
Abstract
G-protein–coupled receptors (GPCRs) are pivotal in cellular responses to the environment and are common drug targets. Identification of selective small molecules acting on single GPCRs is complicated by the shared machinery coupling signal transduction to physiology. Here, we demonstrate a high-content screen using a panel of GPCR assays to identify receptor selective molecules acting within the kinase/phosphatase inhibitor family. A collection of 88 kinase and phosphatase inhibitors was screened against seven agonist-induced GPCR internalization cell models as well as transferrin uptake in human embryonic kidney cells. Molecules acting on a single receptor were identified through excluding pan-specific compounds affecting housekeeping endocytosis or disrupting internalization of multiple receptors. We identified compounds acting on a sole GPCR from activities in a broad range of chemical structures that could not be easily sorted by conventional means. Selective analysis can therefore rapidly select compounds selectively affecting GPCR activity with specificity to one receptor class through high-content screening.
Keywords
Introduction
Phenotypic screens exploit automated cell imaging to identify small molecules that can alter cellular protein localization and are of increasing interest as high-throughput and forward genetic screens for drug and pathway component identification. 1 In chemical genetics, small molecules active toward cell-based assays are identified through cell-based screening with little or no a priori information on their targets. Identifying these targets remains a challenge,2,3 as does reducing the number of candidates to be tested. Computational methods have been developed to define classes of active molecules relative to their activity toward single or potentially multiple targets and their structure, 4 but a cell-based approach would exploit the assays themselves to select promising candidates.
We present such an alternative: an experimental method to select compounds specifically affecting activation of specific G-protein–coupled receptors (GPCRs). Using a panel of fluorescent GPCR fusion proteins and a collection of small-molecule inhibitors of cellular kinase and phosphatase activity, we constructed a selective exclusion screen. This was designed to identify two classes of compounds: those affecting housekeeping endocytic machinery, or multiple GPCRs (pan-specific), and those affecting only one (GPCR specific) in the same cellular genetic background. The screen was based on measurement of agonist-induced receptor internalization using high-content screening and cellular image analysis.
GPCRs mediate signal transduction and physiological homeostasis in systems as diverse as blood pressure regulation, odor recognition, and visual perception. 5 Given their importance as regulators, GPCRs are the target of more than 45% of all prescription drugs,6,7 many of which exploit disruption of receptor-ligand interactions using antagonists or artificial agonists.
Agonist-induced internalization is a property of many GPCRs, such as the endothelin A and μ-opioid receptors,8,9 and upon ligand binding, these GPCRs leave the plasma membrane and are internalized. GPCR internalization is physiologically important, for it controls signaling via removal of receptors from the cell surface. Activated receptors are rapidly desensitized from G-proteins through phosphorylation by G-protein receptor kinases and the subsequent recruitment of the arrestin adaptor proteins. 10 Arrestins promote the internalization of the receptor into plasma membrane–derived vesicles, either caveolae or clathrin-coated vesicles, which pinch off into the cell interior to deliver the receptor into the endosomal organelles.10,11 Once internalized, GPCRs have several endocytic itineraries and may be transported for degradation, rapidly recycled to the cell surface, or resensitized and recycled from the pericentriolar recycling compartment. Receptor location is likely to be important for cell responses and is suited to automated cell-based high-content screening.
We demonstrate the usefulness of a selective screen for selecting small molecules acting on a single GPCR pathway ( Fig. 1A ). Starting from seven GPCR cell lines and 88 molecules, less than 10% of the molecules affected GPCR internalization without affecting housekeeping endocytosis, defined by transferrin internalization or pan-specific effects on multiple receptors. In this selected set, single molecules were identified that affected only the muscarinic acetylcholine receptor 2 (KN-93) and not other GPCRs in the panel. This selective high-content screen provided a rapid means to pare down potential candidates through exploiting the cell biology of GPCRs and also adopting the genetic principle of a selective screen.
