velocity data measured as a function of methyl-butyrate concentration at 30 °C in 96-well microtiter plates (black, flat-bottomed, polystyrene plates; Greiner Bio-One GmbH). After 10 min of preincubation at 30 °C, the perhydrolytic reaction was initiated by the addition of purified protease or variant (0.15 µM) to the substrate solution containing methyl-butyrate (10–200 mM), hydrogen peroxide (100 mM), sodium bromide (100 mM), and APCC (0.5 mM) in sodium phosphate buffer (100 mM, pH 7.5; final volume 100 µL). Fluorescence was measured at 382 nm (ex.) and 445 nm (em.) (gain 84; 30 °C) using an Infinite M1000. The initial velocity data were fitted using GraphPad Prism software (GraphPad Software; hyperbolic fitting). The kcat was calculated from the ratio of Vmax and protease concentration.
Results
The Results section is divided into three parts to introduce the developed APCC detection system for quantification of peroxycarboxylic acid formation in a 96-/384-well plate format and to validate its application for directed protease evolution. In the first section, the principle of the APCC detection system is summarized. The second section provides data on the APCC performance. Investigated parameters comprise effects of sodium bromide concentration on APCC sensitivity, influence of hydrogen peroxide on PAA detection, and influence of hypobromite and PAA on HCC fluorescence. The third section describes the validation of the APCC detection system in a directed evolution experiment comprising site saturation mutagenesis of position G165 and screening two 96-well microtiter plates (192 clones) for increased perhydrolytic activity toward methyl-butyrate.
Principle of APCC Detection System
