
Abstract
Protein degradation via the ubiquitin-proteasome pathway is important for a diverse number of cellular processes ranging from cell signaling to development. Disruption of the ubiquitin pathway occurs in a variety of human diseases, including several cancers and neurological disorders. Excessive proteolysis of tumor suppressor proteins, such as p27, occurs in numerous aggressive human tumors. To discover small-molecule inhibitors that potentially prevent p27 degradation, we developed a series of screening assays, including a cell-based screen of a small-molecule compound library and two novel nucleotide exchange assays. Several small-molecule inhibitors, including NSC624206, were identified and subsequently verified to prevent p27 ubiquitination in vitro. The mechanism of NSC624206 inhibition of p27 ubiquitination was further unraveled using the nucleotide exchange assays and shown to be due to antagonizing ubiquitin activating enzyme (E1). We determined that NSC624206 and PYR-41, a recently reported inhibitor of ubiquitin E1, specifically block ubiquitin-thioester formation but have no effect on ubiquitin adenylation. These studies reveal a novel E1 inhibitor that targets a specific step of the E1 activation reaction. NSC624206 could, therefore, be potentially useful for the control of excessive ubiquitin-mediated proteolysis in vivo.
Introduction
Protein ubiquitination and subsequent degradation regulates almost every aspect of eukaryotic cellular function. 1 A failure to remove certain proteins or, conversely, excessive protein degradation has been implicated in many human diseases, including cancer and disorders of the brain. Consequently, there is considerable interest in targeting the ubiquitin proteasome system (UPS) for the development of therapeutics. 2 Early success with the proteasome inhibitor bortezomib, carried out by Millennium Pharmaceuticals (Cambridge, MA) and used to treat relapsed multiple myelomas, has validated the UPS as a target for inhibition of cancer and opened up new avenues to look for other candidate targets and molecules.3,4 The fact that bortezomib inhibits the entire ubiquitin system indicates that relatively nonselective inhibitors of the UPS might prove useful to combat cancer, encouraging the exploration of additional points of intervention within the pathway.
Cyclin-dependent kinase inhibitor p27 is mainly regulated at the level of protein stability. 5 In many cancers, control of p27 levels is perturbed, resulting in increased polyubiquitination via the SCFSkp2 E3 ubiquitin ligase and subsequent proteasomal degradation. 6 The resulting increase in CDK2 activity initiates a cascade of biological events that culminates in the promotion of G1/S progression and increased cell growth rate. A drastic reduction of p27 caused by excessive degradation has been detected in approximately 50% of all human cancers, and decreased p27 expression correlates with poor prognoses in cancer patients. 7 In an attempt to identify substances that would block p27 degradation and regulate the cell cycle, Nickeleit et al. 8 introduced a cyclic peptide, argyrin A, that inhibited the catalytic activity of the proteasome. Also, Chen et al. 9 reported a compound, named Compound A (CpdA), that prevented incorporation of Skp2 into the SCFSkp2 ligase complex, causing a reduction of p27 ubiquitination in vitro. Inhibition of SCFSkp2 also induced G1/S cell cycle arrest and caspase-independent apoptosis in several myeloma cell lines. 9 Thus, small-molecule inhibitors that promote stabilization of p27 are desirable and could potentially have therapeutic value.
Sitting on top of the ubiquitination cascade is the ubiquitin-activating enzyme E1. 1 Two E1 enzymes have been described; however, UBA1 appears to be the E1 enzyme responsible for the majority of ubiquitin-mediated processes.10,11 Although E1 is a particularly attractive target for drug discovery, few efforts have been devoted to developing ubiquitin E1 inhibitors. Recently, Yang et al. 12 reported the first small-molecule inhibitor of E1, the pyrazone derivative PYR-41, although the mechanism by which PYR-41 inhibits E1 remains unknown. In addition to inhibiting ubiquitination, PYR-41 caused an increase of p53 in cancer cells upon treatment, resulting in p53-mediated apoptosis. 12 Similarly, the nitric oxide prodrug JS-K reduced E1 activity by decreasing total cellular ubiquitination and elevating expression of wild-type p53, a critical step for the stimulation of an anticancer response. 13 Also, MLN4924 is a small molecule that potently inhibits the NEDD8 activating enzyme (NAE), which regulates the activity of the cullin family of proteins. 14 In their report, Soucy et al. 14 show that the inhibition of the NAE pathway triggers apoptosis in cancer cells through the deregulation of DNA synthesis during S-phase. When MLN4924 was evaluated in mice, a significant growth suppression of lung tumor xenografts was observed at doses that were well tolerated. A closer look at the mechanism of MLN4924 inhibition suggested that MLN4924 covalently attaches to NEDD8, mimicking a NEDD8 adenylate, but one that is incapable of driving the reaction forward, thereby blocking the activity of NAE. 15 Altogether, these reports provide a proof of concept that E1 inhibitors can serve as promising cancer therapeutics.
The general mechanism of E1 activation is well established. 1 First, adenosine triphosphate (ATP) initiates the reaction by conjugating ubiquitin to E1, producing an E1-ubiquitin-adenylate and pyrophosphate (PPi). In the second step, a cysteine thiol group attacks the adenylated ubiquitin, which directs the formation of a thioester bond between the C-terminus of ubiquitin and the E1 and the subsequent release of adenosine monophosphate (AMP). In the third step, another ATP and ubiquitin are recruited to the adenylation site, generating a fully loaded E1 that carries two molecules of activated ubiquitin (covalent thioester and noncovalent adenylate, respectively).16,17 In this form, E1 associates with an E2 (ubiquitin-conjugating enzyme) to ultimately pass the ubiquitin to a target protein via ubiquitin ligase complexes (E3s).
Here we report the discovery, synthesis, and initial characterization of an asymmetric disulfide small-molecule compound NSC624206, later synthesized and referred to as
Materials and Methods
Construction of Kip16, a GFP-p27-Expressing Cell Line
Mink lung epithelial cells expressing GFP-p27 were generated by retroviral-mediated gene transfer. Briefly, pBabe-GFP-p27 amphotropic virus was made by cotransfecting pBabe-GFP-p27-Puro with pCL-Ampho in 293T cells. Viral supernatant was collected and used to infect the mink lung epithelial cell line Mv 1 Lu (CCL-64) from ATCC in the presence of 8 µg/mL polybrene. Puromycin was added at 5 µg/mL, and stable clones were selected. Each clone was subcultured and tested for GFP-p27 expression in the presence or absence of 10 µM MG132 (Calbiochem, San Diego, CA) for 24 h. Clones expressing high levels of green fluorescent protein (GFP) in the presence of MG132 but low or nondetectable GFP in its absence were expanded. Immunoblotting using an anti-p27 antibody (Santa Cruz Biotechnology, Santa Cruz, CA) was used to confirm the expression of the GFP-p27 fusion protein and stabilization of GFP-p27 upon MG132 treatment. Several clones were picked and characterized. Kip16, which showed the most dynamic change in GFP-p27 levels in response to MG132 treatment while exhibiting low background fluorescence, was selected for further studies.