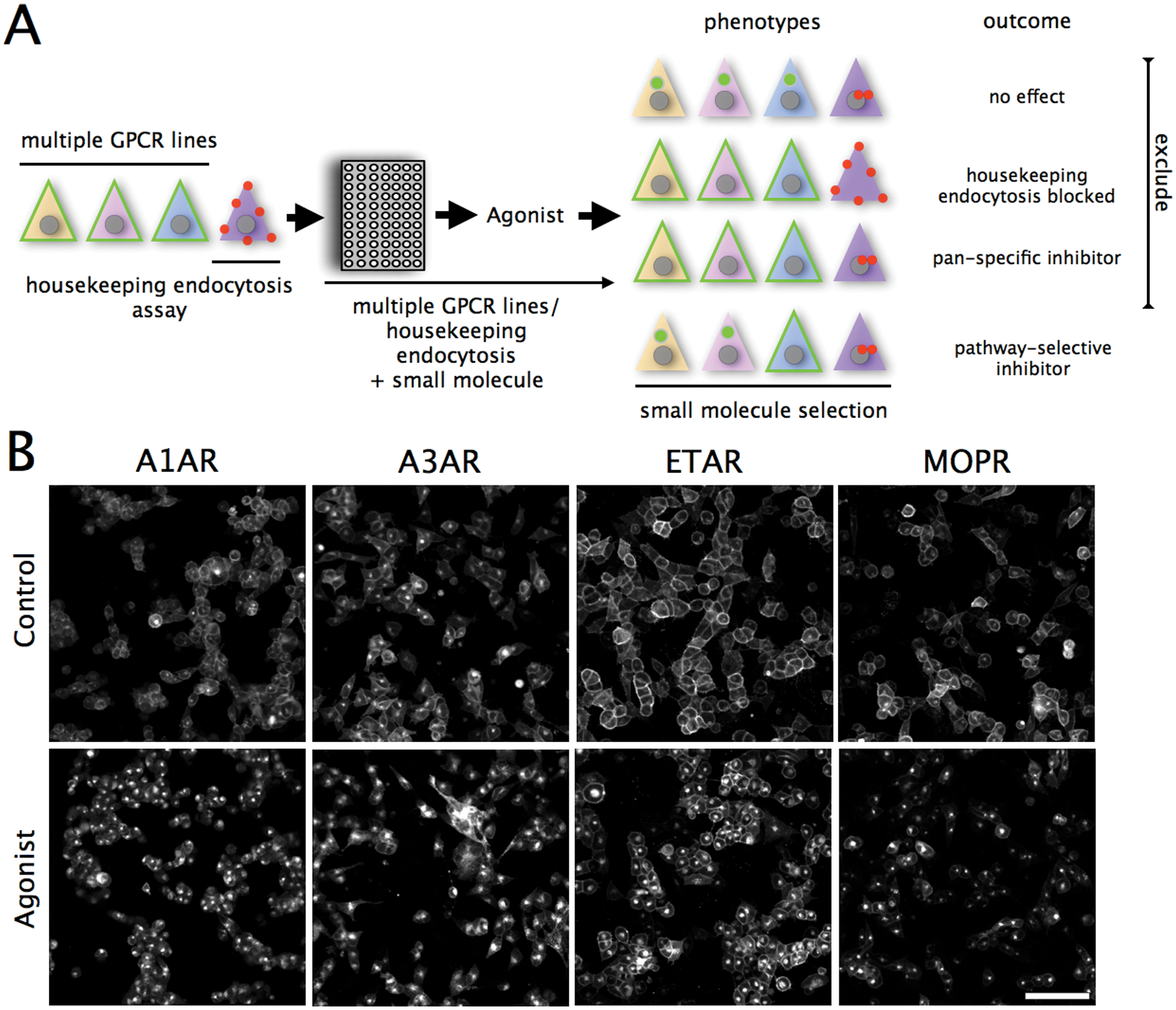
(A) Compound subtractive screen scheme. Cell lines expressing G-protein–coupled receptors (GPCRs) are treated, stimulated with small molecules, stimulated with agonist, imaged, and receptor internalization quantified by image analysis. Phenotypes are classified as having no effect, blocking housekeeping endocytosis (transferrin uptake), and affecting multiple or single GCPRS. Selective molecules are defined as not affecting housekeeping endocytosis, or more than one GPCR; all others are excluded as pan-specific. (B) Agonist-induced GPCR-EGFP fusion protein internalization in cell lines expressing the A1 adenosine receptor (A1AR), A3 adenosine receptor (A3AR), endothelin A receptor (ETAR), or the μ-opioid receptor (MOPR). To demonstrate internalization, cell lines were serum starved for 120 min then stimulated with agonist or DMSO for 60 min and imaged. Scale bar = 50 μm.