Figure 1 shows the APCC detection system for quantifying peroxycarboxylic acid concentrations. First, sodium bromide is oxidized to hypobromite by peroxycarboxylic acid, which is converted into a carboxylic acid. Subsequently, the fluorogenic substrate APCC is O-dearylated by hypobromite, releasing the fluorescent HCC and 4-imino-2,5-cyclohexadien-1-on. The excitation and emission spectra of the commercially available fluorescent product, HCC, showed maxima at 382 and 445 nm. An identical spectrum was obtained for HCC generated in the APCC detection system. Conversions in sodium acetate buffer (100 mM, pH 5.0) and sodium phosphate buffer (100 mM, pH 7.5 and pH 9.0) showed that the APCC assay can be used for the detection of peroxycarboxylic acids in a pH range from 5 to 9 with a maximized fluorescence intensity at pH 9.0.
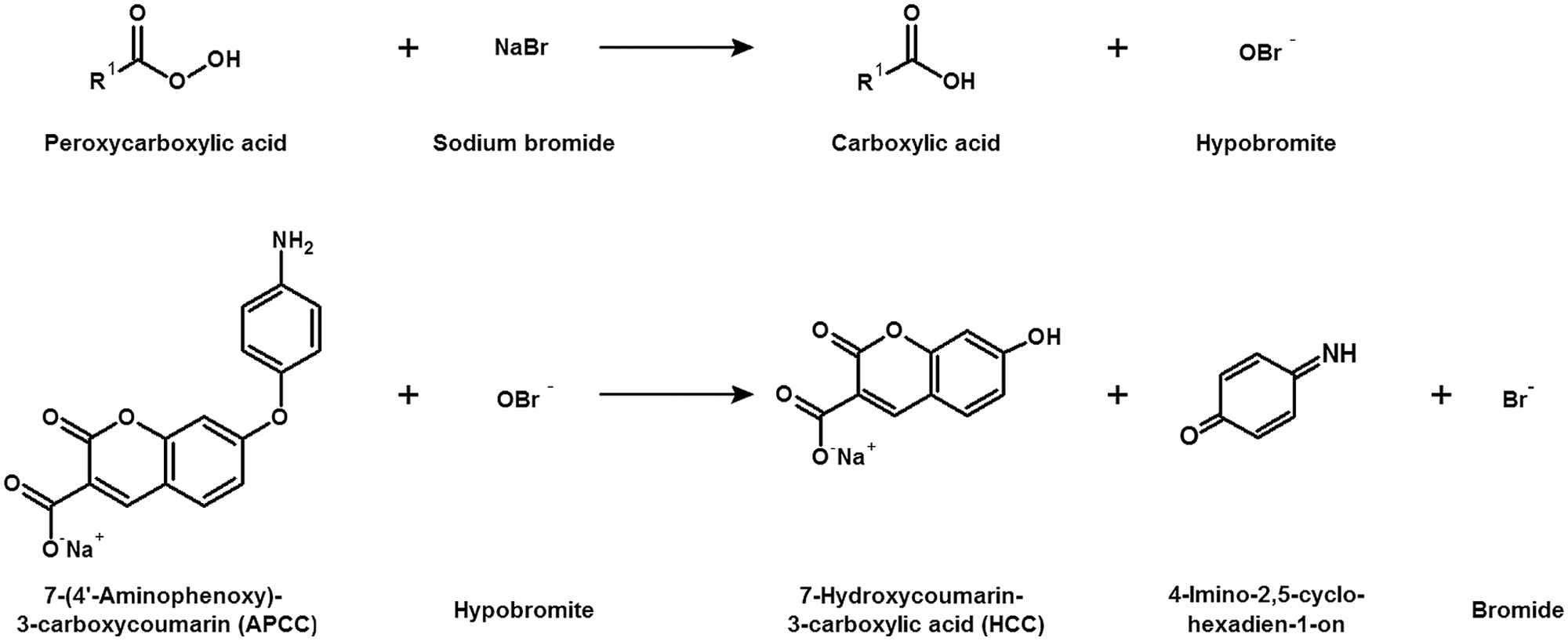
Principle of 7-(4′-aminophenoxy)-3-carboxy coumarin (APCC) detection system. Sodium bromide is oxidized to hypobromite by peroxycarboxylic acid, which is converted into a carboxylic acid. Subsequently, the fluorogenic substrate APCC is O-dearylated by hypobromite, releasing the fluorescent 7-hydroxycoumarin-3-carboxylic acid (HCC) and 4-imino-2,5-cyclohexadien-1-on.
Performance Criteria of APCC Assay
Effect of sodium bromide concentration on the APCC assay sensitivity
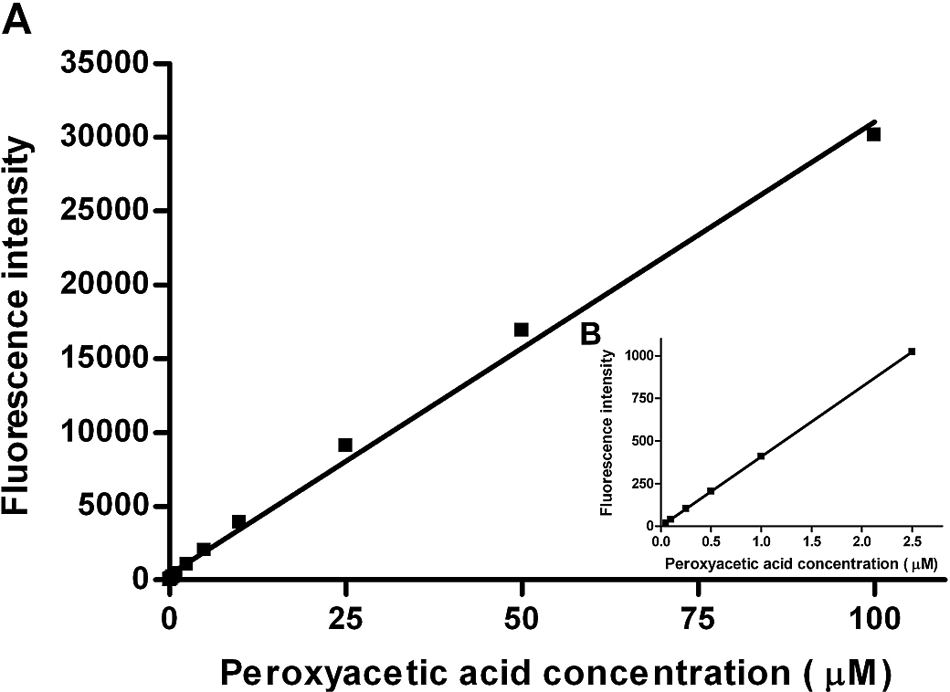
Figure 2 shows the influence of different sodium bromide concentrations (10 [■], 50 [▲], 100 [▼], and 400 [◊] mM) on peroxyacetic acid detection in the presence of APCC (0.5 mM; solubility limit in aqueous solution). Linearity between fluorescence signals to peroxyacetic acid concentrations (0.05–100 µM) is given for all four sodium bromide concentrations. A sodium bromide concentration of 50 mM is sufficient to maximize HCC product formation. In total, 100 mM sodium bromide was finally selected as standard for the APCC assay and for recording the calibration curves. The detection limit of the APCC system was determined to be 0.05 µM PAA with a linear range from 0.05 to 100 µM ( Fig. 3 ) in the presence of standard sodium bromide (100 mM) and APCC (0.5 mM) concentrations.