Screening for Inhibitors of p27 Proteolysis in Kip16
Kip16 cells were dispensed into 96-well flat clear-bottomed plates at 40 000 cells/well in 100 µL medium and incubated overnight at 37 °C in a humidified 5% CO2 atmosphere. Compounds were then added to a final concentration of 3 or 30 µM in 300 µL fresh medium. The first column of the plate received 0.3% DMSO only (negative control), whereas the cells in the last column were treated with 1 µM MG132 (positive control). Eighty compounds per plate were directly transferred from the National Cancer Institute (NCI) stock plate (see Compound Library section) using a multichannel pipette. After 24 h of incubation, the medium was removed and cells were washed twice with phosphate-buffered saline (PBS). Cells were left in the second PBS wash, and GFP fluorescence was quantified on a Tecan Safire 2 plate reader (Tecan, San Jose, CA) with the following settings: measurement mode: fluorescence bottom, emission wavelength/bandwidth: 510/12 nm, excitation wavelength/bandwidth: 485/12 nm, 40-µs integration time. After the reading, PBS was removed, and the cells were fixed with 4% paraformaldehyde for 15 min and stored at 4 °C for microscopy evaluation. GFP-positive cells were visualized with a GFP filter set using a 10× objective on a Nikon Eclipse TE2000-S (Nikon, Tokyo, Japan) equipped with a Roper Scientific (Trenton, NJ) Photometrics camera.
Compound Library
The compounds tested in this study were provided in a gratis fashion by the NCI/National Institutes of Health (NIH) Developmental Therapeutics Program (DTP). They are available at http://dtp.cancer.gov. We received 3161 compounds that were selected from the DTP Open Repository collection of 140 000 small molecules encompassing four focus sets: Challenge Set (57 compounds with unknown cell killing mechanism), Natural Product Set (235 compounds), Diversity Set (1990 compounds), and Mechanistic Set (879) compounds. The Challenge Set is no longer available, and the Natural Product and Diversity Sets have been replaced with the Natural Product Set II and the Diversity Set II. The compounds were supplied in 96-well plates as 20 µL of 10- or 1-mM DMSO stocks and stored at –20 °C.
Expression and Purification of E1 and Cdc34 (E2)
Human E1 was expressed with an N-terminal glutathione S-transferase (GST) tag fusion by means of recombinant baculovirus expression in Hi5 insect cells using the pFastBacHTA vector purchased from Invitrogen (Carlsbad, CA). The cells were lysed by sonication in the presence of protease inhibitors in a buffer containing 200 mM NaCl, 50 mM Tris-HCl (pH 7.5), 1% NP40, 1 mM dithiothreitol (DTT), and 1 mM EDTA. Cleared lysate was incubated with glutathione beads (Amersham Biosciences, Pittsburgh, PA) for 1 h at 4 °C. After several washes with lysis buffer, untagged E1 was produced by thrombin cleavage. The protein solution was passed through a gel filtration column (S200 from Amersham Biosciences), and E1 concentration and purity were evaluated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and Coomassie Blue gel staining. The purity was generally greater than 90%, and purified E1 was aliquoted and stored at –80 °C after quick freezing in liquid nitrogen. N-terminal hexahistidine (His)–tagged human Cdc34 was cloned into the pQE-30 vector from Qiagen (Valencia, CA) and expressed in Escherichia coli. His-Cdc34 was purified by nickel–nitriloacetic acid (Ni-NTA) chromatography followed by ion exchange and size exclusion chromatography. The purity and concentration of His-Cdc34 were determined by SDS-PAGE analysis.
E1/E2 Thioester Activity Assay
The thioester reaction was performed at 30 °C for 5 min in a volume of 5 µL by combining 40 nM ubiquitin E1, 1 µM fluorescein-ubiquitin (U-590; Boston Biochem, Cambridge, MA), 100 nM Cdc34 (E2), 0.5 µL 20× energy regeneration system (20×ER) (10 mM ATP, 20 mM Tris-HCl [pH 7.4], 100 mM MgCl2, 200 mM creatine phosphate, 2 mg/mL creatine phosphokinase, 10% glycerol) in thioester assay buffer containing 20 mM Tris (pH 7.6), 50 mM NaCl, and 10 mM MgCl2. The reaction was stopped with an equal volume of SDS-PAGE loading buffer, plus or minus DTT, and the proteins were resolved using 12% gels that were run on ice to prevent the reduction of E1-ubiquitin due to the heat generated during electrophoresis. Thioester bond formation was visualized by scanning the gel using Typhoon scanner 9400 (GE Healthcare, Piscataway, NJ) that was set to fluorescence mode (532 nM).
In Vitro Ubiquitination of p27 and TRF1
Mouse p27, cloned into pCS2, was translated in vitro in a reticulocyte lysate system (Promega, Madison, WI) in the presence of [35S] methionine. p27 was phosphorylated by purified recombinant CDK2-CyclinE as described in Ungermannova et al. 18 Then, 5 µL of the phosphorylation reaction was incubated with a ubiquitination mixture containing 100 nM ubiquitin E1, 200 nM His-Cdc34 (E2), 100 nM SCFSkp2 (E3), 50 nM Cks1, 10 µM ubiquitin (Sigma Aldrich, St. Louis, MO), 10 µM methylated ubiquitin (Boston Biochem), 1 µL 20×ER, 1 µM ubiquitin aldehyde, and 1 µM MG132 in a total volume of 15 µL. The reaction was quenched after 30 min in a 30 °C water bath by addition of 4× SDS sample buffer. The products of ubiquitination were resolved on a 12% polyacrylamide gel that was destained in 45% methanol, 10% acetic acid, and they were dried and exposed overnight to a PhosphorImager screen for imaging using a Typhoon scanner (GE Healthcare). In vitro ubiquitination of TRF1 was carried out as described in Zeng et al. 19
[32P]-AMP: [α-32P]-ATP and [32P]-PPi:[γ-32P]-ATP Exchange Assays
The reaction mixture contained, in a final volume of 10 µL, 50 mM Tris-HCl (pH 7.5), 150 mM NaCl, 10 mM MgCl2 (reaction assay buffer), 150 nM E1, 100 µM cold ATP, 2 mM AMP, 1 µM [α-32P]-ATP or [γ-32P]-ATP (PerkinElmer, Waltham, MA), and 500 µM sodium pyrophosphate (PPi Na salt). Then, 5 µM ubiquitin was added to the mixture to initiate the ATP:AMP exchange. After incubation at 37 °C for 10 min, the reactions were quenched with EDTA, and 0.5-µL aliquots of the reaction mixtures were spotted on thin-layer chromatography (TLC) polyethylenimine-modified cellulose plates purchased from Avantor (Center Valley, PA) (~1.5 cm from the bottom of the plate and 0.5 cm apart) and developed in filtered 0.34 M potassium phosphate (pH 7.0) for 20 min in a glass jar. The TLC plates were air dried for 10 min, covered in plastic wrap, and then exposed to a PhosphorImager plate for 5 to 10 min. The separation of radiolabeled nucleotides was visualized using a Typhoon scanner. ImageJ (NIH, Bethesda, MD) was used to quantify the amounts of AMP and ATP produced, and the count values were inserted into the formula AMP/(AMP + ATP) to determine the ratio of the two nucleotides. Experiments were performed in duplicates, and the concentration–response curves were generated by nonlinear least squares regression analysis of the data using GraphPad Prism (GraphPad Software, San Diego, CA).
