
Abstract
Genome-scale small interfering RNA (siRNA) screens have become an increasingly popular approach to new target identification and pathway elucidation. However, the large data sets generated from siRNA screens have demonstrated high false-positive rates and the requirement for extensive experimental triage to distinguish true hits. A number of groups have independently reported the presence of siRNAs with identical seed sequences among their top screening hits. Based on these observations, we have developed a comprehensive technique for detecting and visualizing seed-based off-target effects in siRNA screening data. This is accomplished by analyzing the behavior of siRNAs that share identical seed sequences, which we refer to as common seed analysis (CSA). By applying these techniques to primary screening data of the Wnt pathway, we identify 158 distinct seed sequences that have a statistically significant effect on the assay. The promiscuous seed sequences identified in this manner can then be discounted in the analysis of follow-up experiments using single siRNAs. The ability to detect off-target effects when sufficient numbers of siRNAs share a common seed has significant implications for the design of siRNA screening experiments, data analysis, hit selection, and library design.
Introduction
Small interfering RNA (siRNA) screening has grown to be a popular strategy1–3 for discovering new drug targets and elucidating biological pathways. To date, the large-scale screening of siRNA libraries has yielded interesting yet contradictory results. 4 Although many of these genome-wide screens have led to notable discoveries,5,6 skepticism of high-throughput siRNA approaches has been fueled by studies that show small overlap in the genes identified by different screens interrogating the same biology, 4 suggesting high false-positive rates. The low overlap in results from these screens is likely to be a consequence of several factors, including real biological differences in the cellular backgrounds used, variance due to the technical implementation of the assay and screen, and the use of siRNA reagents generated by independent design algorithms. To improve the success rate and impact of siRNA screens, identifying the main sources of false-positives and finding an efficient means to differentiate true-positives early in the experimental process is critical.
It has been well established that siRNA duplexes have both on-target (to reduce expression of intended gene) as well as off-target activities (to reduce expression of nonintended genes). 7 A significant portion of false-positives from siRNA screens is likely due to off-target effects. 8 siRNA off-target effects have been linked to the mechanism of action for miRNAs, 9 in which a short sequence on the 5′ end of the RNAi duplex (the “seed region,” bases 2–8) is complementary to the 3′UTRs of multiple mRNAs, causing degradation of their associated transcripts.7,10 Because sequence matches as short as six nucleotides can result in down-regulation of a transcript, the number of off-target effects due to a single siRNA can number in the hundreds. Consistent with this, we show that seed-based off-target effects have significantly more influence on our screen of the Wnt pathway than on-target effects do.
To date, a workflow to distinguish true-positives from off-target false-positives that requires extensive postscreen experimental triage has been proposed and widely accepted. 11 These steps typically include testing of additional siRNA sequences, confirmation in multiple cell lines, and the matching of phenotypic assay results to changes in mRNA levels. Other groups have developed methodologies to address off-target effects by treating them as random occurrences and attempting to mitigate them by gathering data from multiple siRNAs targeting the same gene at the primary screen level. 12 Alternatively, approaches exist in which siRNAs containing seed regions that are present in a large number of 3′UTRs are avoided altogether. 10
To complement these existing approaches and to help address the high incidence of “promiscuous” seeds among top screening hits, we demonstrate a methodology called common seed analysis (CSA; software supplied in supplementary material), which uses siRNA seed region biases observed in the primary screening data to visualize and eliminate results from confirmation screens in which statistically significant off-target effects are present. CSA can be applied as part of the hit-prioritization process to eliminate clear off-target effects from consideration. Furthermore, this method can be readily implemented using existing siRNA screening libraries and does not require specific seed hexamer selection. We have applied this methodology to identify off-target effects in the context of a full-genome siRNA screen designed to identify new targets in the Wnt pathway by assaying for β-catenin, a signature gene in that pathway. 13
Materials and Methods
Genome-Scale siRNA Screen
To validate CSA for detection of siRNA off-targets, we applied the methodology to data generated previously in a genome-scale siRNA screen to identify genes involved in the regulation of the Wnt/β-catenin pathway.13,14 We describe the details of that screen only briefly and refer the reader to Chung et al. 14 for a more complete description of the methods used. The primary screen was run against both whole-genome (pools) and druggable (singles and pools) siRNA libraries using a human HT1080 sarcoma cell line engineered with a luciferase reporter coupled to a β-catenin promoter. This β-catenin-Luc reporter system was activated with conditioned media containing Wnt-3a. This assay was chosen to test the CSA methodology because it exhibited excellent transfection efficiency and had a robust assay window that allowed for miniaturization to a 1536-well nanoplate format. Miniaturization of the assay significantly increased plate throughput so that replicates were logistically feasible, even at the genome-scale primary screening stage.