Materials and Methods
Chemicals
All fine chemicals were purchased from Sigma-Aldrich (St. Louis, MO). Fluorophores and their reactive forms were purchased from Molecular Probes (Eugene, OR). DRAQ5 was from BioStatus (Shepshed, UK). Kinase and phosphatase inhibitors were purchased as 95% to 99% pure 10 mM stock solutions in dimethylsulfoxide or water (Biomol, Hamburg, Germany). Stock solutions and formatted assay plates were stored at −20°C.
Cell Lines and Cell Culture
Human embryonic kidney (HEK) 293 cells were cultivated in Dulbecco’s modified eagle’s medium/F12 (Invitrogen, Carlsbad, CA) supplemented with 10% fetal calf serum, 1% penicillin streptomycin, and 1% geneticin. GPCR receptor cDNA clones (cDNA.org) were fused to the EGFP coding sequence of the pEGFP-N1 vector (Clontech, Palo Alto, CA) and ligated in the pCDNA3.1 (–) vector (ADORA1:NM_000674, ADORA3:NM_000677, EDNRA:AY275462, OPRK1:NM_00912.3, OPRM1:NM_000914.3, CHRM2:NM_000739.2, CHRM5:NM_012125.3). The resulting GPCR-EGFP DNA was cloned, sequence verified, and used for transfection of HEK293 cells using standard protocols. Each recombinant clone was obtained through several cycles of selection and limiting dilutions. For screening, cells were passed onto fibronectin-coated coverslip-bottomed 96-well plates (Greiner, Longwood, CO) at a density of 3 × 104 cells/well, 24 h in advance.
Cell Imaging
Cell-based assays were imaged using automated four-color excitation/emission point scanning confocal systems (ImageXpress ULTRA, Molecular Devices, Sunnyvale, CA) with a 40× ELWD 0.6 NA Nikon objective. Images were analyzed using metaXpress and Acuity express (Molecular Devices). Typically, we acquired four images per well/plate.
Cell-Based Screening
Compounds in DMSO or H2O were diluted into 1% serum 1 mM HEPES pH 7.4 buffered media at the working concentration (0.1 nM to 10 μM) and warmed to 37 °C prior to screening.
For the GPCR internalization assay, recombinant HEK cells cultivated in fibronectin-coated 96-well plates were washed in phosphate-buffered saline supplemented with calcium and magnesium. The cells were then serum deprived and compound treated by incubation for 120 min with 10 μM compounds in low serum (1% fetal bovine serum) assay medium at 37 °C, 5% CO2. The compound solutions were then supplemented with agonist while maintaining compound concentration at 10 μM. The cells were further incubated for 60 min at 37 °C, 5% CO2.
Poststimulation, cells were washed twice with ice-cold phosphate-buffered saline supplemented with calcium and magnesium and fixed in 4% (w/v) paraformaldehyde in phosphate-buffered saline supplemented with calcium and magnesium for 10 min. Cells were washed twice with phosphate-buffered saline supplemented with calcium and magnesium and stained with 2.5 μM DRAQ5 (Biostatus, Leicestershire, UK) prior to image acquisition.
Agonist EC50 titrations for each GPCR line were performed on serum-deprived cells, as above.
Transferrin internalization was measured after 120 min preincubation of wild-type HEK293 cells with compounds and 10 μM Syto42, followed by the addition of 100 μg/mL Alexa-633 transferrin in 1% serum assay medium and 60-min incubation at 37 °C and 5% CO2. Postinternalization, cells were washed and fixed as above without the addition of DRAQ5 prior to imaging.