Western Blotting
For Western blot analysis, total protein extracts were prepared by lysing cells in lysis buffer (50 mM Tris–Cl [pH 8.0], 5 mM EDTA, 150 mM NaCl, 1% NP-40, 0.1% SDS, and 1 mM phenylmethylsulfonyl fluoride). Then, 50 µg of total soluble proteins was separated by SDS-PAGE. Proteins were transferred to nitrocellulose membrane, and the membrane was blocked for 1 h with 4% nonfat milk, followed by overnight incubation at 4 °C with primary antibodies against acetylated histone H3 (1:1000; #06-599, Upstate, Charlottesville, VA), p27 (1:1000; SC-53871, Santa Cruz), Skp2 (1:1000; H-435, Santa Cruz), or Ezrin (1:5000; E-8897, Sigma Aldrich). Membranes were then incubated with peroxidase-conjugated secondary antibodies for 1 h at room temperature. Detection was performed using Super Signal WestDura purchased from Thermo Scientific. The expression of ezrin was used as a loading control.
Results
Screening for Inhibitors of p27 Proteolysis Using a Cell-Based Assay
A hallmark of many advanced cancers is the excessive degradation of the cyclin-dependent kinase inhibitor p27, which is directed by SCFSkp2-mediated ubiquitination.
20
Therefore, preventing p27 degradation represents a rational approach in cancer therapeutics. To facilitate the identification and characterization of small-molecule inhibitors, we generated a mink lung epithelial cell line (Kip16) stably expressing p27 that was cloned in frame with GFP. The resulting N-terminal GFP-p27 fusion, detectable by fluorescence microscopy and by microplate reader fluorescence quantification, was used to determine the levels of p27 expression upon treatment of cells with a library of 3161 small-molecule compounds provided by the DTP of the National Cancer Institute. An observed increase in GFP fluorescence suggests that a compound increases p27 expression. Kip16 cells were seeded onto a 96-well plate, where the cells in the first and last columns were treated with the vehicle (DMSO) and the proteasome inhibitor MG132, respectively. Cells treated with MG132 responded with a uniform overexpression of GFP-p27 (
Fig. 1A
,
C
). As expected, no increase in GFP-p27 levels was observed in response to DMSO. To assess the effect of compounds on GFP-p27 expression, cells were treated with 3 µM of each compound for 24 h, followed by a fluorescence readout on a Safire II plate reader (Tecan). As an example,
Figure 1A
shows the screening results after treatment with compounds from NCI plate 4149. Compounds 6E and 6G were regarded as possible hits because of an observed increase in fluorescence compared to the MG132 and DMSO controls. Overall, we screened 40 plates and identified 15 possible hit compounds with a hit rate of 0.5% (