Assay results had a normal Gaussian distribution across the library and were scored based on Z*-score, which is founded on the median absolute deviation-based hit selection strategy described by Chung. 15 Following primary screening in triplicate, 1337 (6%) of the siRNA pools were selected from the whole-genome screen for further confirmation based on criteria that included potency, activity in a different cell line (data not shown), and input from biologists. The corresponding siRNA pools were then denconvoluted to their three component single siRNAs and retested in triplicate in the original assay.
siRNA Library
The custom whole-genome and druggable siRNA libraries used were synthesized by Sigma-Proligo, and all duplexes underwent quality control testing. The whole-genome library contained 22 102 pools (three siRNAs targeting the same gene in each pool), whereas the druggable library of 6605 siRNA pools was screened in two formats, both as pools and as individual siRNAs. The design algorithm used minimizes off-target effects 16 and increases siRNA efficiency. siRNA duplexes contain features including sequence asymmetry, repeat masking, masking of the 5′UTR, and restrictions on location in the 3′UTR and have undergone sequence alignment to eliminate siRNAs with at least 17 bp of complementarity to other genes. In addition, common sequence was targeted in genes with multiple splice forms.
Statistical Analysis
Previous work on detecting off-target seeds in siRNA screens has focused on finding seeds that are overrepresented in selected hits. 9 As a result, the seeds detected may depend on the threshold set for hits in the screen. We chose instead to analyze all seeds present in three or more pools in the screen and to evaluate each for evidence that it induces a bias in the assay results. Choosing an appropriate statistical test for bias in a continuous variable requires that we know if the data are normally distributed. In this case, use of the Shapiro-Wilk 17 test of normality revealed that some of the seed groups were nonnormal, leading us to use the nonparametric Kolmogorov-Smirnov statistical test to detect seed bias. Using Kolmogorov-Smirnov provides a p value for the null hypothesis that the distribution of assay values obtained for all pools containing a given seed is the same as the background distribution of assay results. When this p value is very small, we can conclude that the presence of the seed is causing a bias in assay results. Because this statistical test is applied a large number of times (to all seeds that are contained in three or more pools), the p values were adjusted to account for multiple comparison bias using Benjamini-Hochberg 18 to limit our false discovery rate to 0.01.
Results
Differentiating Sources of Variability
Genome-scale siRNA primary screens are typically implemented using pools of 3 or 4 siRNAs, each of which have independent sequences designed to target the same gene. 19 The rationale behind this strategy is to maximize the chance of at least one siRNA in the pool being active. In most cases, the behavior of the pool follows that of the most highly active single siRNA in the pool. 20 Therefore, it is reasonable to conclude that screening in pools of siRNA duplexes maximizes the possibility of identifying on-target hits in the assay. However, the use of pooled siRNAs in screens can also be confounding because of the possibility of multiple off-target effects being caused by the different oligonucleotides tested, which could negatively affect data reproducibility.
The overall quality of results from genome-scale siRNA screening assays can be affected by a wide variety of factors. 11 In addition to the prevalence of seed-based off-target effects, variability in transfection efficiency, long duration of the assays (typically 4–6 d), and day-to-day inconsistencies in cell viability are among factors that lead to significant variability in siRNA screening data. As a result, it is important to delineate individual sources of variance whenever possible. An example of such differentiation is the ability to distinguish technical reproducibility from inherent biological variance. 21 One way to accomplish this is through assessment of technical replicate correlation during primary screening. The results of one such assessment from the Wnt/β-catenin-Luc primary screen can be seen in Figure 1A . In this example, the Wnt/β-catenin-Luc reporter assay exhibits very good technical reproducibility, as determined by an R value of 0.94 for replicates 1 and 2. Not shown are replicates 2 and 3, which have an R of 0.88, and replicates 1 and 3, which have an R of 0.90.