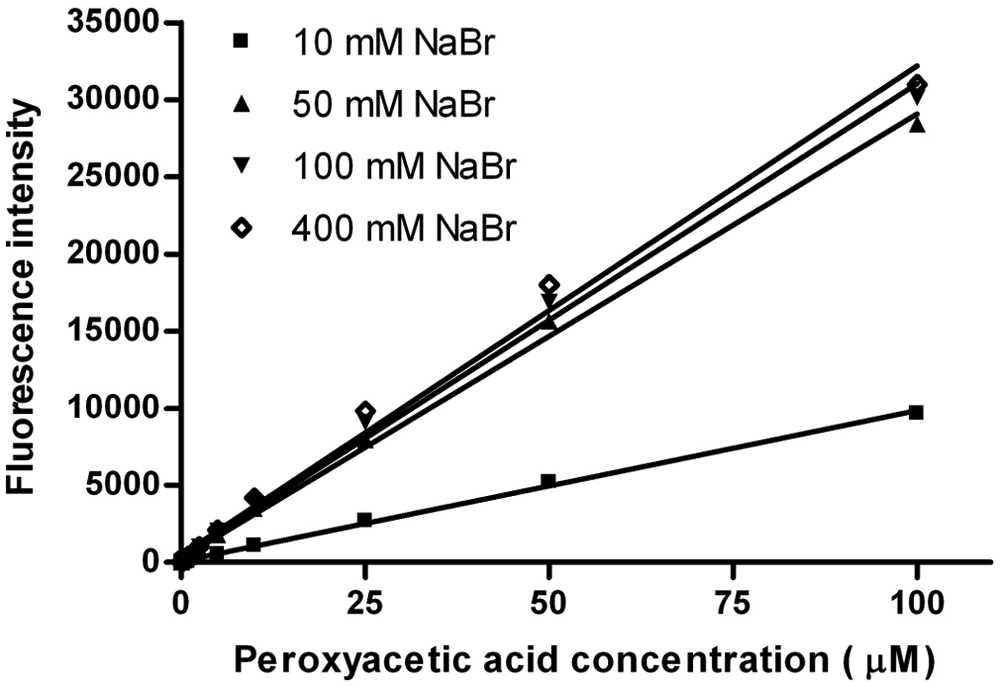
Linearity of fluorescence intensity to peroxyacetic acid concentration (0.05–100 µM) for different sodium bromide concentrations in the absence of protease with 7-(4′-aminophenoxy)-3-carboxy coumarin (APCC; 0.5 mM) in sodium phosphate buffer (100 mM, pH 7.5; final volume 100 µL). The reported values are the average of three measurements with average deviations from the mean values. The error values do not exceed the marker symbols.