Fig. 1B
). The following criteria were used to define a compound as a hit: (1) there was an increase in GFP-p27 levels by more than 65% of the mean signal generated by MG132 read by the plate reader, (2) there was no change in cell viability observed by microscopy, and (3) the expression of GFP-p27 was homogeneous and robust (similar to MG132-treated cells) when examined by the microscope. The average signal-to-background ratio (S/B) was 3, and Z′ factor was 0.6, which suggests that this assay is adequate for high-throughput screening (HTS;
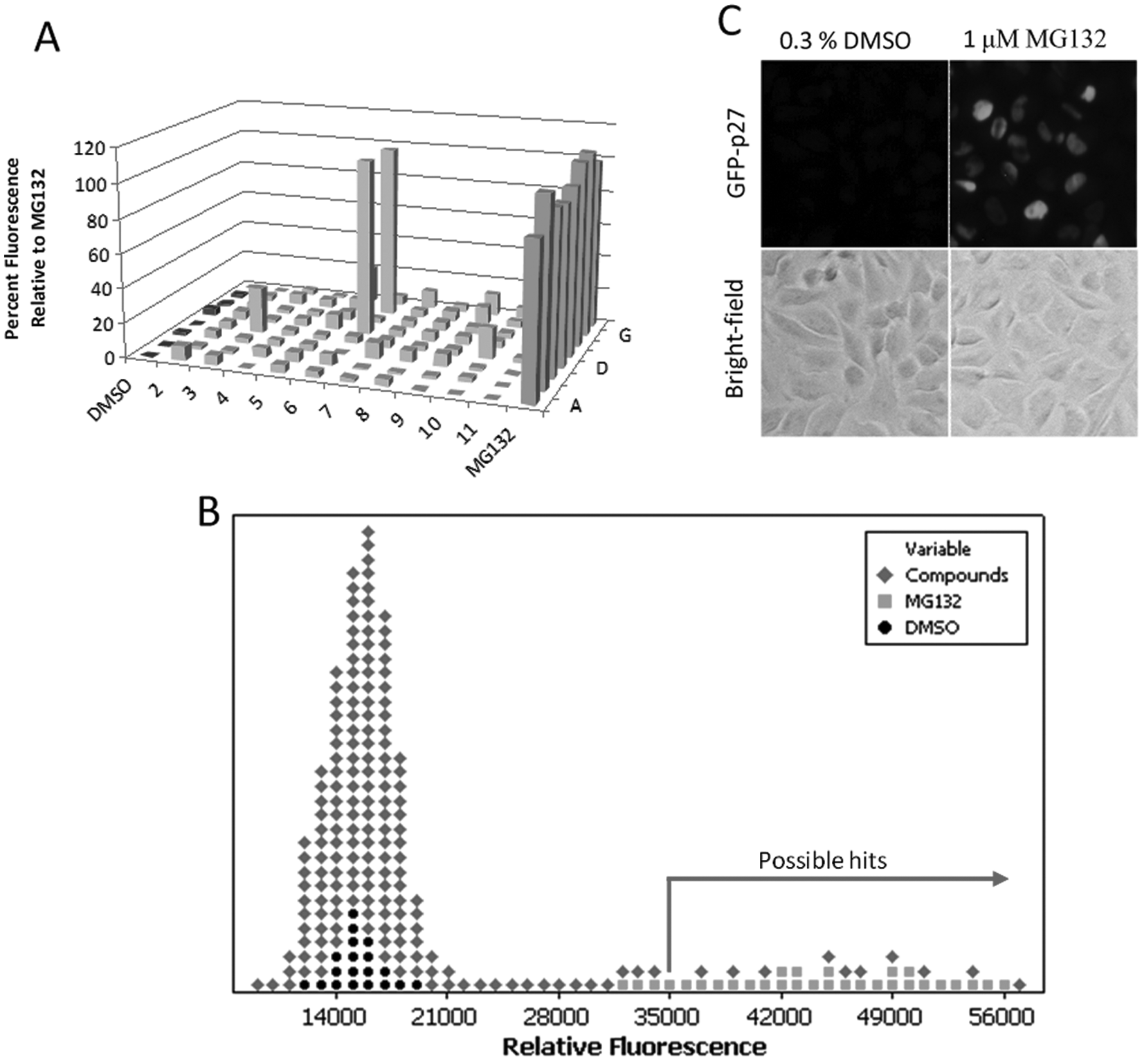
Identification of inhibitors of p27 proteolysis from a library of small molecules using a cell-based assay. (
Effect of Hits on p27 Ubiquitination In Vitro
Although screening compounds using cell-based assays are more physiologically relevant than in vitro–based assays, compounds can potentially affect multiple pathways, giving rise to a significant number of false positives. Hence, a further validation of the hits in a secondary assay is necessary to assess the effect of the compounds on the intended target. Because our primary interest was to identify inhibitors of p27 ubiquitination, we tested the hit compounds in a totally reconstituted in vitro ubiquitination of p27 by SCFSkp2.18,22 The reaction was assembled with recombinant purified components that were added to an in vitro translated [35S]-labeled p27 in the presence of 300 µM of the hit compound or DMSO. As shown in
Figure 2A
, although DMSO had no effect on the attachment of ubiquitin onto p27, four small-molecule compounds reduced p27 ubiquitination, including NSC659390, NSC169942, NSC624206, and NSC363182 (
Fig. 2B
). To confirm that all four compounds are bona fide inhibitors of p27 ubiquitination, concentration-inhibition data were collected from the p27 ubiquitination reactions using a 300- to 30-µM concentration gradient. All but NSC624206 performed poorly in inhibiting p27 ubiquitination at tested concentrations (data not shown). We next measured inhibition by NSC624206 at lower concentrations (1–100 µM) and detected reduction of p27 polyubiquitination up to 1 µM of the compound (
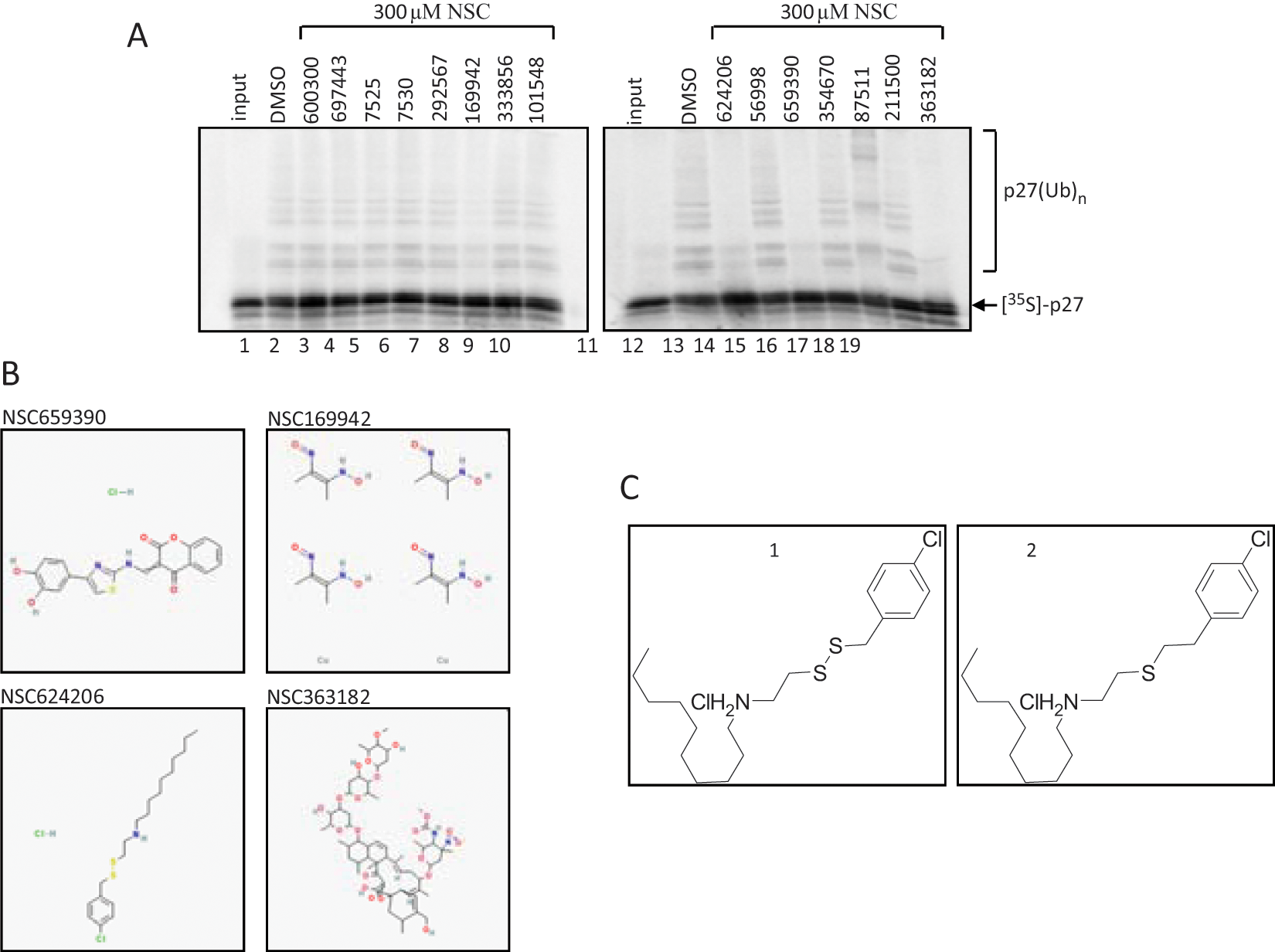
Secondary screening of compounds identified in the cell-based assay. (
Structure and Activity Investigation of NSC624206 and Its Sulfide Analog