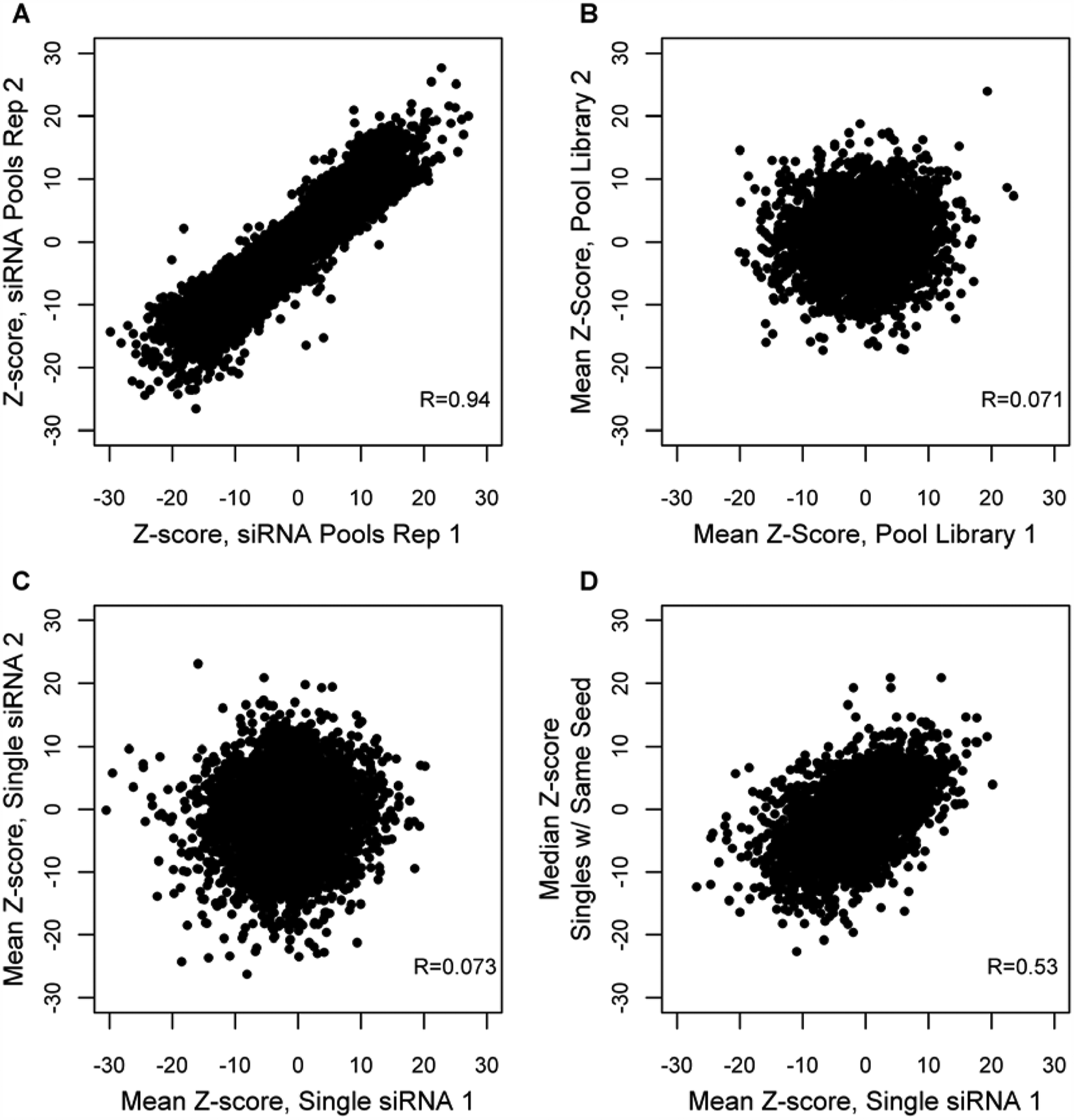
Sources of variance in Wnt primary screening data. (A) Correlation of technical replicates for the Wnt/β-catenin-Luc reporter assay, replicates 1 and 2 shown. (B) Correlation of siRNA pools designed against 3086 genes but having no common sequences. (C) Correlation of different single siRNA duplexes designed against the same genes, consisting of 6564 total points (genes). (D) Assay results for siRNA library 1 versus the median Z-score for all siRNAs from library 2 that have the same seed sequence but are designed to target different genes.
In addition to assessing technical variance seen in the Wnt/β-catenin-Luc siRNA screening assay, we were interested in determining how well different siRNA pools designed to target the same gene correlated. For this purpose, we compared the results for 3086 common genes from the whole-genome and druggable libraries in the Wnt/β-catenin-Luc reporter assay ( Fig. 1B ). We further restricted the analysis to include only those pools that did not contain any of the same siRNA sequences. Surprisingly, there is almost no correlation between assay results for different pools designed against the same genes (R value is 0.071). The lack of correlation between siRNA pools intended to target the same gene (despite the good technical reproducibility of the assay) demonstrates that although we can expect consistent results from different experiments using the same siRNA reagents, altering the siRNA sequence introduces substantial variability to our results. In accordance with this observation, recent work 22 demonstrated a very similar pattern of high technical reproducibility when using the same siRNA pools but extremely poor correlation between different libraries. Therefore, we can conclude that this effect is not a problem specific to a particular library or assay but rather a general phenomenon of siRNA screening.
To determine if this lack of correlation between siRNA pools targeting the same gene is caused by screening using pools (and seeing the effects of multiple off-target interactions) instead of individual siRNAs, we tested the siRNA duplexes making up the pools of the druggable library as deconvoluted singles. Because these siRNAs have base sequences designed for different regions of the mRNA of the target gene, they are in effect distinct reagents. When the different single siRNAs targeting the same gene were tested, an extremely weak correlation was observed ( Fig. 1C ), similar to the results obtained when testing the corresponding pools. As a result, we next explored the possibility that we were witnessing sequence-dependent siRNA off-target effects.