(
Effect of hypobromite, PAA, and hydrogen peroxide on fluorescence intensity of HCC
In a first control experiment, the influence of hypobromite on the formed fluorophore HCC (0.05–25 µM) was investigated. Hypobromite is produced by peroxyacetic acid in the presence of sodium bromide (100 mM) in sodium phosphate buffer (100 mM, pH 7.5) at concentration levels matching at least the HCC (0.05–25 µM) concentration. Fluorescence was measured after 10 min, showing a reduction of less than 5% compared with fluorescence measured in the absence of hypobromite (see
In a second control experiment, hydrogen peroxide (30 mM and 100 mM) in the presence of HCC (0.05–25 µM) showed less than 5% reduction of the fluorescence intensity compared with HCC in the absence of hydrogen peroxide (see
In a third control experiment, the PAA concentration was gradually increased up to a 20 molar excess in the presence of HCC with no effect on fluorescence intensity (see
Effect of hydrogen peroxide on peroxyacetic acid detection
Figure 4 shows the influence of hydrogen peroxide, in the absence of protease, on the fluorescence generated from the conversion of APCC to HCC. Surprisingly, the HCC product formation is reduced when hydrogen peroxide concentration is increased for all investigated PAA concentrations (0.05–100 µM). For all four hydrogen peroxide concentrations (1 mM, 10 mM, 30 mM, and 100 mM), a linear trend is observed between fluorescence and PAA concentration. The reduced HCC fluorescence is, as controls above show, not caused directly by HCC bleaching from hydrogen peroxide or by the used PAA concentrations with a molar ratio up to 5. The sensitivity decreases up to 10-fold (from 0.05–0.5 µM) in the presence of hydrogen peroxide (100 mM). However, PAA formation can be detected in the presence of more than 10 000–fold molar excess of hydrogen peroxide.
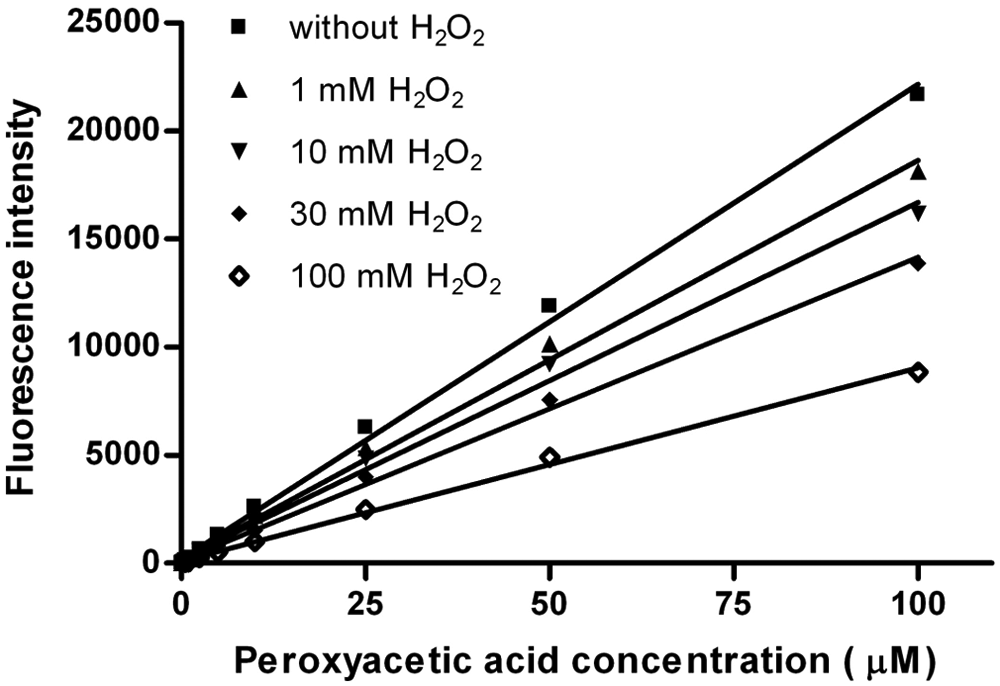
Linearity of fluorescence to peroxyacetic acid concentration (0.05–100 µM) for different concentrations of hydrogen peroxide in absence of protease with 7-(4′-aminophenoxy)-3-carboxy coumarin (APCC; 0.5 mM) and sodium bromide (100 mM) in sodium phosphate buffer (100 mM, pH 7.5; final volume 100 µL). The reported values are the average of three measurements with average deviations from the mean values. The error values do not exceed the marker symbols.