Curve Fitting and Analysis
Agonist-induced GPCR internalization was analyzed using algorithms that recognize cell boundaries, segment the cells, and perform cell-by-cell determination of the number, size, and intensity of internal fluorescent compartments. These algorithms are commercially available and run in the MetaXpress environment (Molecular Devices). Measurements were expressed as the mean and standard deviation for all cells in four images per well, per treatment, and exported to a spreadsheet. We selected the measurement output for each GPCR that gave the highest goodness of fit for agonist EC50 titrations, as determined by R2 as the metric for that assay.
Image analysis data were analyzed using PRISM (Graphpad, La Jolla, CA). Curve fitting was performed using sigmoidal dose-response fitting of triplicate titrations.
Results and Discussion
Our rationale to screen for molecules affecting a single GPCR pathway was to test a kinase/phosphatase inhibitor collection against seven GPCR models and then select compounds selectively affecting GPCR(s). The selection scheme is shown in Figure 1A and exploited the principle of first eliminating compounds with no effect, then those disturbing endocytosis, and finally excluding pan-specific molecules affecting more than one GPCR.
To evaluate this approach, we first constructed and validated cell lines expressing fluorescent GPCR fusion proteins to the enhanced green fluorescent protein (
Fig. 1B
;
To identify kinase/phosphatase inhibitors disrupting GPCR internalization, each of the panel of GPCR-GFP cell lines was screened against a collection of 88 compounds and trafficking inhibitors (
Fig. 2A
;
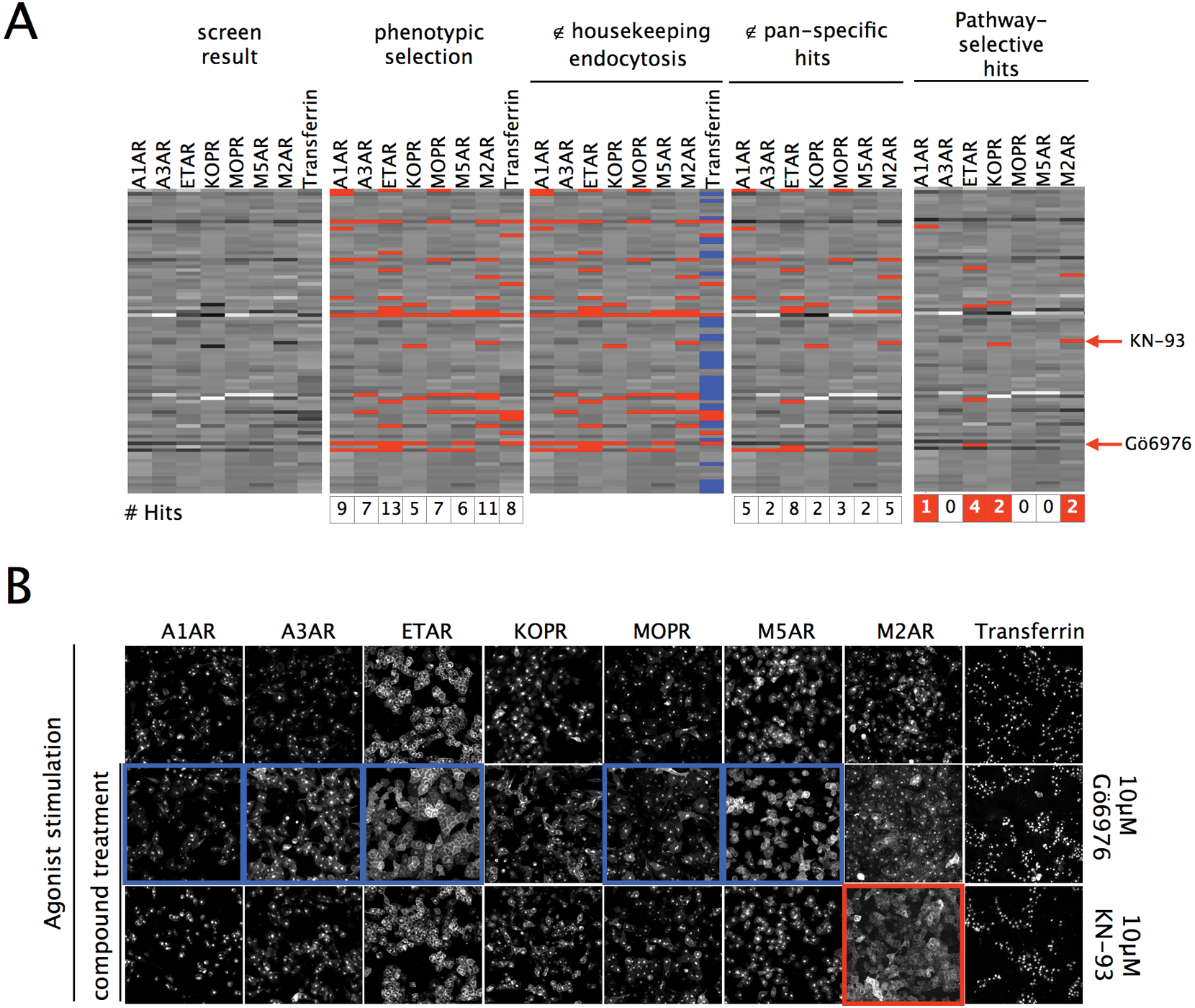
(A) Seven stable cell lines expressing G-protein–coupled receptor (GPCR)–green fluorescent protein (GFP) fusion proteins to each of the following GPCRs: A1AR, A3AR, ETAR, KOPR, MOPR, M5AR, and M2AR, and an assay of wild-type cell transferrin uptake was treated with a collection of 88 kinase/phosphatase inhibitors then agonist stimulated. Data were normalized, center reduced, and plotted against a linear scale from black (minimum) to white (maximum). Hits affecting GPCRs were defined as greater than two standard deviations from the mean (phenotypic selection: red overlay), and any hits also affecting transferrin (>0.6 standard deviations, blue overlay panel) were excluded (∉ housekeeping endocytosis panel). Molecules affecting more than one GPCR were excluded (∉pan-specific hits) to identify molecules affecting single GPCRs (pathway selective hits). The number of hits in each step of the subtractive screen is indicated at the base of the screening panels. The pan-specific hit Gö6976 (∉) and the M2AR-specific hit KN-93 (∉) are indicated. (B) Confocal images of the effects of a pan-specific compound Gö6976 and a selected M2AR-specific compound KN-93 are shown for the entire assay panel after agonist stimulation. Neither molecule affected housekeeping endocytosis (right panel). Gö6976 is defined as pan-specific because it affected more than half of the assays (blue), whereas the selected molecule KN-93 affected only internalization of the M2AR (red). Scale bar = 50 μm.