Seed-Dependent Off-Target Effects
It has been well established that siRNA off-target effects are caused by the seed sequence of the siRNA (bases 2–8 of the guide strand).7,10 Previous work demonstrated an enrichment of a specific seed region among the top screening hits. 23 Furthermore, in Sudbery et al., 9 a statistical test was applied to identify 17 hexamer and 13 heptamer seed sequences enriched in high-scoring siRNAs. To assess how much impact off-target effects are having on the outcome of a screen relative to on-target effects, we examined the correlation of results from siRNA singles having the same seed sequence but designed to target different genes. In this case, we are able to see a much more robust correlation (R = 0.53; Fig. 1D ) than that observed between siRNAs with different seed sequences targeting the same gene (R = 0.073; Fig. 1C ). By comparing correlation coefficients, we estimate that the seed sequence has at least 10-fold more influence on siRNA activity than its designed on-target effect in this screen. If this trend holds for other assays, it helps to explain why false-positives are commonplace in siRNA screening results.
To identify specific seed sequences that are causing off-target effects in the assay, we began by surveying the entire data set to find common biases. Because the number of siRNAs in a screen (~66 000 for a whole genome screen) is larger than the number of possible hexamer sequences (46 = 4096), many seed sequences occur in more than one siRNA. In cases where the seed sequence occurs frequently in the library, we can apply a statistical test (see the Materials and Methods section) to determine if pools containing a siRNA with that particular seed have a biased distribution in comparison with all other pools.
In total, there were 158 statistically significant seed biases detected in the screen (
To verify that we can infer seed bias in single siRNAs based on data from pooled siRNAs, we examined the 12 seeds that were the most statistically significant examples in which five or more of the pools containing the seed sequence had been deconvoluted to singles ( Fig. 2A ). For these seeds, we confirmed that the bias and directionality observed in the pools is present in the singles with the seed sequences and absent with the other members of the pool ( Fig. 2B ).
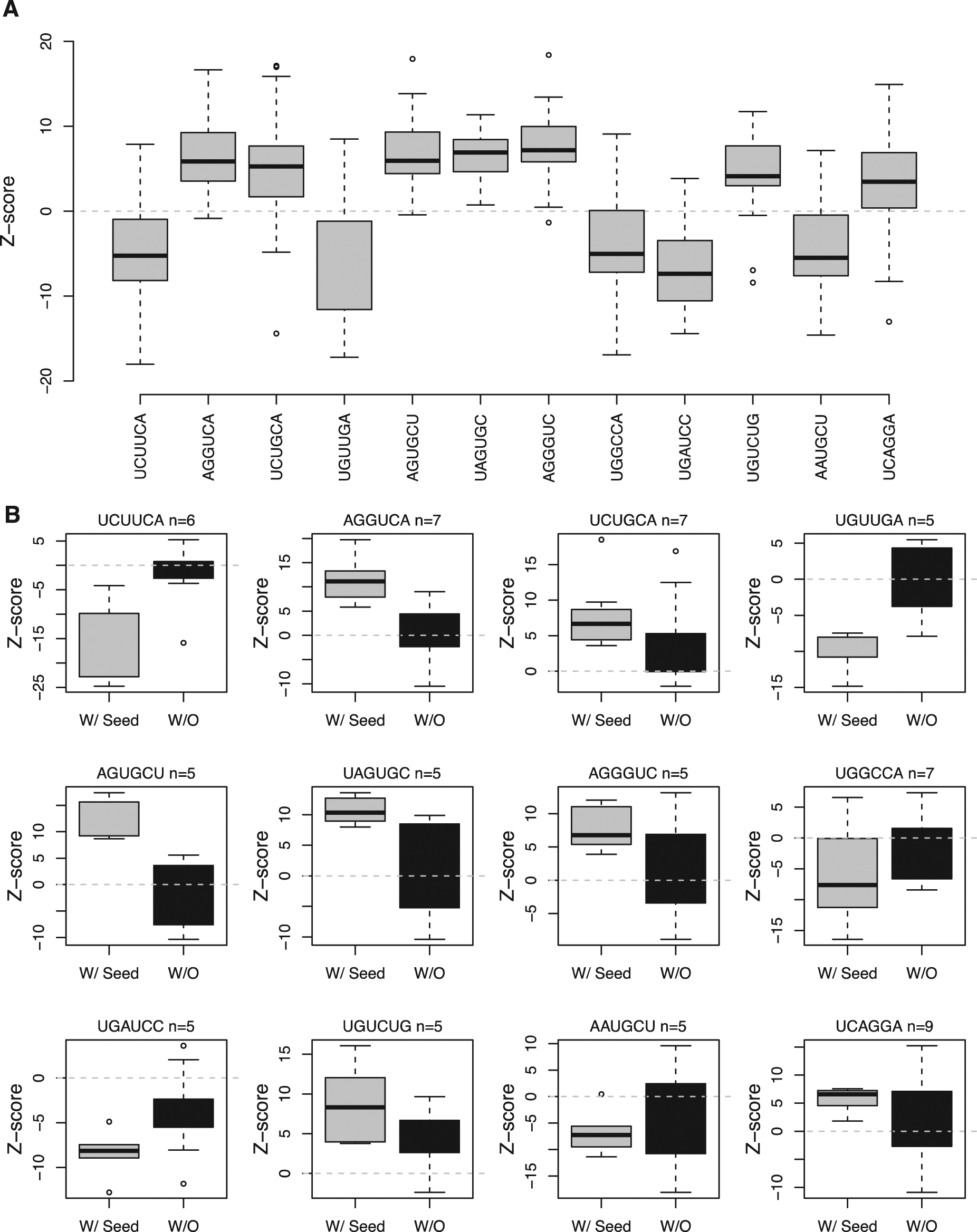
Detecting false-positive signals from siRNA seeds. (A) Box plot showing the activity of all pools containing 12 of the 158 statistically significant seeds. These 12 seeds were examined because they were the most statistically significant examples in which more than five of the pools containing the seed sequence had been deconvoluted to singles. Presence of these seeds in any of the three siRNAs in a pool significantly biases the assay results in either a positive or negative direction (mean of all samples is ~0). (B) Box plots showing the activity of siRNAs containing 1 of the 12 seeds (gray) side by side with the median activity of other siRNAs containing different seeds but targeting the same gene (black). All of the single siRNAs containing a specific off-target seed maintain the bias and directionality observed in pools at the primary screening stage, whereas the siRNAs from the same pools without the off-target seeds are distributed around zero.