Validation of APCC Detection System in a Directed Evolution Experiment
Key performance parameters for identifying improved variants in the microtiter plate–based screening system are the standard deviation and the linear detection range of the screening system. The standard deviation of the screening system has been reduced by optimizing growth and screening conditions to 11% in the 384-well microtiter plate format ( Fig. 5A ). The optimized setup uses LB media with potassium phosphate buffer (90 mM, pH 7.6), 24-h expression, 2-h incubation for inhibition of catalase, and 5-fold dilution of supernatant with reaction buffer prior to activity determination. The detection system was validated by identifying variants of subtilisin Carlsberg with increased perhydrolytic activity by screening a site saturation mutagenesis library. Modeling studies on subtilisin Carlsberg suggested that position G165 might have an effect on substrate binding. Site saturation library at position G165 was generated and screened toward increased perhydrolytic activity using methyl-butyrate as a substrate. Variant M1 (G165L) showed a 2.9-fold increased perhydrolytic activity and a 7-fold decreased proteolytic activity compared with the parent in the cell culture supernatant ( Fig. 5B ).
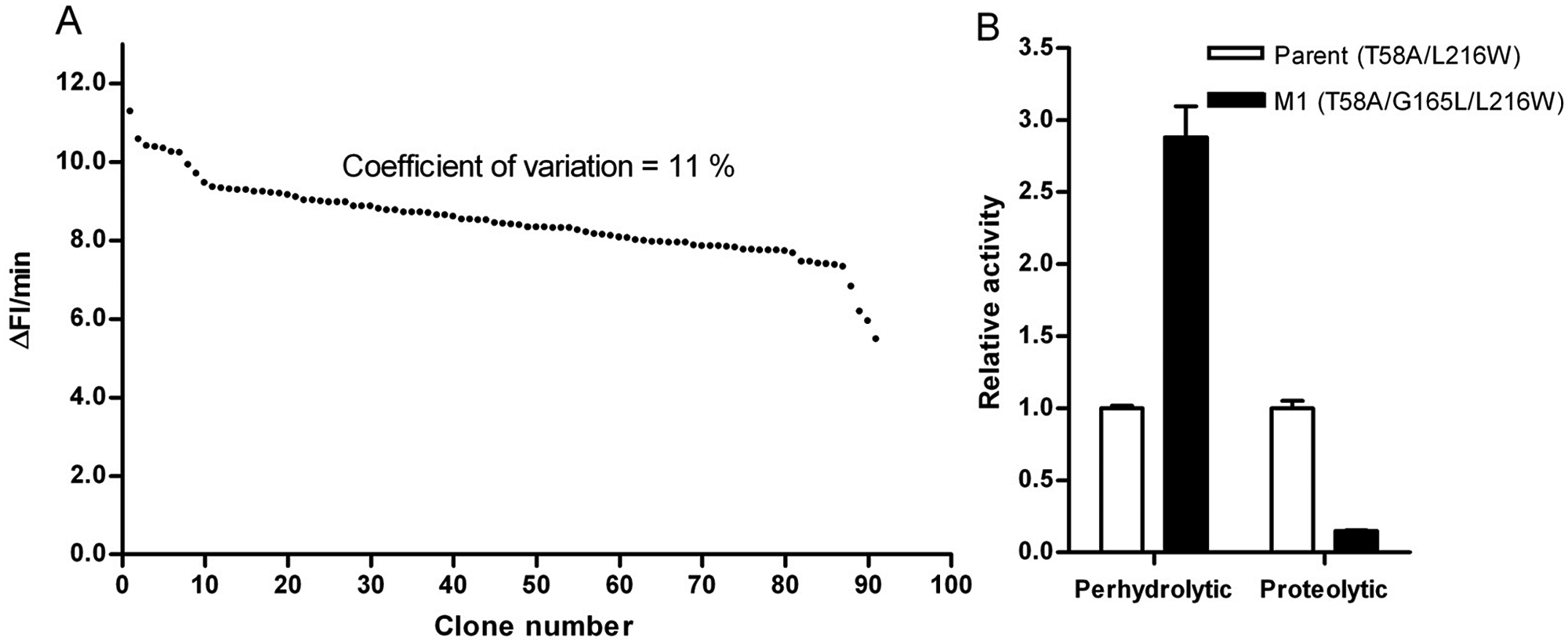
(
Subtilisin Carlsberg as well as variants T58A/L216W and M1 (T58A/G165L/L216W) were expressed in shaking-flask cultures and purified using a two-step procedure as described in Materials and Methods, and obtained purity of the variants was above 90% (see
Kinetic Parameters of Subtilisin Carlsberg and Variants for Conversion of Methyl-Butyrate to Peroxybutyric Acid and Methanol in the Presence of 100 mM Hydrogen Peroxide

The reported values are average value of three measurements, and average deviations from the mean values are shown.