Molecules affecting GPCRs were selected using two criteria: the first selection step removed molecules affecting the housekeeping transferrin pathway by excluding any hit from the screen that altered the transferrin assay by >0.6 standard deviations; this excluded >65% of the hits for each GPCR assay, for example, for the A3AR ( Fig. 2A ). The second round then eliminated pan-specific molecules affecting more than one GPCR ( Fig. 1B , final panel) to ultimately identify those acting on a single receptor. GPCR-specific molecules were identified acting on the M2AR (KN-93, damnacanthal, AG-879), ETAR (wortmannin), and KOPR (ML-7); pan-specific molecules included the protein kinase C inhibitor Gö6976 (A1AR, ETAR, MOPR, M5AR). Pan-specific effects on more than three GPCR assays strongly correlated with effects on transferrin internalization.
This analysis identified the majority of hits for individual GPCR high-content screening as nonspecific/pan-specific. The subtractive screen selectively reduced the hits by excluding >90% overall and reduced the mean hit rate per assay to about 1%. Molecules affecting single assays were found for five of eight assays examined (four GPCRs and transferrin) based on their phenotype, as shown in
Figure 2
, for an example pan-specific inhibitor (Gö6976) as compared with a molecule acting on the M2AR alone (KN93). Analysis showed that >90% of molecules affecting a single pathway also affected other assays, as shown for molecules affecting >3 GPCRs that disrupted transferrin uptake in >85% of cases. This yielded 10% of screened hits as active toward a single assay. Single compound structures acting on sole pathways were identified for the muscarinic acetyl choline receptor 2 (KN93). KN-93 is a CAM kinase II inhibitor and arrested muscarinic acetyl choline receptor 2 at the cell surface but did not affect the closely related muscarinic acetyl choline receptor 5. To exclude antagonism of the receptor-agonist interaction, we determined the half maximally inhibitory concentration (IC50) as 0.6 μM for KN-93 acting on the M2AR stimulated with 100 μM carbachol (
Together, these data demonstrate that although screens on one receptor target may identify many molecules, most of these are pan-specific, but these can be efficiently sorted into specific versus pan-specific hits through a subtractive screen examining multiple GPCR responses.