Common Seed Analysis Visualization
Although we can eliminate many off-target seeds using statistical analysis, Figure 1D indicates that off-target effects are pervasive and may be present even when not rising to the level of statistical significance after correcting for multiple comparisons. Therefore, we developed a visualization of seed bias so that genes can be assessed on a case-by-case basis. This allows researchers to make reasonable judgments about the strength of evidence for a particular hit. This workflow can be seen in Figure 3 and examples of its use for four representative genes can be seen in the strip plots in Figure 4 . Figure 4A shows an example in which three single siRNAs all recapitulate the phenotype observed for the pool, which would ordinarily be viewed as a confirmation. However, examining assay results from pools targeting different genes that contain the same seed sequence as each of the three singles reveals statistically significant bias, calling into question the results of all three singles. In the case of Figure 4B , two siRNAs have statistically significant off-target effects in opposite directions, whereas the third (UAUCCC) is not statistically significant but remains suspicious given that all the siRNA pools containing that seed had a positive directionality in the assay. Figure 4C exhibits no statistically significant off-target effects after correction for multiple comparisons. However, the two most active siRNAs contain seeds that were only associated with down-regulation of the reporter in other pools. Although there is no visible bias in the third seed and it still has the down-regulation phenotype observed in the pool, a single siRNA generating a phenotype is considered relatively meager evidence for the target gene’s involvement in the pathway. Figure 4D shows an example in which there are no statistically significant off-target effects or suspicious trends in the directionality of seed hexamers. Thus, if we did not already know that down-regulating Wnt5B would inhibit the reporter system used, this would be deemed a strong candidate for follow-up studies. These examples indicate the importance both of statistical analysis and data visualization to mitigate the influence of off-target effects in siRNA screening results.
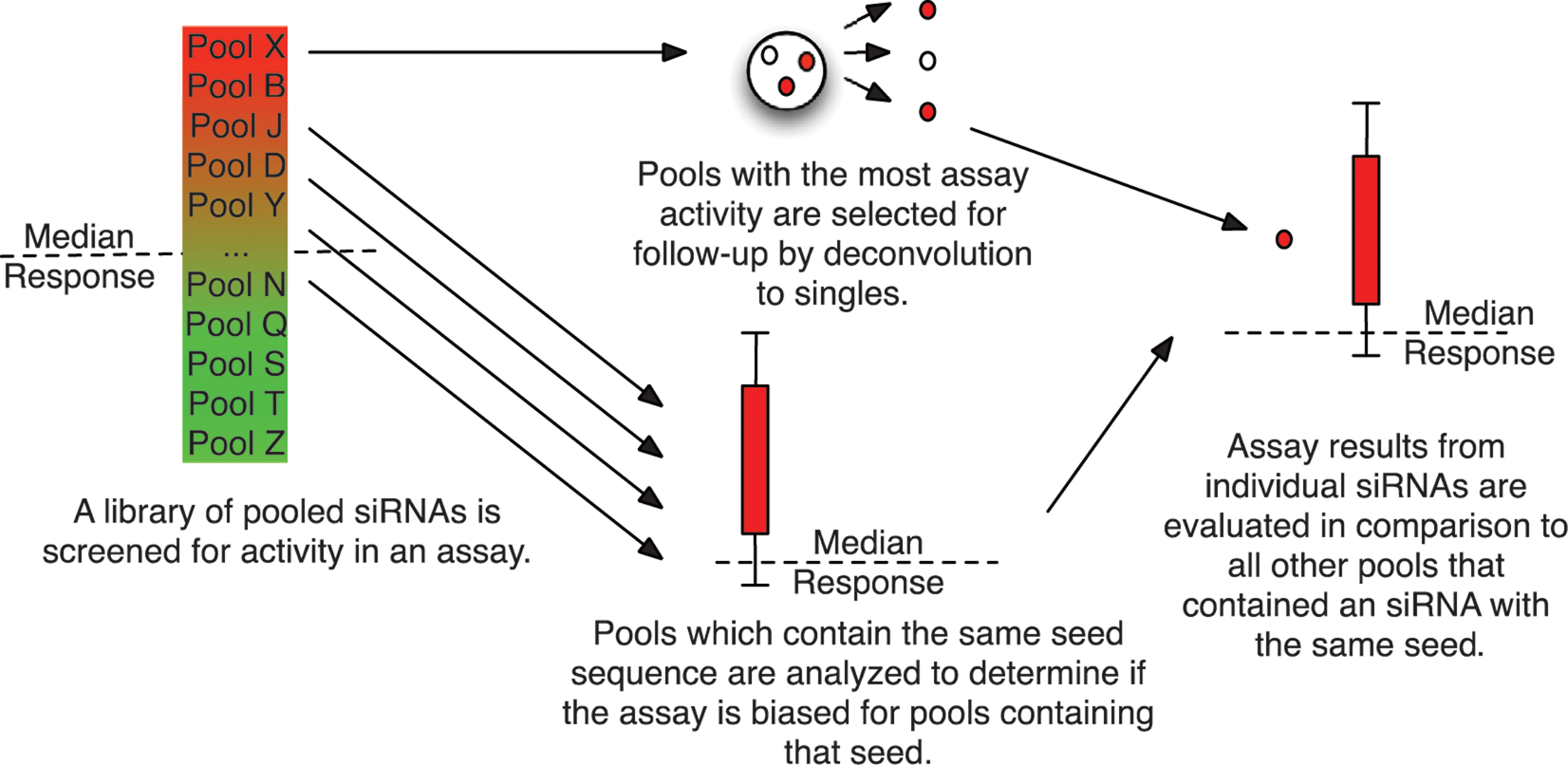
Workflow for detecting off-target effects. Assay results from the initial screen of pooled siRNAs are used to select pools for follow-up (via deconvolution to singles) and to analyze seed-based effects on the assay. The seed analysis of the pools and the results from single siRNAs are then combined to evaluate if the activity observed is likely due to on-target or off-target (seed-based) effects.
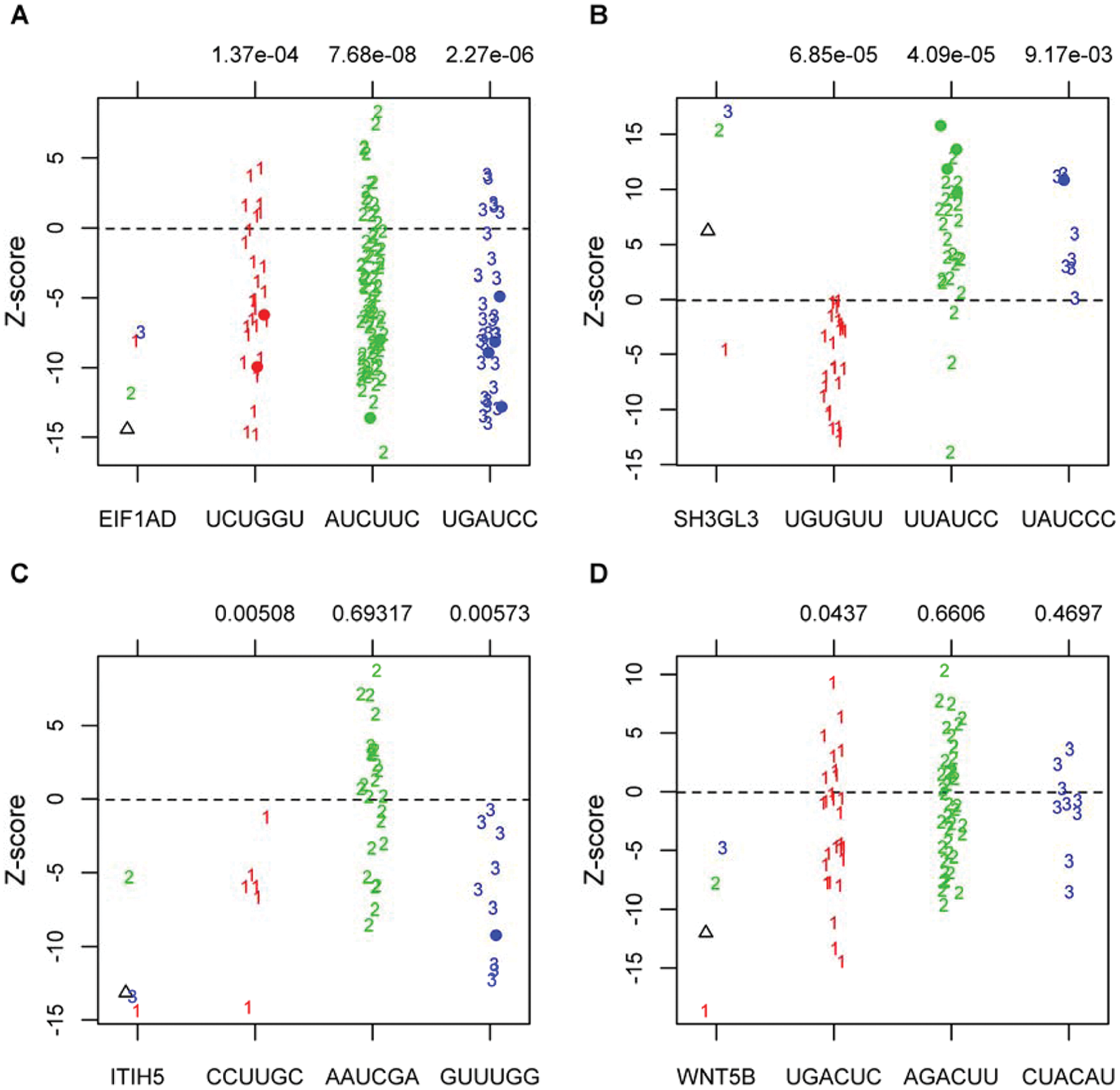
Off-target plots. Off-target plots for the deconvolution of four pools selected for follow-up in the confirmation screen: (A) EIF1AD, (B) SH3GL3, (C) ITIH5, and (D) WNT5B. The left-most column plots both the pool result (triangle) and singles deconvoluted from that pool (numbers) for each gene. The three columns on the right show assay results for pools (numbers) and singles (dots) that contain the same seed