Even though the in vitro ubiquitination assay provides a direct assessment of the inhibitory activity of the compound, it offers limited information about what step within the reaction is hindered. An inhibitor of p27 ubiquitination may act at the substrate level by preventing phosphorylation of p27 by CDK2-CyclinE, disturb the transfer of ubiquitin onto E1 or E2, or operate as an allosteric effector impeding protein-protein interactions or other functional processes within the ubiquitination complex. In order for us to understand where in the reaction NSC624206 is acting, we synthesized the compound, as the amount provided by the NCI was rather limited. Resynthesis also enables us to further validate the results. The synthetically reproduced NSC624206 compound is referred to as
1 Inhibits Ubiquitin E1 In Vitro
To uncover which step in the ubiquitination reaction is inhibited by
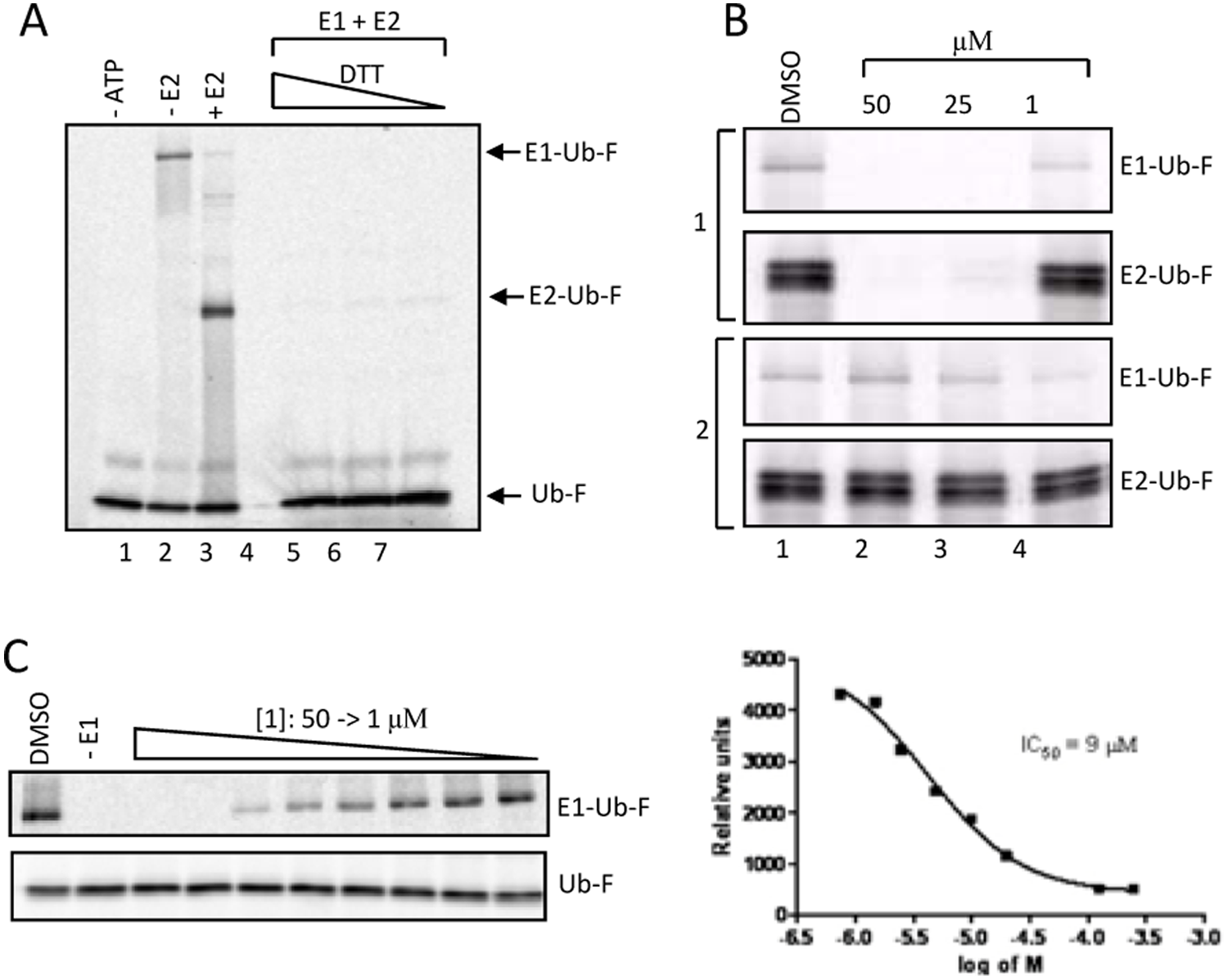
Because
Mechanisms of E1 Inhibition In Vitro
To date, there are very few low µM inhibitors of ubiquitin E1 in the literature. PYR-41 (IC50 = 5 µM) is the first E1 inhibitor in this category. MLN4924, a potent NEDD8 E1 inhibitor (IC50 = 4.7 nM), also targets ubiquitin E1 (IC50 = 1.5 µM). Although the mechanism of MLN4924 is known, the mechanism of PYR-41 is unknown. To determine the mechanism of E1 inhibition by PYR-41 and
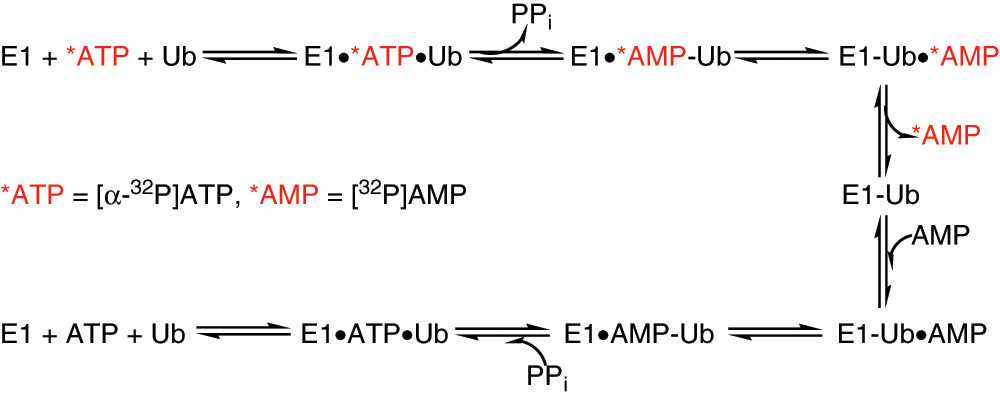
Reaction steps within E1 activation tested by [α-32P]-AMP:[α-32P]-ATP exchange assay. AMP, adenosine monophosphate; ATP, adenosine triphosphate.
We first examined the nucleotide exchange under steady-state conditions where reactions were supplied with 100 µM of [α-32P]-ATP, 5 µM ubiquitin, and 1 µM E1. As expected, no detectable [32P]-AMP was produced since the enzyme only underwent a single turnover. When the nucleotide exchange assay was performed in molar excess of cold pyrophosphate and AMP, a rapid release of labeled AMP was observed on a TLC plate where [α-32P]-ATP remained at the origin and [32P]-AMP moved toward the solvent front ( Fig. 5 ). We carried out the ATP:AMP assay with varying time and E1 concentration in an attempt to evaluate sensitivity and linearity of the reaction. AMP formation was observed at 1 min after the exchange reaction was initiated by adding ubiquitin. At 60 min, most of the labeled ATP was converted to AMP; however, the [32P]-AMP increased linearly at early time points ( Fig. 5A ). In a separate experiment, we varied E1 concentrations ranging from 1 µM to 62.5 nM and observed the most accumulation of labeled AMP when 1 µM E1 was used. Using the 10-min time point, the assay was linear to a concentration of 250 nM E1 ( Fig. 5B ). On the basis of these results and for future ATP:AMP exchange experiments, we chose to carry out the exchange assay for 10 min in the presence of 150 nM E1.