Phenotypic screening has the advantage that small molecules can be identified that perturb cell phenotypes with little or no a priori information on their targets or mode of action. 2 High-content screening13,14 has been applied to several cell pathways such as p53 signaling, 15 neurite outgrowth, 16 TGF signaling, 17 and correction of the cystic fibrosis defect. 18 High-content screens often operate on single pathways, but screening results are convolved by off-target side effects of compounds that are therapeutically undesirable.
Here, we demonstrated how molecules could be selected that act on a sole GPCR in a high-content screen. The rationale applied the principle of a subtractive screen, as used in classical genetics, to high-content compound screening. By excluding compounds affecting multiple GPCRs, a handful of compounds were identified affecting specific single GPCRs with no a priori knowledge on the compound-receptor interaction.
To demonstrate the usefulness of this screening rationale ( Fig. 1A ), a panel of cell-based assays was screened against a collection of 88 kinase and phosphatase inhibitors using high-content image analysis. A screening strategy based on the exclusion of molecules that act on housekeeping pathways or similar pathways permits the removal of those molecules with the highest likelihood of pan-specific modes of action. “Hits” were identified as those that affected one GPCR without affecting transferrin uptake. When the assays are all performed in the same cell background, where each molecule acts against the same set of proteins, the assay itself acts as a discriminate filter, for it defines where these interact with a given set of signaling pathways.
Classical phenotypic screens examine compound effects on one cell-based assay, and the data indicate that secondary screening hits against multiple biologically related assays greatly increases the likelihood that hits are selective and do not affect housekeeping or common machinery shared between GPCRs.
GPCR biology provided a set of similar assays that permit correlated screening to identify hits. Given the similarity of the assays analyzed here and the low number of molecules, using a subtractive screen was a metric for isolating the most selective molecules from screening experiments, with the highest relative activity toward the biological pathways resolved by any single given assay. The majority of the molecules identified in single screens (greater than 90%) affected either basic endocytic housekeeping or were pan-specific (e.g., Gö6976). This indicated that subtractive screening greatly increases the potential value of the molecules selected.
Using existing kinase and phosphatase inhibitors created a challenging test case for this analysis: these molecules presumably act on the same protein pathways in each assay studied for they are exposed to the same cellular background (HEK cells) and have not been chemically optimized for selectivity toward an assay, so they have pleiotropic effects. Surprisingly, we did find compound selectivity for half of the assays when they were compared with the entire panel tested.
This approach is of interest for drug discovery using high-content screening because unbiased small-molecule selection will result in the isolation of candidate molecules that indirectly affect several pathways. Combining several assays with subtractive secondary screening provided a rapid approach for compound selection and furthermore for structural optimization from a confined set of starting compounds, an approach that may benefit drug development.
Pathway-selective analysis may have applications in the field of phenotypic chemical genetic screening, as a method for isolating the most pathway-specific molecules from cell-based screening. This may improve the identification of small-molecule targets from phenotypic screens by significantly narrowing the number of molecules to be examined and increasing the efficiency of high-content screening campaigns.
Footnotes
Abbreviations: A1AR, adenosine receptor 1; A3AR, adenosine receptor 3; ETAR, endothelin A receptor; Et-1, endothelin-1; kOPr, kappa opioid receptor; muOPr, μ-opioid receptor; M2AR, muscarinic acetylcholine receptor 2; M5AR, muscarinic acetylcholine receptor.
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korean government (MEST; No. 2011-00244), Gyeonggi-do, KISTI, and by parliamentary grant and Department of Science and Technology support in South Africa (to N.E.).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.