Discussion
The first fluorescent-based screening system for detection of peroxycarboxylic acids in a microtiter plate was developed as part of an HTS platform for peroxycarboxylic acid–producing enzymes. The APCC method allows a continuous detection of peroxycarboxylic acids and relies on the detection of a generated fluorescent coumarin compound (see Fig. 1 ). To ensure that the screening system can be used for directed evolution, a first proof of concept was done to improve perhydrolytic activity of subtilisin Carlsberg toward methyl-butyrate. Main performance criteria for HTS systems are a low standard deviation, reproducibility, simplicity, high sensitivity, and low interference of media or reaction compounds on enzymatic activity. To determine the sensitivity of the APCC assay, peroxyacetic acid concentrations were quantified in the presence of varied concentrations of sodium bromide and APCC. In addition, the fluorescent detection product, HCC, was “treated” with bleaching agents (hydrogen peroxide, peroxyacetic acid, and hypobromite) to determine its stability.
A main challenge of peroxycarboxylic acid detection systems for directed enzyme evolution is interference of cell lysate/supernatant components with the detection system. Most peroxycarboxylic acid detection methods require enzyme purification before peroxycarboxylic acid determination, and only a few are downscaled to a microtiter plate format ( Table 1 : no. 9 25 and 11 27 ). The validated APCC screening system for directed perhydrolase evolution is the first reported continuous assay that enables detection of perhydrolytic activity without enzyme purification and has been optimized for medium- and high-throughput screening in the microtiter plate format. The standard deviation of the APCC detection system is 11% under optimized conditions. Screening systems with a standard deviation lower than 14% are routinely and successfully used in directed evolution experiments. 33 Subtilisin Carlsberg is furthermore the first protease engineered through a directed evolution experiment toward increased perhydrolytic activity, which offers novel opportunities for bleaching applications. The protease variant, T58A/G165L/L216W, identified in the current work showed a 9.4-fold increased specificity constant of perhydrolytic activity compared with the wild type. The kcat value of M1 reached (11.3 s−1) for catalyzed perhydrolysis of methyl-butyrate is already close to the kcat value (13 s−1) reported for the perhydrolysis of acetic acid of a recently engineered esterase. 13
The screening platform was developed in this work to improve protease and likely other hydrolases (e.g., lipases or esterases) having perhydrolytic activity or find new enzymes with peroxycarboxylic acid production by screening metagenome libraries. The wide pH range (5–9) and robustness (sodium bromide concentration, oxidative HCC stability) of the APCC detection system offer a broad flexibility in terms of assay conditions. In summary, we hope that the APCC screening system will extensively be employed in directed evolution generating hydrolases for industrial applications in bleaching.
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the German government through the Bundesministerium für Bildung and Forschung (Bioindustrie-2021, FKZ0315250) and Henkel AG & Co. KGaA.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.