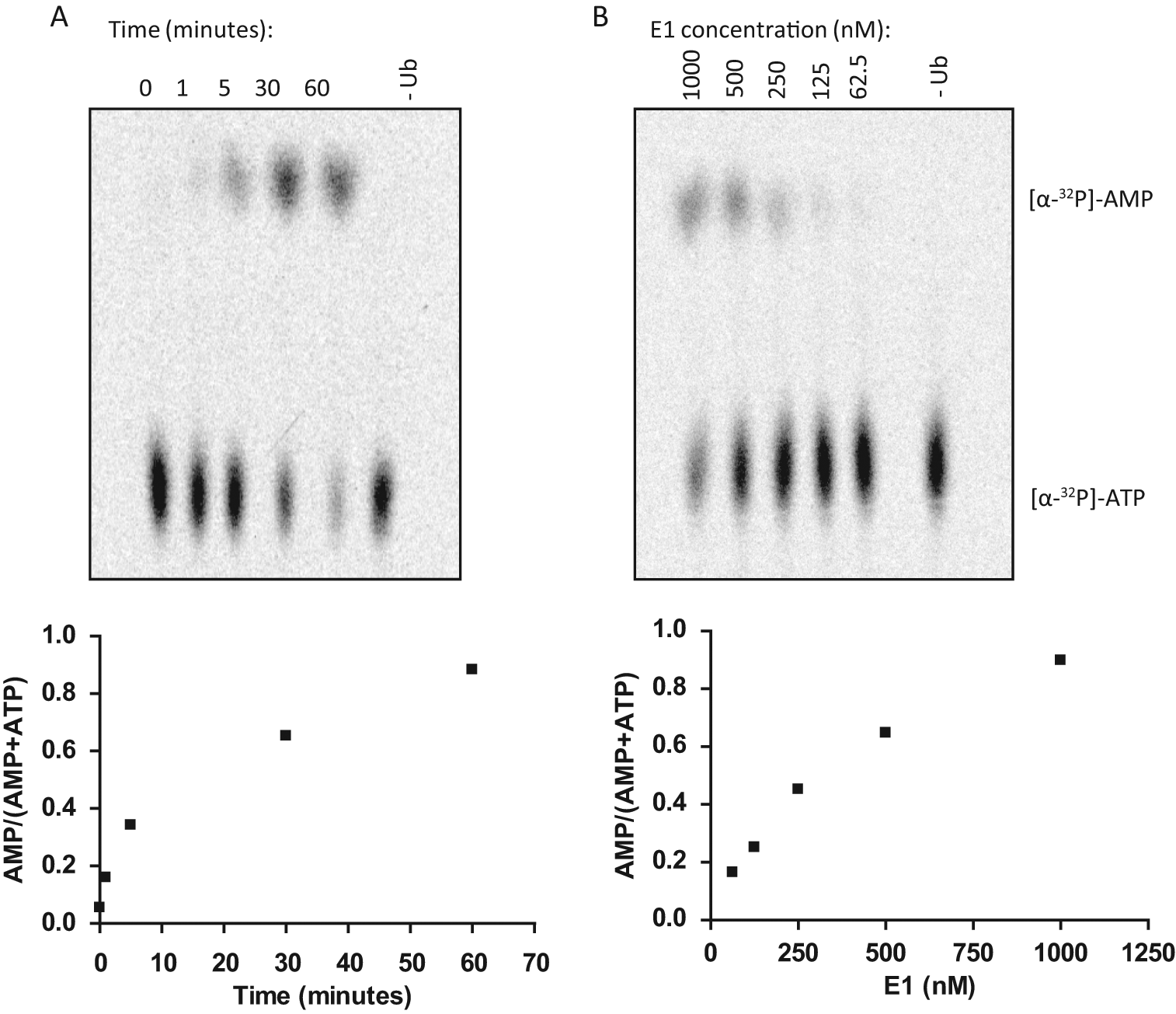
Development of nucleotide exchange assay to monitor ubiquitin-dependent ATP:AMP exchange during E1 activation. (
Using the same reaction conditions and substituting [α-32P]-ATP with [γ-32P]-ATP, we promoted [32P]-PPi:[γ-32P]-ATP exchange, where γ-labeled PPi remained at the origin and ATP traveled toward the solvent front. The main advantages of this assay over conventionally used E1 activation assays include the accuracy in measuring E1 activity with the reduced time, ease of use, and deconvolution of each step within the E1 activation reaction.
Since a ratiometric approach to data analysis can give an accurate estimation of relative amounts of the two labeled nucleotides, we used the nucleotide exchange assays to provide insight into the mechanism by which
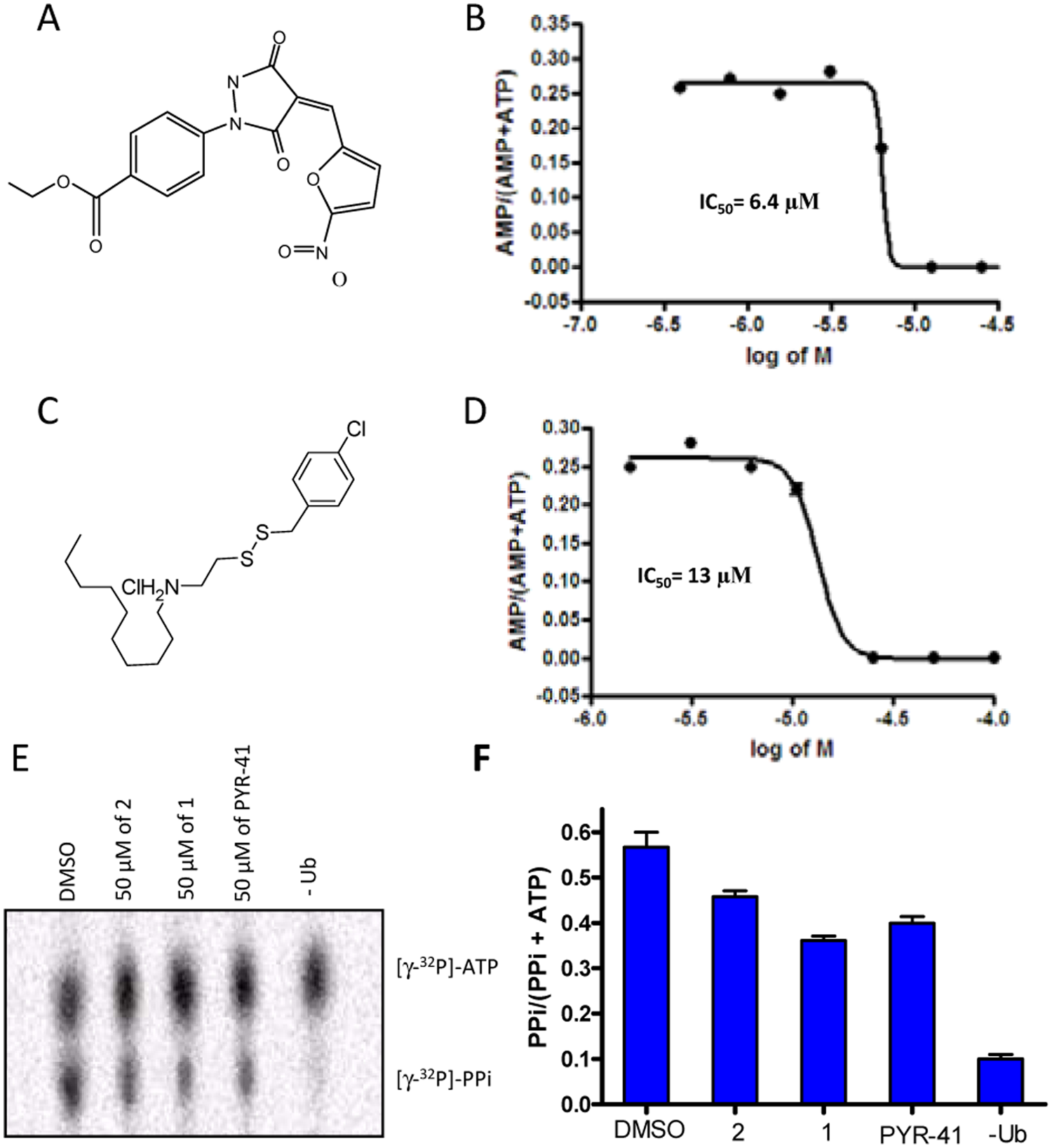
Determination of IC50 for PYR-41 and 1 using the ATP:AMP exchange assay. (
We next tested PYR-41 and
Here we demonstrated that the ATP:AMP exchange assay is advantageous to use to estimate IC50 values for compounds that perturb conjugation of ubiquitin onto E1. The assays enable us to reveal the exact effects of the hits on specific steps in the E1 reaction. The fact that we can perform tens of reactions per day makes radioactive nucleotide exchange a compelling secondary screen assay.