sequence as one of the single siRNAs from the pool of interest, color-coded and numbered to match. The seed sequence is below each column, and above the column is the statistical significance of the bias for that seed. These p values are not controlled for multiple comparisons.
Base Preferences for Off-Target Seeds
In addition to helping eliminate false-positives from screening results, common seed analysis may aid in our understanding of siRNA off-target effects. Figure 5 shows the bias in nucleotide base #1 of the seed sequence (base #2 of the guide strand), comparing statistically significant off-target seed sequences and the distribution of bases at that position in the entire library. There is a statistically significant bias in this position (p value = 1.761e-11 using Fisher’s exact test), preferring Uracil at this position to other nucleotides. Whether this trend could be used to design siRNAs with fewer off-target effects will depend on the reason for the effect. If it is a preference linked specifically to the mechanism of off-targeting, then Uracil could be selected against. However, if the preference is due to improved RISC loading for siRNAs with a Uracil at base #1 of the seed, any design attempts here to reduce off-target effects would also correspondingly reduce siRNA efficacy against the intended target.
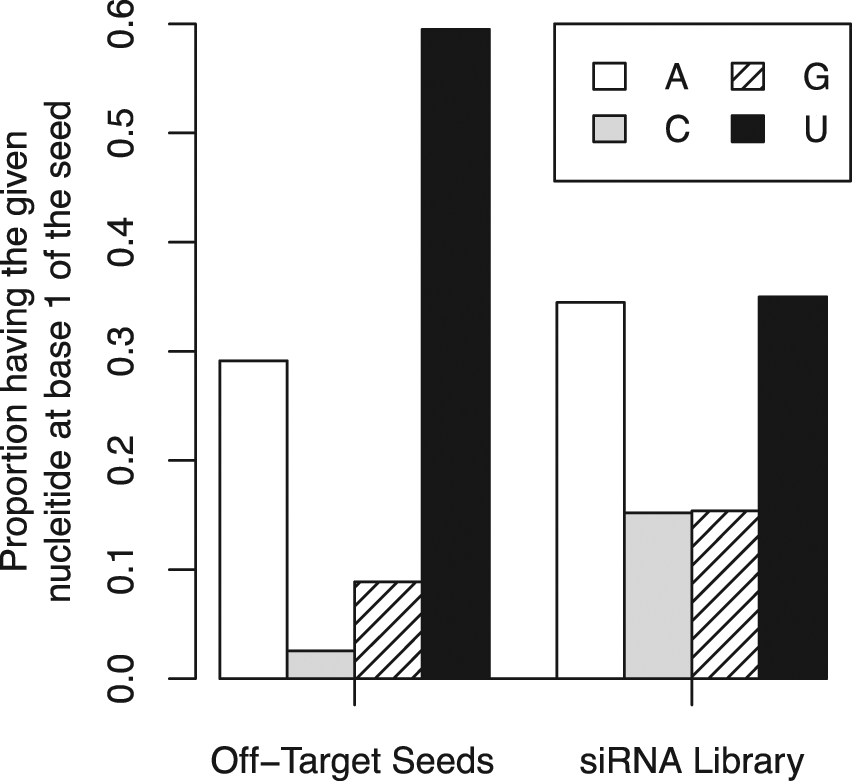
Bias in seed composition. Comparison of the frequency of each nucleotide at base 1 of the seed sequence for seeds identified as having a statistically significant effect on the assay versus the entire siRNA library.