Compound 1 Induces Accumulation of p27 in Cells
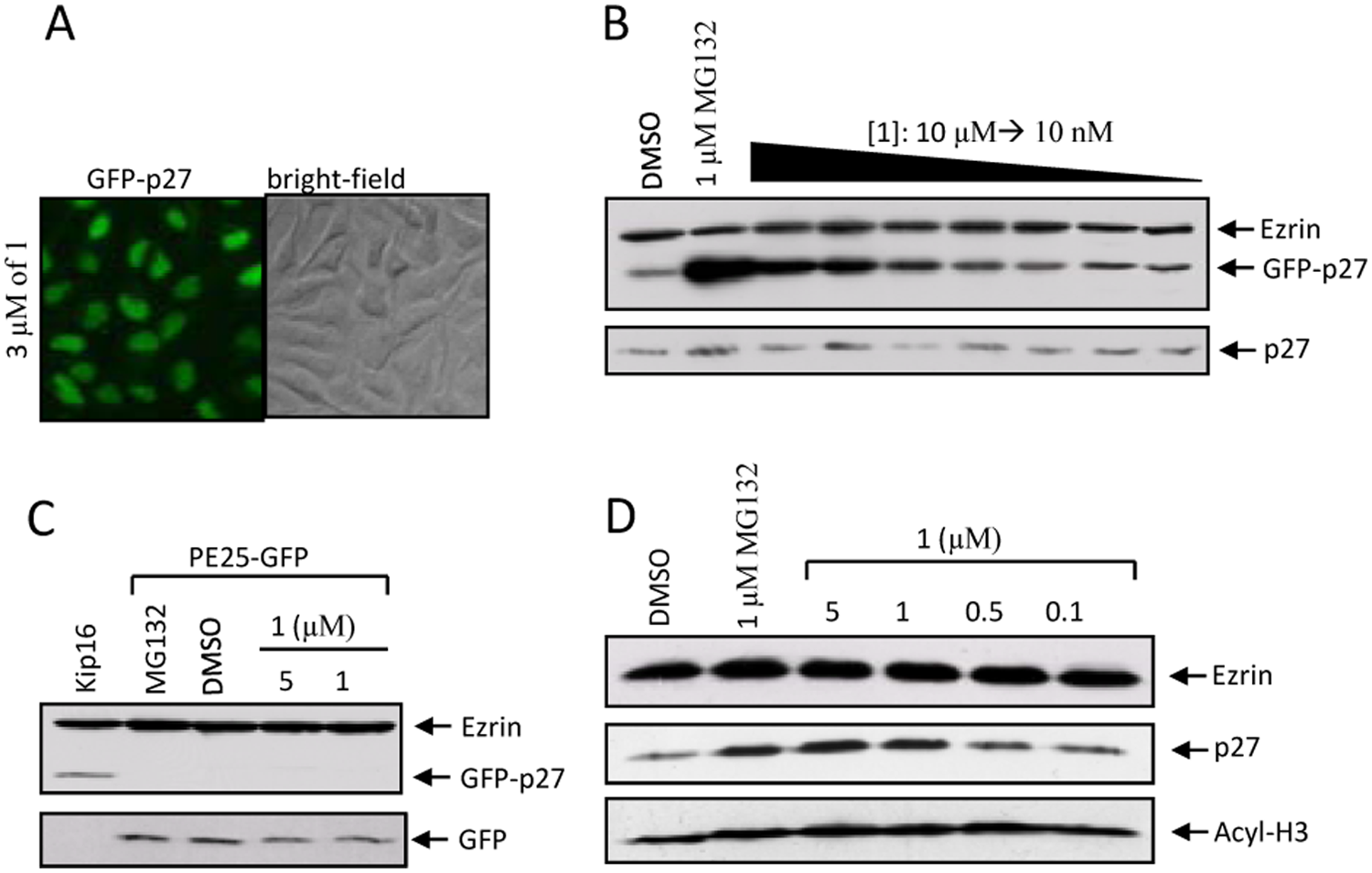
Discussion
There has been a considerable interest in developing inhibitors targeting the ubiquitin-proteasome system. Here we describe our efforts in identifying novel small-molecule inhibitors that perturb the ubiquitin-proteasome system using GFP-p27 as a reporter protein. Through the screening of a small-molecule compound library, we have discovered that NSC624206 inhibits p27 ubiquitination in vitro and stabilizes p27 expression in cells. Furthermore, we have shown that NSC624206 inhibits the ubiquitin activating enzyme E1. Mechanistically, we have demonstrated that NSC624206 and PYR-41 specifically inhibit the ubiquitin-thioester formation step of the E1 activation reaction without having an effect on ubiquitin adenylation.
Results from the small-molecule screen lead to the identification of 15 compounds that prevented p27 degradation in cells. It is important to note that the mechanism of inhibition is unclear. For example, the compounds used in our screen could directly impair the function of the proteasome, perturb specific events in the ubiquitination pathway, or promote the activity of deubiquitination enzymes. The observed upregulation of the substrate may also be explained on the level of transcriptional upregulation. For example, the compound could inhibit histone deacetylase, as in the case of suberoylanilide hydroxamic acid (SAHA), which leads to transcriptional activation and subsequent upregulation of protein production. 23 Thus, although the primary screen does not suggest a mechanism of action of the compounds, it also does not rely on any mechanistic assumption. Instead, these examples demonstrate that assays using reporter substrates are in fact functional assays that recapitulate the intricacy of the system in a single readout. Still, why the cells accumulated p27 in the presence of certain compounds was of interest. Therefore, a secondary in vitro screen that isolated the ubiquitination pathway was applied to the 15 compounds identified from the primary screen. Four of the 15 top hits showed inhibitory potential in the secondary p27 in vitro ubiquitination screen. Careful dissection of the p27 ubiquitination cascade revealed that one of the four identified compounds, NSC624206, had in vitro inhibitory activity against the ubiquitin activating enzyme E1. Degradation of p27Kip1 is known to be regulated by several cellular signaling pathways and ubiquitin E3 ligases. In this study, we focused on the characterization of NSC624206. The mechanism(s) of inhibition of p27Kip1 ubiquitination by the other three compounds identified needs to be further characterized in future studies. It is possible that these compounds may have inhibitory effects on other components of p27 ubiquitination such as E2, the E3 ligase specific to p27, or a component of the E3 ligase.
Whereas the catalysis of ubiquitin adenylation was unaffected, as measured by the [P]-PPi:[γ-32P]-ATP exchange assay, the production of AMP was greatly decreased, indicating that NSC624206 prevented the formation of the thioester bond between ubiquitin and the active site of the E1. Hence, we can rationalize that it is the nucleophilic nature of the active site cysteine that promotes an electrophilic attack on one of the sulfur atoms of the compound, which induces the displacement of the compound via a thiol-disulfide exchange reaction and ultimately leads to loss of E1 enzymatic activity (
Fig. 8
). This model is consistent with the observation that
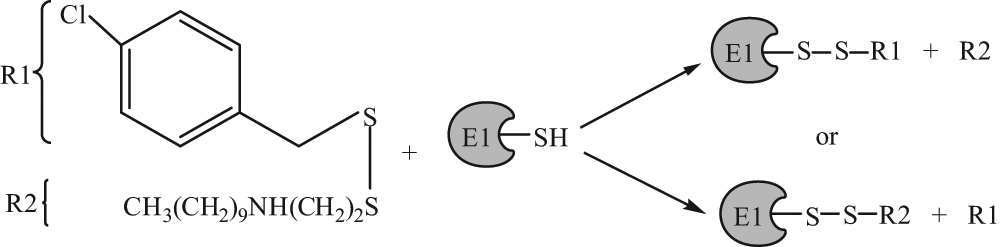
Proposed reaction mechanism of
In general, the thiol of cysteine residues can participate in oxidation reduction reactions. Also, cysteine can coordinate metal ions, as seen in zinc finger proteins, or it can react with electrophiles due to its nucleophilic character. Modification of cysteine by all of these processes strongly alters the properties of the related protein, which may be exploited in designing pharmacological agents. For example, several compounds that target tubulin, a protein whose cysteine residue upon oxidation prevents tubulin from polymerizing, 24 have been reported to covalently modify Cys-239 via a nucleophilic aromatic substitution reaction while exerting a cytotoxic effect in many cancers. 25 In addition, the asymmetric 1-methylpropyl-2-imidazolyl disulfide (IV-4), also known as PX-12, inhibits the small redox protein thioredoxin-1 (Trx-1) through irreversible thioalkylation of a critical cysteine located outside of the catalytic site of the enzyme. 26 Trx-1 is overexpressed in tumors; it promotes growth, apoptosis, and angiogenesis and is associated with decreased patient survival. 27 It is encouraging to report that PX-12 has progressed into phase II clinical trials for the treatment of advanced pancreatic carcinoma. Further findings about PX-12 suggest that this compound also affects tubulin polymerization and causes inhibition of papain and ficin, cysteine proteases, indicating a wider range of PX-12 activity. 28 Numerous cellular proteins rely on their active cysteines to function properly, so it is not surprising that PX-12 or the similarly configured disulfide compound presented here could engage in nonspecific activities. This should not undervalue the usefulness of such compounds, as indicated by the early clinical trial success of PX-12, thus validating this approach for drug discovery. Given these precedents, there is a possibility that NSC624206 could be further developed as a lead compound for E1 therapeutics in vivo.
A few ubiquitin E1 inhibitors have been reported in the literature. Panepophenanthrin, a natural compound derived from the mushroom strain Panus rudis, inhibits E1 in vitro with an IC50 ~72 µM. 29 Himeic acid A, derived from the marine fungus Aspergillus, has also been shown to inhibit E1 in vitro with IC50 ~50 µM. 30 PYR-41 inhibits ubiquitin E1 both in vitro and in cells. 12 The IC50 of PYR-41 is around 5 µM and thus more potent than the compounds described here. However, the exact mechanism of panepophenanthrin, himeric acid A, and PYR-41 inhibition is not known. Our findings suggest that PYR-41 blocked the formation of the thioester linkage between the active site cysteine of E1 and ubiquitin while not disturbing the release of PPi during adenylation. During the characterization of PYR-41, Yang and coworkers 12 also suggested that PYR-41 could potentially link to the E1 through its active site cysteine, as the addition of excess reduced glutathione eliminated restored E1 activity. Given the structure of PYR-41, it is tempting to invoke inhibition by hetero-conjugate addition of an E1 cysteine residue to the α,β-unsaturated pyrazolidinedione.
Our experimental results suggest that NSC624206 is not simply a thiol-reacting reagent. There is considerable selectivity of this compound in targeting the ubiquitin E1 enzyme. NSC624206 does not significantly inhibit ubiquitin conjugating enzymes (E2s), which also have catalytic Cys residues that accept activated ubiquitin via a thioester bond. However, not all E2s are insensitive to NSC624206. For example, Cdc34 is modestly inhibited by NSC624206. There are four classes of E2 enzymes. Classification of E2s is based on whether there are additional extensions to the catalytic core. 31 UbcH5a and UBE2G2 (UBC7) belong to class I, which contains only the catalytic domain. Cdc34 is a class III E2 with C-terminal extensions. Based on the structure of yeast ubiquitin E1 and E1-E2 interfaces of other ubiquitin-like systems,32–34 it has been predicted that E1 (Uba1) will undergo a distinct ubiquitin-fold domain (UFD) movement and conformation change during the E1-E2 handoff. 33 The C-terminus of Cdc34 has been shown to interact with ubiquitin. 35 It is tempting to speculate that ubiquitin E1 transfer between E1 and Cdc34 involves distinct conformation changes. NSC624206 may perturb this transition. It will be interesting to test the effect of NSC624206 on E1-E2 ubiquitin transfer with all classes of E2s in future studies to determine if NSC624206 can functionally discriminate between different classes of E2s.
Mechanistically, NSC624206 had an in vitro inhibitory activity against the ubiquitin E1/E2 pathway that resulted in the reduction of p27 and TRF1 polyubiquitination in vitro. Moreover, NSC624206 stabilized endogenous p27 in HepG2 cells without having an effect on transcriptional activity. It is therefore possible that NSC624206 might operate via the inhibition of E1-Cdc34 inside of cells; however, further studies are necessary to better delineate whether the observed increase of cellular p27 is due to cellular E1/E2 inhibition.
Footnotes
Acknowledgements
We thank Kaiser Danny Tran and Craig Manahan for excellent technical help. We thank Johannes Rudolph for discussions.
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
The authors disclosed receipt of the following financial support for the research and/or authorship of this article: This work was supported by a grant from the National Institutes of Health (CA107089) to Xuedong Liu.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.