Cross-Assay Conservation of Off-Target Seeds
Because there is significant cross-talk between different biological pathways, we might expect that seeds that have an effect on one assay might also effect other assays. If these promiscuous seeds could be identified, they could be eliminated from future siRNA library designs. To briefly address this issue, we compared the 158 off-target hexamer seeds identified in our assay with 17 hexamer seeds identified by Sudbery et al. 9 as being overrepresented in hits for a different assay of a different pathway. There was only one hexamer sequence in common between the two sets, ACUUGA, which is approximately the frequency of overlap we would expect by chance. Although this small comparison is insufficient to rule out the existence of seeds that effect a broad range of assays, we do not yet see evidence to support this conjecture.
Discussion
The ability of large-scale siRNA screens to successfully identify new targets and elucidate the components of biological pathways clearly hinges on appropriate interpretation of the data. Although the development of improved statistical methods coupled with the testing of additional siRNAs has certainly been a logical step toward minimizing off-target effects in siRNA screens, a more comprehensive approach incorporating what is known regarding the specific nature of off-target effects is presented here. CSA incorporates the sequence-driven concept of hexamer seed-region matches dictating the activity of many siRNAs in the context of a screen.
Given the high incidence of seed-based off-target effects in siRNA screens, integration of CSA with existing methodologies such as seed complement frequency (SCF) 10 is a reasonable strategy. In Anderson et al., 10 the authors concluded that the frequency of a particular seed sequence in the 3′UTR transcriptome explains variations in off-target signature size between different siRNAs targeting the same gene. However, despite preferentially selecting siRNAs with low seed complement frequencies, some off-target effects are still likely to be present. Whereas SCF provides a framework for selecting siRNAs less likely to have a large off-target signature, CSA makes it possible to detect off-target effects when they do occur and eliminate them from the analysis. Thus, it is logical to combine SCF and CSA methodologies to reduce the influence of off-target effects by addressing multiple aspects of siRNA screening including sequence design, off-target detection, and visualization.
Because of the preponderance of off-target hits relative to true positives, incorporation of seed-region sequence information on the scale of a whole-genome siRNA screen will likely lead to a significant reduction in false-positive rates. A reduction in off-target hit rates during siRNA high-throughput screening would not only bring forward genes that are less likely to be assay artifacts but also reduce the time and resources required to follow up target identification screens in general as the success rate would be significantly greater. However, the incorporation of this strategy would necessitate changes to our standard workflow. Although primary screens are conducted in pools of three siRNAs targeting the same gene, deconvolution of hits to single siRNAs is necessary to detect seed-based off-target effects with maximal efficiency. Once the three-component siRNA singles have been deconvoluted and retested in the assay at hand, a large number of seed regions prove themselves to be nonspecific (see examples in Fig. 4 ), and siRNAs containing these seeds must be eliminated from consideration as “on-target” hits. As a result, following the removal of siRNAs with promiscuous seed regions from consideration, we are faced with a problem of insufficient data to make an activity determination for a large number of genes tested in the assay. To address this problem, we propose expansion of testing to include additional siRNA duplexes targeting each gene that was deemed a hit in the primary screen. Although this workflow alteration increases the amount of experimental work required, it should result in a significantly better proportion of on-target hits resulting from siRNA screens.
In addition to changing the typical workflow of siRNA screens, the implementation of CSA leads to further examination of the libraries used. To increase the ability of CSA to detect promiscuous seeds, libraries need to be designed with a balanced seed composition. This change would provide the opportunity for more seeds to be deemed promiscuous (or not) with sufficient statistical power due to the number of examples upon which its seed-specific activity can be assessed. To maximize the power of siRNA screening-based approaches, considerations such as these should help shape future siRNA library design efforts.
Finally, although the focus of this article was defining a method of off-target detection appropriate for screens conducted in pools and then confirmed in component singles, our results have implications for the economy of screening using pooled siRNAs. Figures 1B and 1C (pools versus pools and singles versus singles, respectively) show little difference in the correlation of results. Because screening in pools does not appear to generate more reproducible results than screening in singles, and detection of off-target effects would arguably be more sensitive when screening in singles, it is unclear what advantage screening in pools has over screening in singles. We hope to more closely examine this issue in future work.
As genome-scale siRNA screens continue to become a more common approach to both new target identification and pathway elucidation, consideration of seed region–based off-target effects is critical. The CSA methodology shown here provides a means for doing so, in a manner that only moderately alters existing siRNA screening workflows. Furthermore, because of the large amount of effort currently being expended on siRNA-based approaches in general, implementation of CSA as an effective means for mitigating off-target effects in large-scale experiments will improve success rates and accelerate progress in the field.
Footnotes
Acknowledgements
The authors wish to thank J. Burchard for discussions on RNAi off-target effects, E. Hudak and R. Liehr for assistance with compound management, A. Kreamer for data upload, C. Wang for performing screening experiments, C. Ohart for robotic support, B. Major (University of Washington) and R. Moon (University of Washington) for cell line development, and B. Roberts, W. Arthur, E. Smith, N. Chung, and M. Cleary for assay development.
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
The authors received no financial support for the research, authorship, and/or publication of this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.